
Determination of N-Carbamylglutamate in Feeds and Animal Products by High Performance Liquid Chromatography Tandem Mass Spectrometry

Abstract
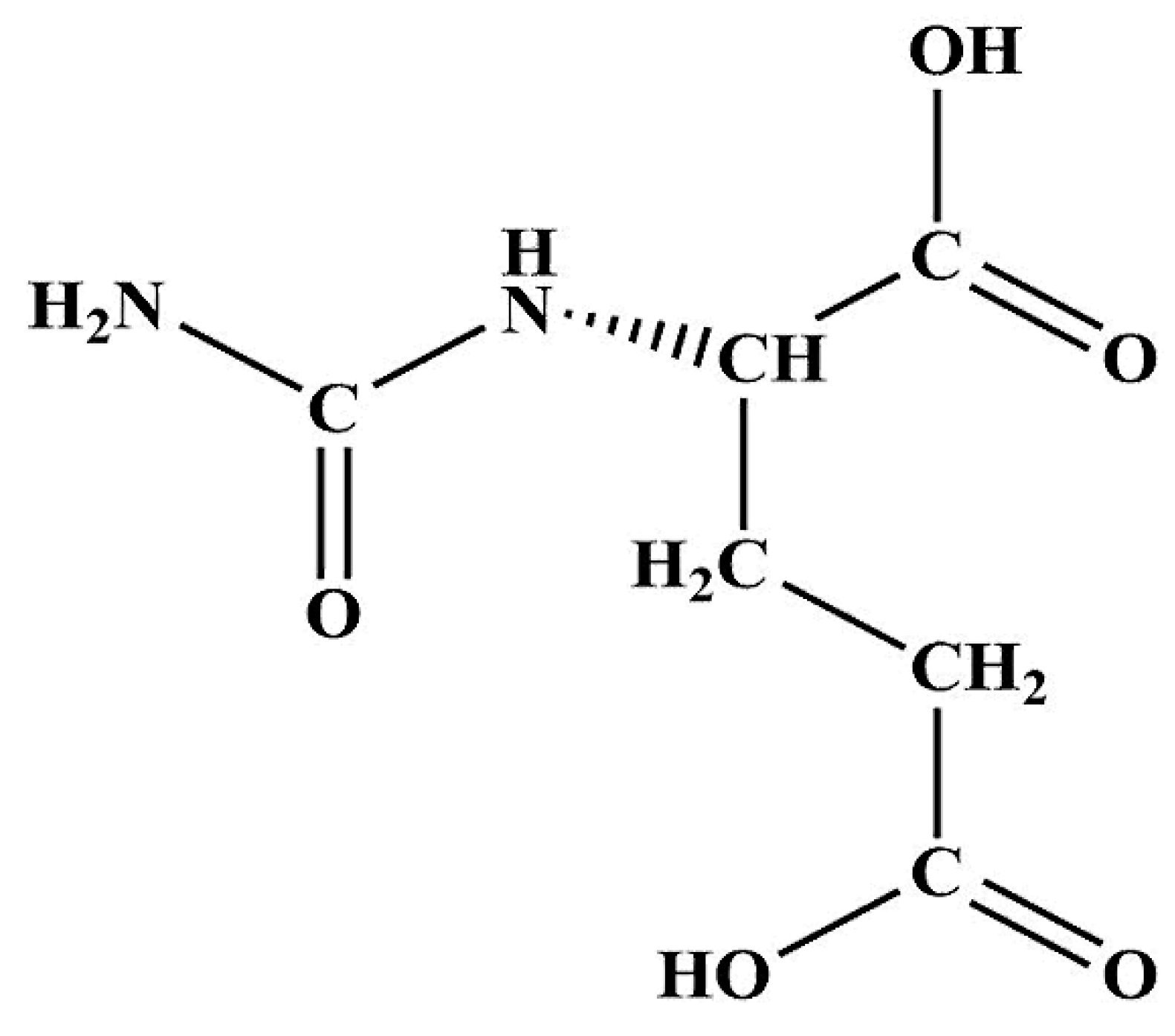
:1. Introduction
2. Results and Discussion
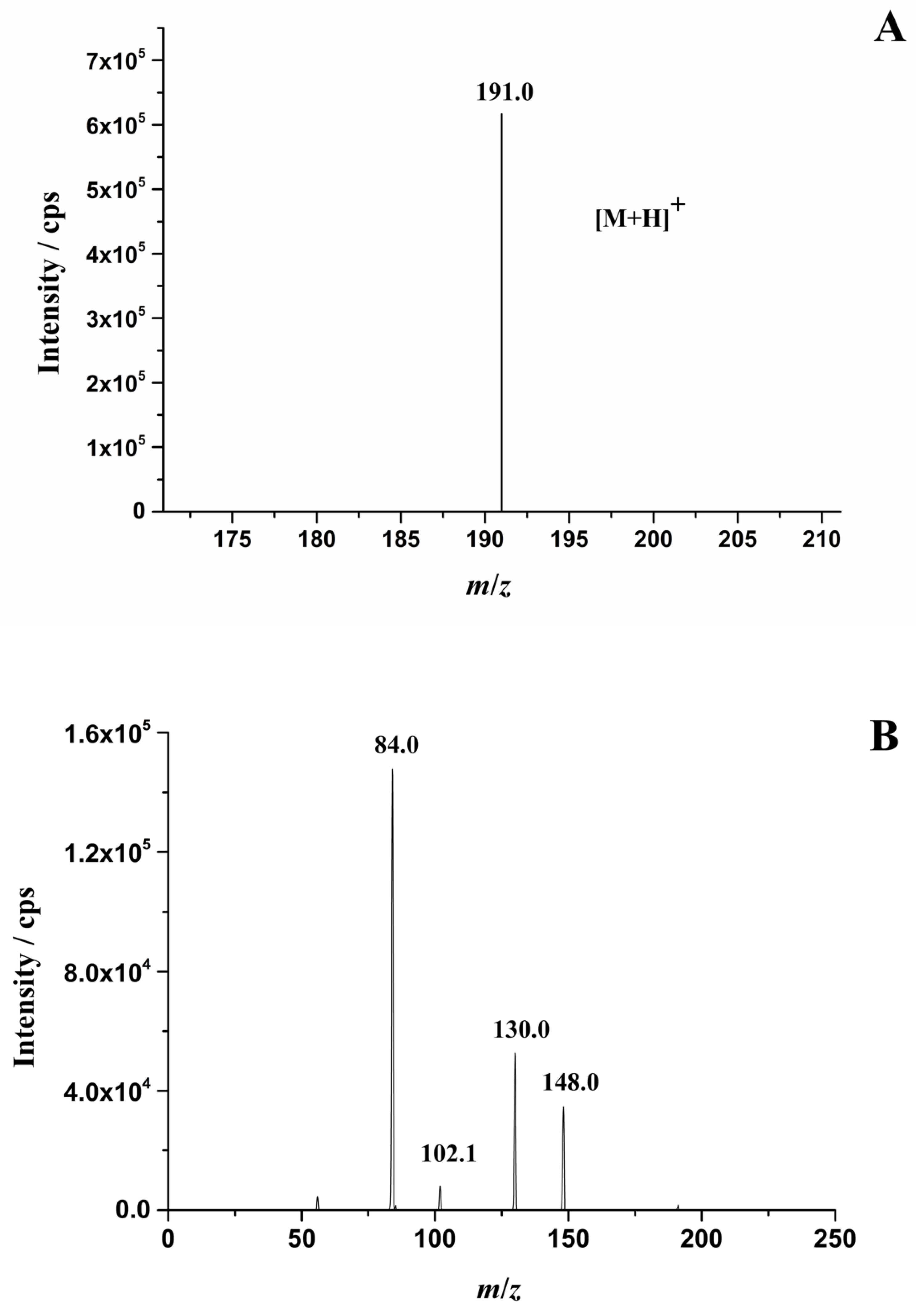
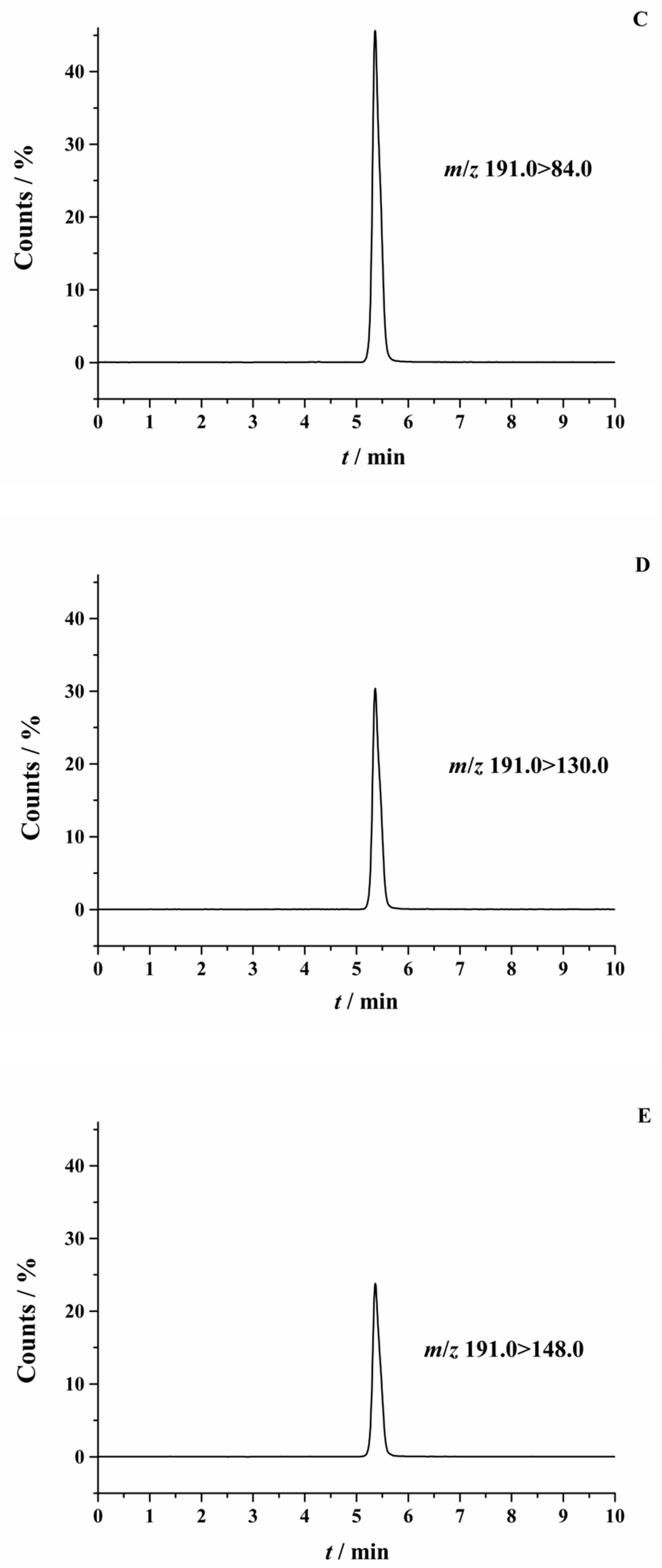
2.1. Optimization of HPLC-MS/MS Conditions
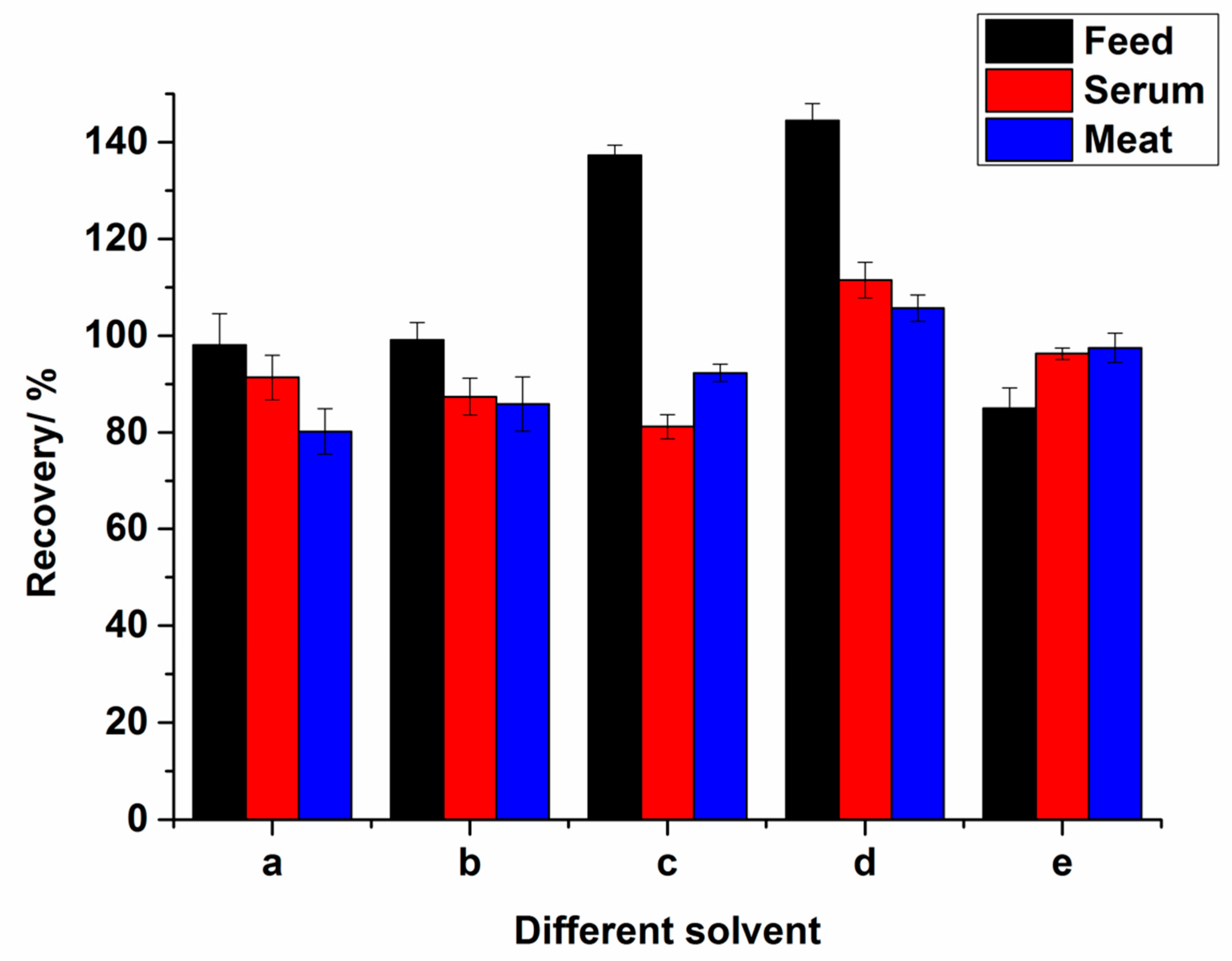
2.2. Optimization of Extract Condition
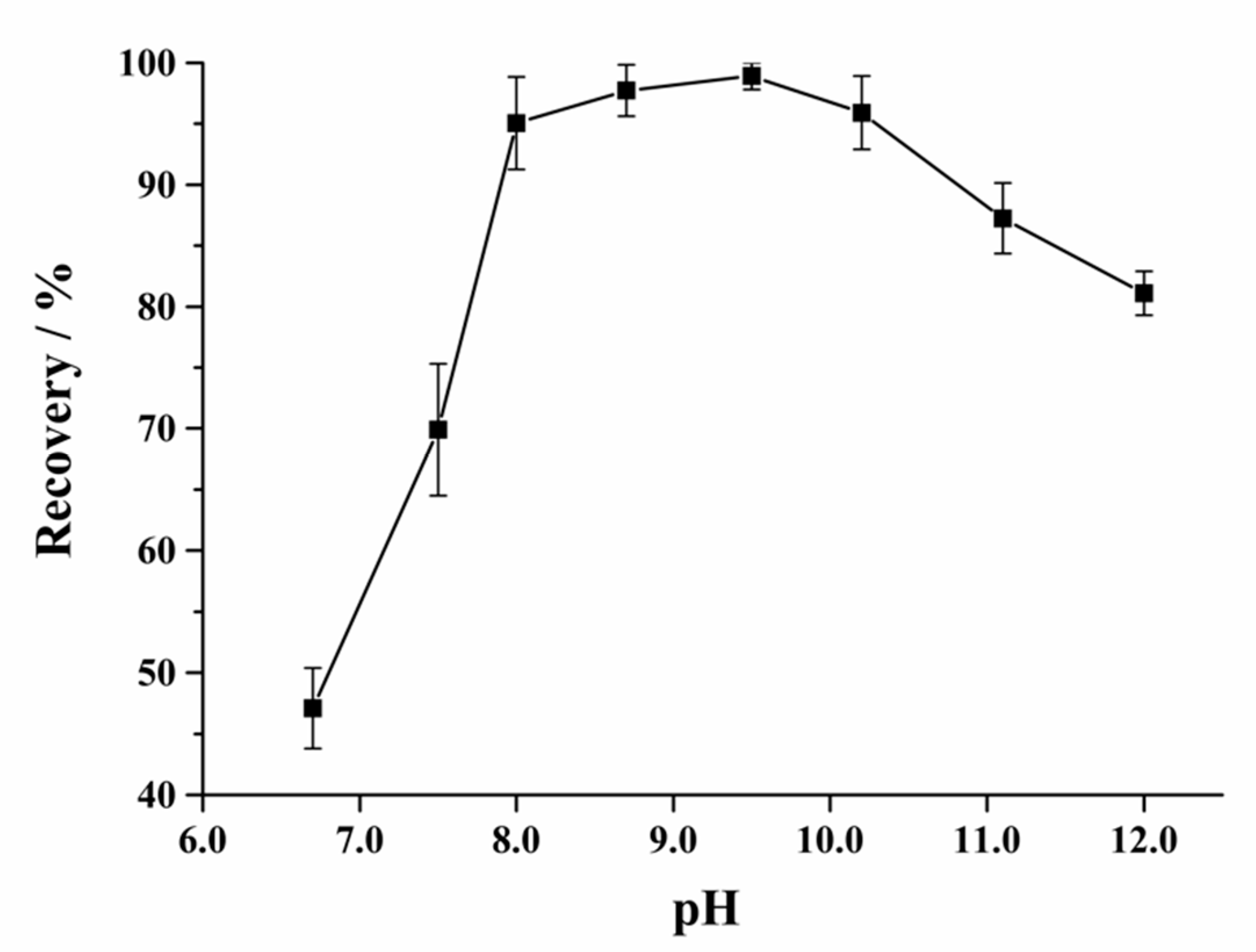
2.3. Optimization of Purification Condition
2.4. Method Validation
2.4.1. Stability and Matrix Effect
2.4.2. Linearity, Precision, Accuracy, LOQ and LOD
2.5. The Analysis of Authentic Samples
3. Materials and Methods
3.1. Materials and Reagents
3.2. Instruments and Apparatus
3.3. Sample Preparation
3.4. Sample Extraction and Purification
3.5. HPLC-MS/MS Conditions
3.6. The Method Validation
3.7. The Analysis of Authentic Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wu, G.; Knabe, D.A.; Kim, S.W. Arginine nutrition in neonatal pigs. J. Nutr. 2004, 134. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.S.; Shen, Z.A.; Barnett, J.F.; Krsmanovic, L.; Myhre, A.; Delaney, B. N-acetyl-glutamic acid: Evaluation of acute and 28-day repeated dose oral toxicity and genotoxicity. Food Chem. Toxicol. 2009, 47, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wan, D.; Xie, C.; Li, T.; Huang, R.; Shu, X.; Ruan, Z.; Deng, Z.; Yin, Y. Acute and sub-acute oral toxicological evaluations and mutagenicity of N-carbamylglutamate (NCG). Regul. Toxicol. Pharmacol. 2015, 73, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Schubiger, G.; Bachmann, C.; Barben, P.; Colombo, J.P.; Tönz, O.; Schüpbach, D. N-Acetylglutamate synthetase deficiency: Diagnosis, management and follow-up of a rare disorder of ammonia detoxication. Eur. J. Pediatr. 1991, 150, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Bachmann, C.; Wermuth, B.; Bordugo, A.; Ferrari, V.; Colombo, J.P.; Zacchello, F. Partial N-acetylglutamate synthetase deficiency: A new case with uncontrollable movement disorders. J. Inherit. Metab. Dis. 1992, 15, 395–398. [Google Scholar] [CrossRef]
- Gebhardt, B.; Vlaho, S.; Fischer, D.; Sewell, A.; Böhles, H. N-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduria. Mol. Genet. Metab. 2003, 79, 303–304. [Google Scholar] [CrossRef]
- Gebhardt, B.; Dittrich, S.; Parbel, S.; Vlaho, S.; Matsika, O.; Bohles, H. N-Carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemia. J. Inherit. Metab. Dis. 2005, 28, 241–244. [Google Scholar] [CrossRef]
- Kalkan, U.S.; Coker, M.; Habif, S.; Ulas, E.S.; Karapinar, B.; Ucar, H.; Kitis, O.; Duran, M. The first use of N-carbamylglutamate in a patient with decompensated maple syrup urine disease. Metab. Brain. Dis. 2009, 28, 241–244. [Google Scholar] [CrossRef]
- Wu, G.; Bazer, F.W.; Davis, T.A.; Jaeger, L.A.; Johnson, G.A.; Kim, S.W.; Knabe, D.A.; Meininger, C.J.; Spencer, T.E.; Yin, Y.L. Important roles for the arginine family of amino acids in swine nutrition and production. Livest. Sci. 2007, 112, 8–22. [Google Scholar] [CrossRef]
- Liu, X.; Wu, X.; Yin, Y.; Liu, Y.; Geng, M.; Yang, H.; Blachier, F.; Wu, G. Effects of dietary l-arginine or N-carbamylglutamate supplementation during late gestation of sows on the miR-15b/16, miR-221/222, VEGFA and eNOS expression in umbilical vein. Amino Acids. 2012, 42, 2111–2119. [Google Scholar] [CrossRef]
- Zhang, B.; Che, L.; Lin, Y.; Zhuo, Y.; Fang, Z.; Xu, S.; Song, J.; Wang, Y.; Liu, Y.; Wang, P.; et al. Effect of dietary N-carbamylglutamate levels on reproductive performance of gilts. Reprod Domest. Anim. 2014, 49, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xiao, L.; Cao, W.; Fang, T.; Jia, G.; Chen, X.; Zhao, H.; Wu, C.; Wang, J. Changes in the metabolome of rats after exposure to arginine and N-carbamylglutamate in combination with diquat, a compound that causes oxidative stress, assessed by 1H NMR spectroscopy. Food Funct. 2016, 7, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Schütz, L.; Morrell, B.; Perego, M.C.; Spicer, L. Effects of N-carbamylglutamate and L-arginine on steroidogenesis and gene expression in bovine granulosa cells. Anim. Reprod. Sci. 2018, 188, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, L.; Wang, Z.; Deng, M.; Nie, H.; Zhang, G.; Ma, T.; Wang, F. N-carbamylglutamate and L-arginine improved maternal and placental development in underfed ewes. Reproduction 2016, 151, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, R.; Meng, C.; Ji, Y.; Sun, L.; Nie, H.; Mao, D.; Wang, F. Effects of dietary supplementation of N-Carbamylglutamate on lactation performance of lactating goats and growth performance of their suckling kidlets. Small Rumin. Res. 2019, 175, 142–148. [Google Scholar] [CrossRef]
- Hu, Y.; Shao, D.; Wang, Q.; Xiao, Y.; Zhao, X.; Shen, Y.; Zhang, S.; Tong, H.; Shi, S. Effects of dietary N-carbamylglutamate supplementation on growth performance, tissue development and blood parameters of yellow-feather broilers. Poult. Sci. 2019, 98, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Wu, C.-L.; Wan, K.; Zhang, M.; Wu, X. Synthesis, Crystal Structure and Spectroscopic Properties of Zn(II) with N-carbamylglutamate Ligand. Chin. J. Struc. Chem. 2015, 34, 423–427. [Google Scholar]
- Tang, S.; Wang, Y.; Wen, P.; Xin, Z. Determination of N-carbamyl-L-glutamic acid in feedstuff by high performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Chinese J. Chromatogr. 2014, 32, 184–188. [Google Scholar] [CrossRef]
- Liu, R.; Hei, W.; He, P.; Li, Z. Simultaneous determination of fifteen illegal dyes in animal feeds and poultry products by ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2011, 879, 2416–2422. [Google Scholar] [CrossRef]
- Yin, B.; Li, T.; Zhang, S.; Li, Z.; He, P. Sensitive analysis of 33 free amino acids in serum, milk, and muscle by ultra-high performance liquid chromatography-quadrupole-qrbitrap high resolution mass spectrometry. Food Anal. Methods 2016, 9, 2814–2823. [Google Scholar] [CrossRef]
- Dhondt, L.; Croubels, S.; De Cock, P.; De Paepe, P.; De Baere, S.; Devreese, M. Development and validation of an ultra-high performance liquid chromatography–tandem mass spectrometry method for the simultaneous determination of iohexol, p-aminohippuric acid and creatinine in porcine and broiler chicken plasma. J. Chromatogr. B 2019, 1117, 77–85. [Google Scholar] [CrossRef] [PubMed]
- De Baere, S.; Devreese, M.; Watteyn, A.; Wyns, H.; Plessers, E.; De Backer, P.; Croubels, S. Development and validation of a liquid chromatography–tandem mass spectrometry method for the quantitative determination of gamithromycin in animal plasma, lung tissue and pulmonary epithelial lining fluid. J. Chromatogr. A. 2015, 1398, 73–82. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrices | Added Concentration (%) | Recoveries (%) | LOD (mg kg−1) | LOQ (mg kg−1) |
|---|---|---|---|---|
| Compound feed | 0.01 | 96.85 (1.3) a | 0.022 | 0.073 |
| 0.05 | 98.89 (2.9) | |||
| 0.1 | 99.24 (5.0) | |||
| Concentrated feed | 0.05 | 107.0 (3.5) | ||
| 0.2 | 106.1 (7.1) | |||
| 1.0 | 97.60 (8.8) | |||
| Premix | 0.5 | 94.27 (6.8) | ||
| 2.0 | 91.90 (3.9) | |||
| 10 | 100.2 (3.6) |
| Matrices | Added Concentration | Recoveries (%) | LOD (μg mL−1) | LOQ (μg mL−1) |
|---|---|---|---|---|
| Serum (μg mL−1) | 0.1 | 94.25(3.8) a | 0.023 | 0.077 |
| 1.0 | 110.2 (2.3) | |||
| 10 | 96.34 (5.1) | |||
| Milk (μg mL−1) | 0.1 | 88.12 (1.8) | 0.014 | 0.047 |
| 1.0 | 102.5 (3.2) | |||
| 10 | 89.58 (4.7) | |||
| Meat (mg kg−1) | 0.05 | 93.84 (7.6) | 0.0038 | 0.013 |
| 0.1 | 101.6 (3.4) | |||
| Kidney (mg kg−1) | 0.05 | 89.14 (3.1) | 0.0069 | 0.023 |
| 0.1 | 93.65 (5.6) | |||
| Liver (mg kg−1) | 0.05 | 94.54 (7.4) | 0.0035 | 0.012 |
| 0.1 | 97.36 (2.9) |
| Sample | Diets Added (%) | NCG Concentration (μg mL−1) | Intra-Day Precision CV (%) | Inter-Day Precision CV (%) |
|---|---|---|---|---|
| Serum b | 0 | ND a | - | - |
| 0.025 | 0.112 | 6.8 | 8.7 | |
| 0.05 | 0.253 | 2.4 | 5.6 | |
| 0.1 | 0.561 | 7.8 | 11 | |
| 0.2 | 1.31 | 12 | 5.1 | |
| Milk | 0 | ND | - | - |
| 20 g/day | 0.0647 | 11 | 7.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Zeng, Z.; Kong, L.; Chen, Y.; He, P. Determination of N-Carbamylglutamate in Feeds and Animal Products by High Performance Liquid Chromatography Tandem Mass Spectrometry. Molecules 2019, 24, 3172. https://doi.org/10.3390/molecules24173172
Ma Y, Zeng Z, Kong L, Chen Y, He P. Determination of N-Carbamylglutamate in Feeds and Animal Products by High Performance Liquid Chromatography Tandem Mass Spectrometry. Molecules. 2019; 24(17):3172. https://doi.org/10.3390/molecules24173172
Chicago/Turabian StyleMa, Yonghang, Zhengcheng Zeng, Lingchang Kong, Yuanxin Chen, and Pingli He. 2019. "Determination of N-Carbamylglutamate in Feeds and Animal Products by High Performance Liquid Chromatography Tandem Mass Spectrometry" Molecules 24, no. 17: 3172. https://doi.org/10.3390/molecules24173172