Simultaneous Quantification and Pharmacokinetic Study of Nine Bioactive Components of Orthosiphon stamineus Benth. Extract in Rat Plasma by UHPLC-MS/MS


 and
and
Abstract
:1. Introduction
2. Results and Discussion
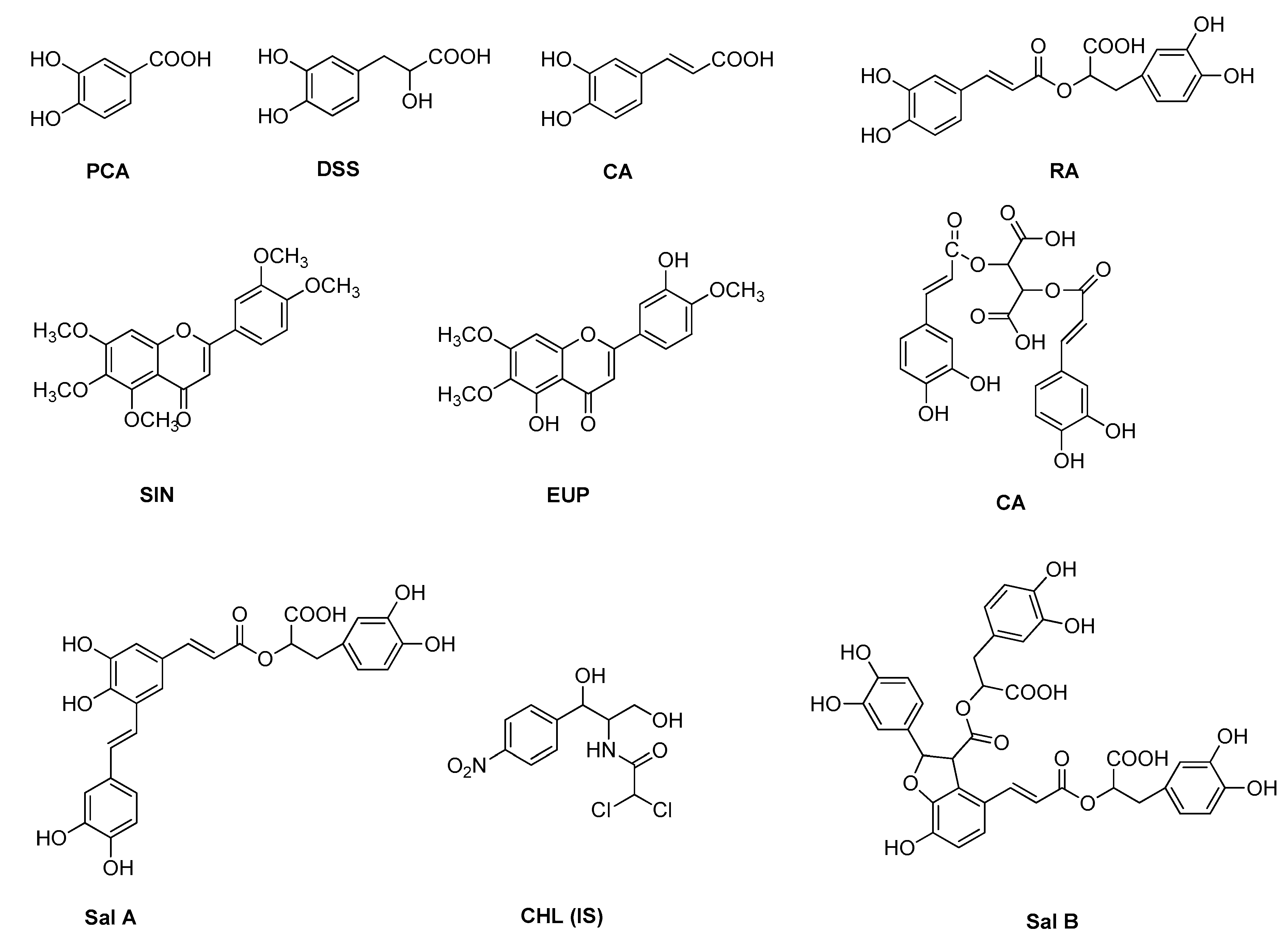
2.1. Method Development
2.2. Method Validation
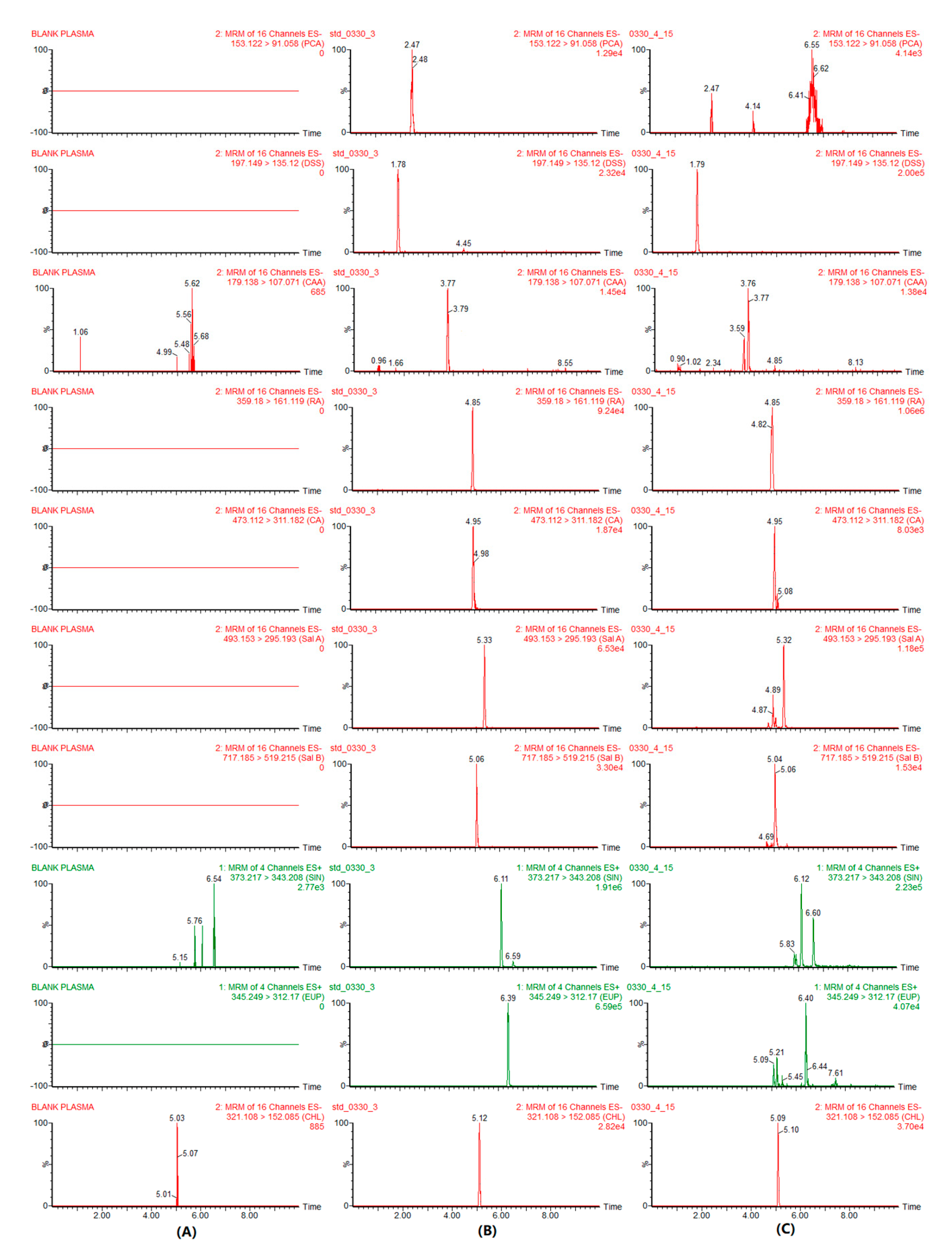
2.2.1. Specificity and Selectivity
2.2.2. Linearity, Precision and Accuracy
2.2.3. Extraction Recoveries and Matrix Effect
2.2.4. Stability
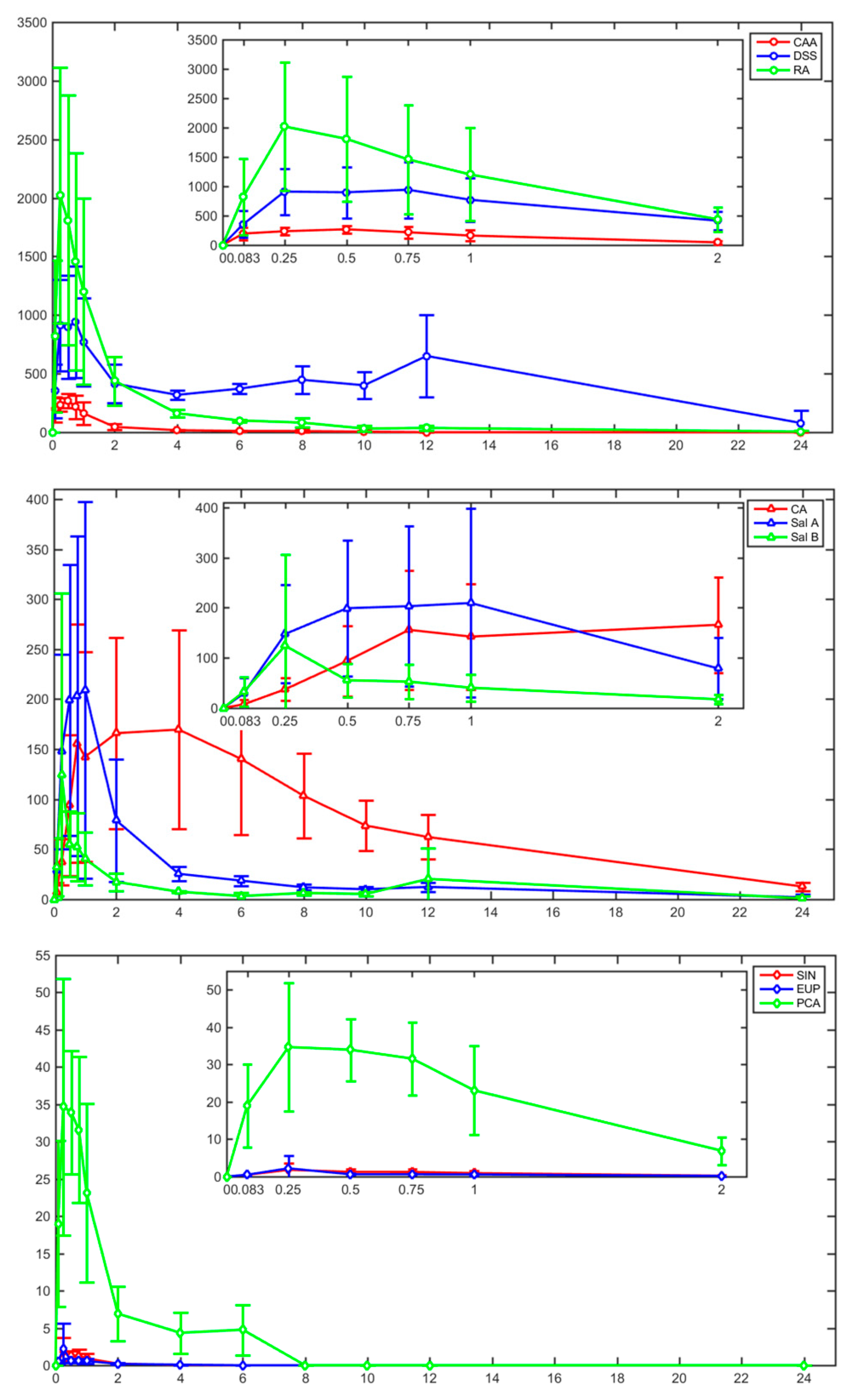
2.3. Pharmacokinetic Study
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of OS Extract
3.3. Equipment and UHPLC-MS/MS Conditions
3.4. Preparation of Calibration Standards and Quality Controls
3.5. Sample Preparation
3.6. Method Validation
3.6.1. Selectivity
3.6.2. Linearity and Lower Limits of Quantification
3.6.3. Precision and Accuracy
3.6.4. Extraction Recovery and Matrix Effect
3.6.5. Stability
3.7. Pharmacokinetic Analysis
3.8. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bai, R.R.; Wu, X.M.; Xu, J.Y. Current natural products with antihypertensive activity. Chin. J. Nat. Med. 2015, 13, 721–729. [Google Scholar] [CrossRef]
- Arafat, O.M.; Tham, S.Y.; Sadikun, A.; Zhari, I.; Haughton, P.J.; Asmawi, M.Z. Studies on diuretic and hypouricemic effects of Orthosiphon stamineus methanol extracts in rats. J. Ethnopharmacol. 2008, 118, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Akowuah, G.A.; Ismail, Z.; Norhayati, I.; Sadikun, A. The effects of different extraction solvents of varying polarities on polyphenols of Orthosiphon stamineus and evaluation of the free radical-scavenging activity. Food Chem. 2005, 93, 311–317. [Google Scholar] [CrossRef]
- Yam, M.F.; Lim, V.; Salman, I.M.; Ameer, O.Z.; Ang, L.F.; Rosidah, N.R.; Abdulkarim, M.F.; Abdullah, G.Z.; Basir, R.; Sadikun, A.; et al. HPLC and anti-inflammatory studies of the flavonoid rich chloroform extract fraction of Orthosiphon Stamineus leaves. Molecules 2010, 15, 4452–4466. [Google Scholar] [CrossRef] [PubMed]
- Adama, Y.; Somchit, M.N.; Sulaiman, M.R.; Nasaruddin, A.A.; Zuraini, A.; Bustamam, A.A.; Zakaria, Z.A. Diuretic properties of Orthosiphon stamineus Benth. J. Ethnopharmacol. 2009, 124, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.A.H.; Yam, M.F.; Ang, L.F.; Mohamed, A.J.; Asmawi, M.Z. Antidiabetic properties and mechanism of action of Orthosiphon stamineus Benth bioactive sub-fraction in streptozotocin-induced diabetic rats. J. Acupunct. Meridian. Stud. 2013, 6, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Shafaei, A.; Khan, M.S.S.; Aisha, A.F.A.; Majid, A.M.S.A.; Hamdan, M.R.; Mordi, M.N.; Ismail, Z. Flavonoids-rich Orthosiphon stamineus extract as new candidate for angiotensin I-converting enzyme inhibition: a molecular docking study. Molecules 2016, 21, 1500. [Google Scholar] [CrossRef] [PubMed]
- Mansora, C.N.A.N.; Latipa, J.; Markom, M. Preparation of Orthosiphon stamineus enriched-extracts and evaluation of their free radical scavenging activity. 2016 UKM FST Postgraduate Colloquium 2016. [Google Scholar] [CrossRef]
- Yam, M.F.; Basir, R.; Asmawi, M.Z.; Ismail, Z. Antioxidant and hepatoprotective effects of Orthosiphon stamineus Benth. standardized extract. Am. J. Chinese Med. 2007, 35, 115–126. [Google Scholar] [CrossRef]
- Dolečková, I.; Rárová, L.; Grúz, J.; Vondrusová, M.; Strnad, M.; Kryštof, V. Antiproliferative and antiangiogenic effects of flavone eupatorin, an active constituent of chloroform extract of Orthosiphon stamineus leaves. Fitoterapia 2012, 83, 1000–1007. [Google Scholar] [CrossRef]
- Hou, B.Y.; Qiang, G.F.; Zhao, Y.R.; Yang, X.Y.; Chen, X.; Yan, Y.; Wang, X.B.; Liu, C.G.; Zhang, L.; Du, G.H. Salvianolic acid A protects against diabetic nephropathy through ameliorating glomerular endothelial dysfunction via inhibiting AGE-RAGE signaling. Cell. Physiol. Biochem. 2017, 44, 2378–2394. [Google Scholar] [CrossRef] [PubMed]
- Sahib, H.B.; Ismail, Z.; Othman, N.H.; Majid, A.M.S. Orthosiphon stamineus Benth. methanolic extract enhances the anti-proliferative effects of tamoxifen on human hormone dependent breast cancer. Int. J. Pharmacol. 2009, 5, 273–276. [Google Scholar]
- Alshawsh, M.A.; Abdulla, M.A.; Ismail, S.; Amin, Z.A.; Qader, S.W.; Hadi, H.A.; Harmal, N.S. Free radical scavenging, antimicrobial and immunomodulatory activities of Orthosiphon stamineus. Molecules 2012, 17, 5385–5395. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zheng, C.; Zheng, Q.; Chen, S.; Shang, Z.; Zhang, H. Salvianolic acid A attenuates early brain injury after subarachnoid hemorrhage in rats by regulating ERK/P38/Nrf2 signaling. Am. J. Transl. Res. 2017, 9, 5643–5652. [Google Scholar] [PubMed]
- Ye, L.; He, Y.; Ye, H.; Liu, X.P.; Yang, L.L.; Cao, Z.W.; Tang, K.L. Pathway-pathway network-based study of the therapeutic mechanisms by which salvianolic acid B regulates cardiovascular diseases. Chin. Sci. Bull. 2012, 57, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Wang, Y.; Du, Q.W.; Liu, Z.G.; Liu, X.B. Cichoric acid reverses insulin resistance and suppresses inflammatory responses in the glucosamine-induced HepG2 cells. J. Agri. Food Chem. 2015, 63, 10903–10913. [Google Scholar] [CrossRef] [PubMed]
- Akowuah, G.A.; Zhari, I.; Norhayati, I.; Sadikun, A.; Khamsah, S. Sinensetin, eupatorin, 3′-hydroxy-5, 6, 7, 4′-tetramethoxyflavone and rosmarinic acid contents and antioxidative effect of Orthosiphon stamineus from Malaysia. Food Chem. 2004, 87, 559–566. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, X.L.; Niu, Y.J.; Diao, Z.J.; Ren, B.; Li, X.Y.; Liu, Z.G.; Liu, X.B. Cichoric acid improved hyperglycaemia and restored muscle injury via activating antioxidant response in MLD-STZ-induced diabetic mice. Food Chem. Toxicol. 2017, 107, 138–149. [Google Scholar] [CrossRef]
- Guan, S.C.; Fan, G.Y. Diterpenoids from aerial parts of Clerodendranthus spicatus and their cytotoxic activity. Helv. Chim. Acta. 2014, 97, 1708–1713. [Google Scholar] [CrossRef]
- Yam, M.F.; Asmawi, M.Z.; Basir, R. An investigation of the anti-inflammatory and analgesic effects of Orthosiphon stamineus leaf extract. J. Med. Food 2008, 11, 362–368. [Google Scholar] [CrossRef]
- Loon, Y.H.; Wong, J.W.; Yap, S.P.; Yuen, K.H. Determination of flavonoids from Orthosiphon stamineus in plasma using a simple HPLC method with ultraviolet detection. J. Chromatogr. B 2005, 816, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Akowuah, G.A.; Zhari, I.; Sadikun, A.; Norhayati, I. HPTLC densitometric analysis of Orthosiphon stamineus leaf extracts and inhibitory effect on xanthine oxidase activity. Pharm. Biol. 2006, 44, 65–70. [Google Scholar] [CrossRef]
- Guo, Z.L.; Liang, X.R.; Xie, Y.Y. Qualitative and quantitative analysis on the chemical constituents in Orthosiphon stamineus Benth. using ultra high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J. Pharmaceut. Biomed. 2019, 164, 135–147. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services Food and Drug Administration. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf (accessed on 21 May 2018).
Sample Availability: Samples of danshensu, protocatechuic acid, caffeic acid, rosmarinic acid, salvianolic acid A, salvianolic acid B, cichoric acid, sinensetin and eupatorin are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Analytes | Calibration Range (ng/mL) | Regression Equation (×10−3) | Correlation Coefficient (r) |
|---|---|---|---|
| Protocatechuic acid (PCA) | 8.70–2900.00 | Y = 2.1779X + 14.8358 | 0.9976 |
| Danshensu (DSS) | 5.04–1680.00 | Y = 10.3631X − 7.6571 | 0.9968 |
| Caffeic acid (CAA) | 6.48–2160.00 | Y = 3.2503X + 17.1602 | 0.9927 |
| Rosmarinic acid (RA) | 3.06–1020.00 | Y = 36.7972X + 21.8624 | 0.9995 |
| Sinensetin (SIN) | 0.345–115.00 | Y = 6473.24X + 1184.08 | 0.9992 |
| Eupatorin (EUP) | 0.47–156.67 | Y = 1900.65X + 292.998 | 0.9998 |
| Cichoric acid (CA) | 4.95–1650.00 | Y = 7.6697X − 15.6481 | 0.9979 |
| Salvianolic acid A (Sal A) | 3.69–1230.00 | Y = 25.7678X + 13.2046 | 0.9915 |
| Salvianolic acid B (Sal B) | 4.20–1400.00 | Y = 11.1706X − 14.6451 | 0.9960 |
| Analytes | Spiked Conc. | Measured Conc.a | Intra-Day | Measured Conc.a | Inter-Day | ||
|---|---|---|---|---|---|---|---|
| (ng/mL) | (ng/mL) | Relative Standard Deviation (RSD, %) | Relative Error (RE, %) | (ng/mL) | (RSD, %) | (RE, %) | |
| PCA | 8.70 | 8.70 ± 0.53 | 6.09 | −4.50 | 8.28 ± 0.47 | 7.01 | −13.60 |
| 290.00 | 290.00 ± 3.93 | 13.54 | −1.30 | 305.58 ± 3.29 | 10.81 | 7.90 | |
| 2900.00 | 2899.98 ± 83.26 | 2.87 | −0.40 | 2884.83 ± 127.48 | 4.42 | −2.70 | |
| DSS | 5.04 | 5.03 ± 1.43 | 2.79 | −0.80 | 4.72 ± 0.79 | 1.67 | −7.80 |
| 168.00 | 168.00 ± 12.33 | 7.33 | 1.20 | 179.95 ± 8.26 | 4.60 | 5.70 | |
| 1680.00 | 1680.00 ± 42.39 | 2.52 | 1.20 | 1668.57 ± 55.24 | 3.31 | −3.70 | |
| CAA | 6.48 | 6.48 ± 3.06 | 4.75 | −0.30 | 6.37 ± 0.31 | 7.12 | −4.80 |
| 216.00 | 216.02 ± 21.89 | 10.13 | 2.40 | 220.12 ± 18.64 | 8.54 | 5.30 | |
| 2160.00 | 2160.00 ± 41.16 | 1.91 | −1.60 | 2156.02 ± 131.56 | 6.11 | −3.60 | |
| RA | 3.06 | 3.07 ± 0.42 | 13.40 | 3.05 | 3.07 ± 0.31 | 10.47 | 1.10 |
| 102.00 | 101.98 ± 9.28 | 9.09 | −0.00 | 101.05 ± 4.36 | 4.30 | −2.00 | |
| 1020.00 | 1019.98 ± 26.22 | 2.57 | −0.50 | 1020.95 ± 20.48 | 2.00 | 0.50 | |
| SIN | 0.345 | 0.32 ± 0.04 | 11.71 | −4.30 | 0.33 ± 0.05 | 5.67 | −2.70 |
| 11.50 | 11.48 ± 0.33 | 2.81 | −3.90 | 11.67 ± 0.27 | 2.24 | 1.90 | |
| 115.00 | 115.00 ± 3.27 | 2.86 | 1.70 | 114.85 ± 1.60 | 1.40 | −3.70 | |
| EUP | 0.47 | 0.45 ± 0.05 | 8.83 | −5.90 | 0.43 ± 0.05 | 9.93 | −10.9 |
| 15.67 | 15.67 ± 1.10 | 7.09 | 1.10 | 16.38 ± 0.33 | 1.85 | 4.10 | |
| 156.67 | 156.68 ± 4.20 | 2.69 | 2.50 | 155.97 ± 4.81 | 3.07 | −4.50 | |
| CA | 4.95 | 4.95 ± 2.39 | 4.83 | 1.00 | 4.85 ± 0.74 | 13.15 | −2.00 |
| 165.00 | 165.02 ± 9.75 | 5.90 | 2.50 | 168.05 ± 4.12 | 2.48 | 6.50 | |
| 1650.00 | 1650.00 ± 47.61 | 2.89 | 0.05 | 1647.05 ± 89.35 | 5.43 | −0.10 | |
| Sal A | 3.69 | 3.67 ± 1.93 | 5.18 | −4.70 | 4.20 ± 0.29 | 8.79 | 14.80 |
| 123.00 | 122.98 ± 33.74 | 2.74 | −4.6 | 122.30 ± 5.31 | 5.31 | −6.46 | |
| 1230.00 | 1230.00 ± 102.21 | 8.31 | 2.90 | 1248.60 ± 32.66 | 2.62 | 1.50 | |
| Sal B | 4.20 | 4.22 ± 2.61 | 6.23 | 1.50 | 4.17 ± 0.45 | 2.13 | −1.40 |
| 140.00 | 140.02 ± 10.29 | 7.34 | 3.50 | 141.58 ± 10.68 | 7.65 | 1.10 | |
| 1400.00 | 1400.00 ± 33.11 | 2.36 | 2.70 | 1398.47 ± 49.31 | 3.53 | −0.07 | |
| Analytes | Spiked Conc. | Recovery | Matrix Effect |
|---|---|---|---|
| (ng/mL) | (Mean ± SD, %) | (Mean ± SD, %) | |
| PCA | 8.70 | 85.49 ± 10.11 | 107.62 ± 9.60 |
| 290.00 | 86.63 ± 4.45 | 106.06 ± 3.91 | |
| 2900.00 | 95.11 ± 9.09 | 100.26 ± 3.22 | |
| DSS | 5.04 | 46.98 ± 11.35 | 114.09 ± 6.76 |
| 168.00 | 47.64 ± 11.37 | 102.35 ± 1.77 | |
| 1680.00 | 57.79 ± 9.39 | 102.36 ± 5.13 | |
| CAA | 6.48 | 64.93 ± 4.69 | 104.40 ± 3.96 |
| 216.00 | 54.04 ± 4.40 | 101.95 ± 1.92 | |
| 2160.00 | 58.52 ± 13.79 | 103.80 ± 2.04 | |
| RA | 3.06 | 89.70 ± 7.51 | 90.50 ± 2.30 |
| 102.00 | 80.54 ± 5.08 | 99.07 ± 0.12 | |
| 1020.00 | 79.22 ± 12.77 | 98.91 ± 2.13 | |
| SIN | 0.345 | 82.10 ± 2.52 | 125.76 ± 8.06 |
| 11.50 | 91.49 ± 4.31 | 109.22 ± 3.06 | |
| 115.00 | 87.19 ± 13.72 | 106.99 ± 2.38 | |
| EUP | 0.47 | 85.24 ± 1.80 | 125.30 ± 10.35 |
| 15.67 | 86.13 ± 2.63 | 115.09 ± 5.92 | |
| 156.67 | 86.13 ± 13.45 | 113.99 ± 2.97 | |
| CA | 4.95 | 83.40 ± 4.27 | 76.13 ± 12.77 |
| 165.00 | 64.36 ± 0.98 | 90.26 ± 8.11 | |
| 1650.00 | 62.16 ± 0.45 | 95.14 ± 1.52 | |
| Sal A | 3.69 | 67.28 ± 2.86 | 67.62 ± 6.33 |
| 123.00 | 76.40 ± 12.52 | 85.52 ± 11.17 | |
| 1230.00 | 67.08 ± 3.27 | 96.81 ± 1.26 | |
| Sal B | 4.20 | 79.22 ± 8.09 | 94.15 ± 4.87 |
| 140.00 | 66.75 ± 2.89 | 98.16 ± 4.85 | |
| 1400.00 | 67.25 ± 4.30 | 96.17 ± 3.66 | |
| IS | 98.8 | 100.69 ± 8.38 | 97.83 ± 7.02 |
| Analytes | Spiked Conc. | 25 °C for 24 h | −80 °C for 20 days | Three Freeze-Thaw Cycles | |||
|---|---|---|---|---|---|---|---|
| (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | |
| PCA | 8.70 | 5.76 | −4.50 | 4.17 | −2.15 | 7.43 | −15.63 |
| 290.00 | 2.73 | 2.36 | 9.54 | 0.33 | 1.92 | 1.73 | |
| 2900.00 | 3.17 | 0.83 | 2.75 | −1.80 | 1.05 | −1.70 | |
| DSS | 5.04 | 12.28 | 4.27 | 6.19 | −5.63 | 4.76 | −2.60 |
| 168.00 | 4.70 | 2.91 | 2.53 | 10.47 | 7.90 | 2.90 | |
| 1680.00 | 4.64 | 1.61 | 0.71 | −2.03 | 1.62 | −0.30 | |
| CAA | 6.48 | 8.15 | −3.02 | 5.31 | −1.65 | 1.53 | −2.27 |
| 216.00 | 4.77 | 2.79 | 2.29 | 2.33 | 4.93 | 2.45 | |
| 2160.00 | 3.11 | 0.47 | 4.55 | −2.73 | 1.30 | −2.37 | |
| RA | 3.06 | 14.53 | −2.63 | 3.20 | 7.23 | 2.19 | −1.50 |
| 102.00 | 2.45 | 8.10 | 5.00 | −5.10 | 1.50 | 1.63 | |
| 1020.00 | 1.48 | 8.13 | 1.84 | −2.33 | 2.47 | −0.13 | |
| SIN | 0.345 | 6.08 | 12.67 | 4.86 | −0.17 | 3.32 | −11.73 |
| 11.50 | 2.71 | 1.61 | 4.96 | 0.33 | 4.14 | 13.00 | |
| 115.00 | 1.21 | 10.03 | 0.43 | −3.37 | 3.39 | −1.27 | |
| EUP | 0.47 | 7.40 | 2.15 | 2.22 | 8.00 | 9.61 | −6.67 |
| 15.67 | 3.87 | 11.52 | 5.93 | 2.00 | 7.35 | 7.37 | |
| 156.67 | 4.83 | 10.37 | 0.30 | −4.57 | 3.47 | −0.73 | |
| CA | 4.95 | 2.36 | 4.93 | 1.83 | 2.38 | 3.22 | −4.03 |
| 165.00 | 5.27 | −1.83 | 3.43 | −5.87 | 4.90 | 4.43 | |
| 1650.00 | 4.18 | −6.40 | 2.22 | 13.47 | 4.73 | −0.47 | |
| Sal A | 3.69 | 1.67 | 1.61 | 3.29 | 3.33 | 1.74 | 3.02 |
| 123.00 | 6.19 | −2.63 | 5.71 | −3.27 | 1.38 | −3.40 | |
| 1230.00 | 6.38 | 0.27 | 4.46 | 0.43 | 1.34 | 3.23 | |
| Sal B | 4.20 | 2.26 | 1.65 | 1.29 | 3.55 | 4.20 | 7.43 |
| 140.00 | 2.10 | 1.63 | 1.11 | −8.23 | 2.18 | −8.23 | |
| 1400.00 | 1.83 | 5.77 | 2.54 | 3.43 | 2.75 | 0.80 | |
| Analytes | AUC(0–t) (ng/mL*h) | AUC(0–∞) (ng/mL*h) | Cmax (ng/mL) | Tmax (h) | t1/2z (h) | MRT(0–t) (h) |
|---|---|---|---|---|---|---|
| PCA | 58.28 ± 21.59 | 69.40 ± 23.90 | 42.77 ± 13.34 | 0.46 ± 0.19 | 1.59 ± 0.77 | 1.76 ± 0.68 |
| DSS | 9421.62 ± 3832.04 | 11921.89 ± 5096.17 | 1008.02 ± 500.41 | 1.95 ± 3.39 | 8.59 ± 5.65 | 9.02 ± 0.95 |
| CAA | 438.64 ± 139.06 | 454.74 ± 127.61 | 298.07 ± 68.84 | 0.36 ± 0.27 | 1.89 ± 1.41 | 1.85 ± 0.56 |
| RA | 3605.09 ± 1571.42 | 3878.59 ± 1531.01 | 2284.82 ± 1213.83 | 0.38 ± 0.21 | 5.58 ± 3.39 | 2.61 ± 0.94 |
| SIN | 1.66 ± 1.16 | 1.76 ± 1.24 | 2.05 ± 1.90 | 0.38 ± 0.21 | 0.59 ± 0.23 | 0.79 ± 0.18 |
| EUP | 1.67 ± 1.01 | 1.83 ± 1.02 | 2.52 ± 3.68 | 0.55 ± 0.33 | 1.13 ± 0.38 | 1.02 ± 0.28 |
| CA | 1895.76 ± 868.25 | 2012.52 ± 838.25 | 186.00 ± 106.77 | 2.79 ± 2.60 | 6.11 ± 2.13 | 7.72 ± 1.14 |
| Sal A | 567.35 ± 361.70 | 652.87 ± 349.37 | 240.48 ± 178.27 | 0.71 ± 0.29 | 7.68 ± 3.60 | 3.77 ± 1.07 |
| Sal B | 194.59 ± 51.93 | 334.03 ± 92.69 | 139.05 ± 175.56 | 2.33 ± 4.74 | 13.50 ± 4.82 | 4.86 ± 3.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Li, B.; Gu, J.; Zhu, P.; Su, F.; Bai, R.; Liang, X.; Xie, Y. Simultaneous Quantification and Pharmacokinetic Study of Nine Bioactive Components of Orthosiphon stamineus Benth. Extract in Rat Plasma by UHPLC-MS/MS. Molecules 2019, 24, 3057. https://doi.org/10.3390/molecules24173057
Guo Z, Li B, Gu J, Zhu P, Su F, Bai R, Liang X, Xie Y. Simultaneous Quantification and Pharmacokinetic Study of Nine Bioactive Components of Orthosiphon stamineus Benth. Extract in Rat Plasma by UHPLC-MS/MS. Molecules. 2019; 24(17):3057. https://doi.org/10.3390/molecules24173057
Chicago/Turabian StyleGuo, Zili, Bo Li, Jinping Gu, Peixi Zhu, Feng Su, Renren Bai, Xianrui Liang, and Yuanyuan Xie. 2019. "Simultaneous Quantification and Pharmacokinetic Study of Nine Bioactive Components of Orthosiphon stamineus Benth. Extract in Rat Plasma by UHPLC-MS/MS" Molecules 24, no. 17: 3057. https://doi.org/10.3390/molecules24173057
APA StyleGuo, Z., Li, B., Gu, J., Zhu, P., Su, F., Bai, R., Liang, X., & Xie, Y. (2019). Simultaneous Quantification and Pharmacokinetic Study of Nine Bioactive Components of Orthosiphon stamineus Benth. Extract in Rat Plasma by UHPLC-MS/MS. Molecules, 24(17), 3057. https://doi.org/10.3390/molecules24173057