
Enzyme Entrapment in Amphiphilic Myristyl-Phenylalanine Hydrogels



Abstract
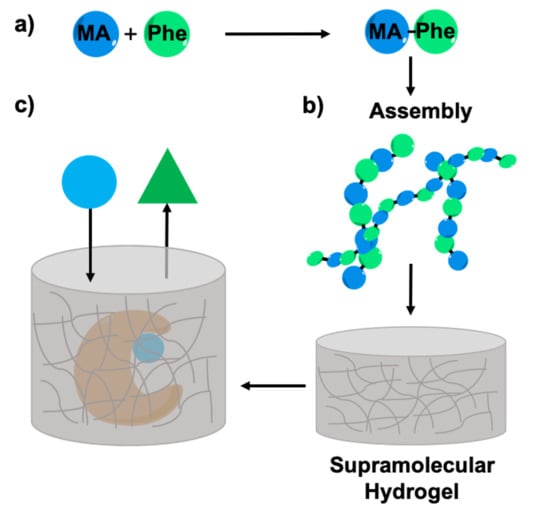
:
1. Introduction
2. Results
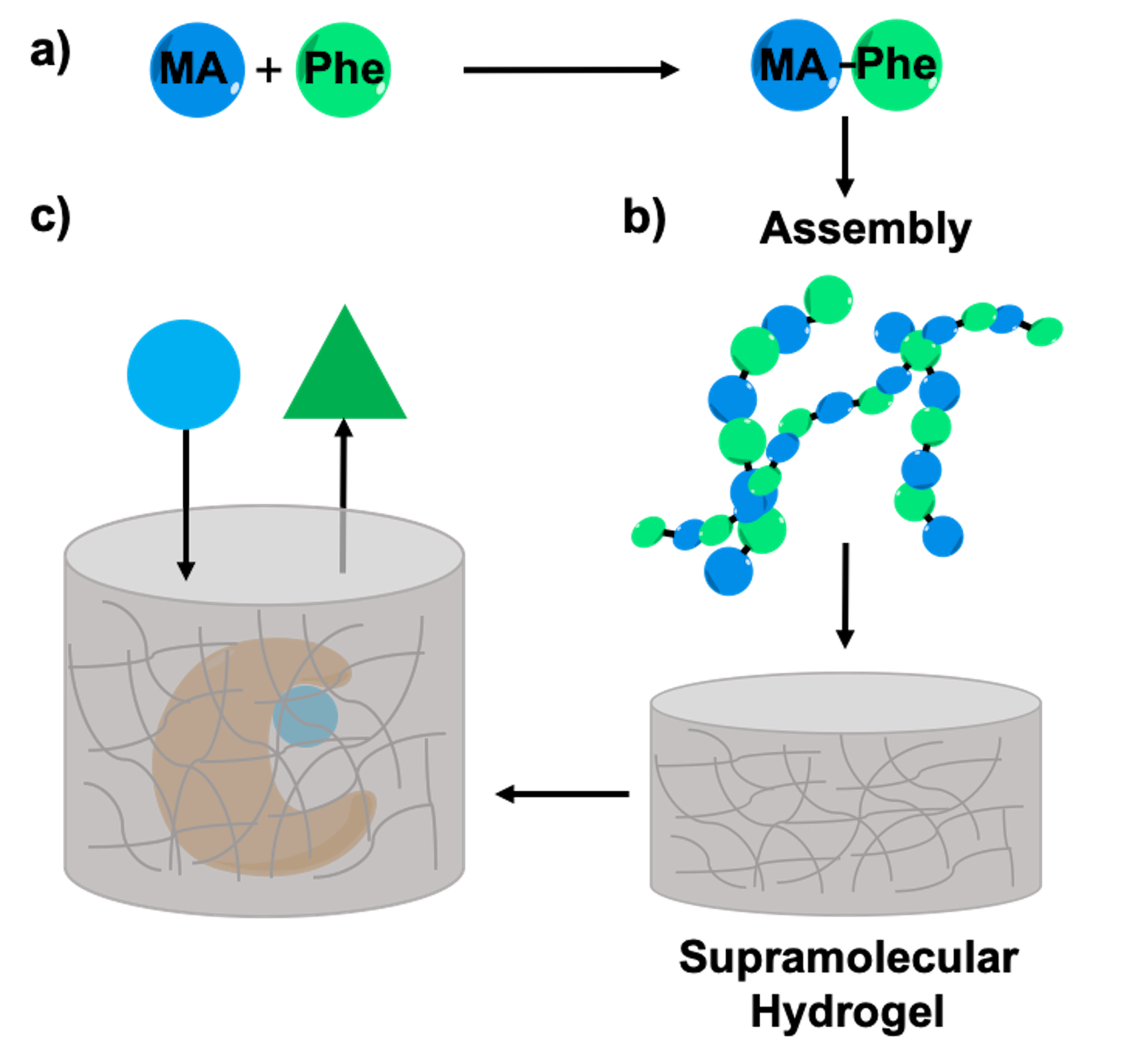
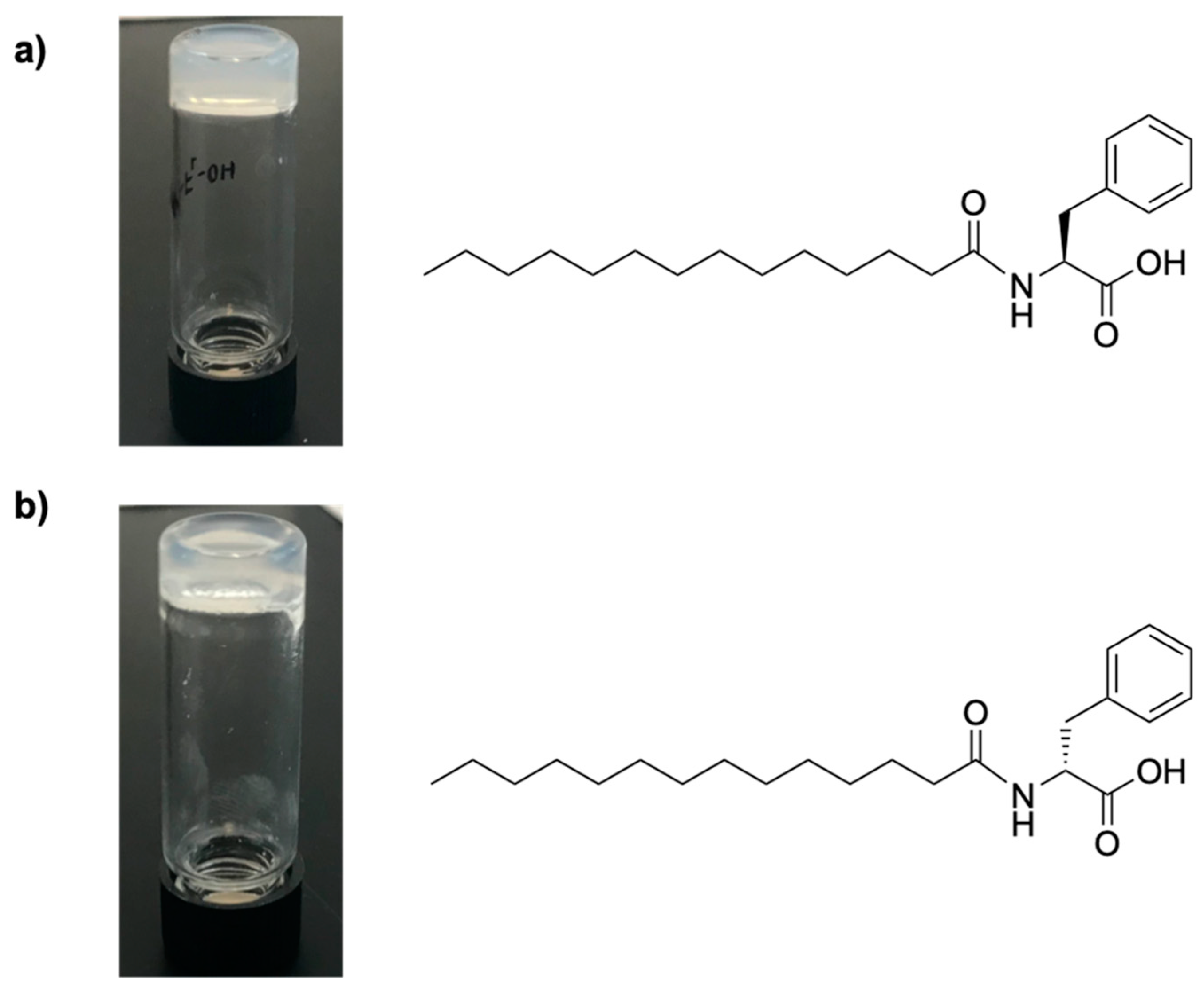
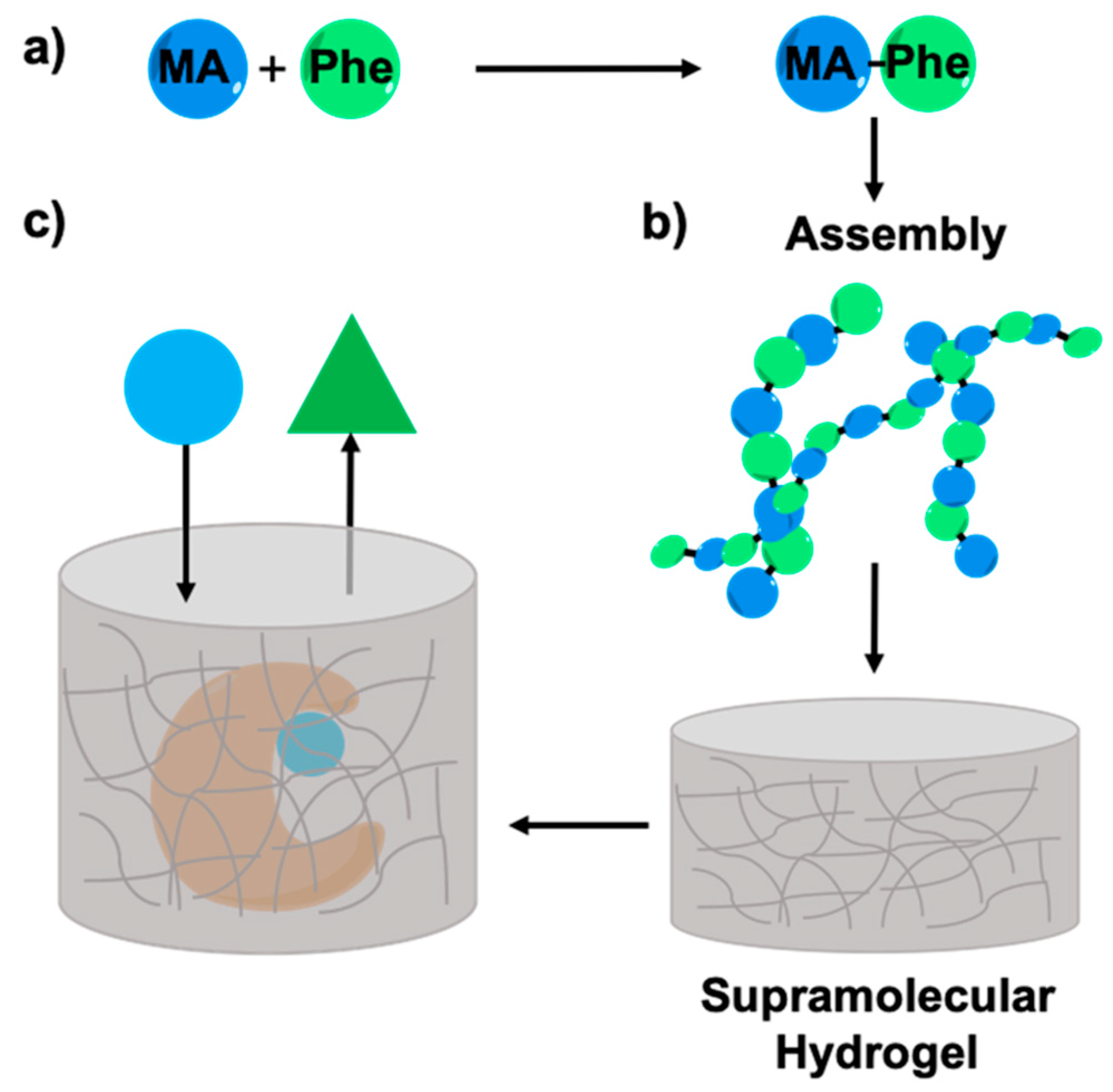
2.1. Synthesis of Myristic Acid-l/d-Phenylalanine (MA-l/d-Phe-OH)
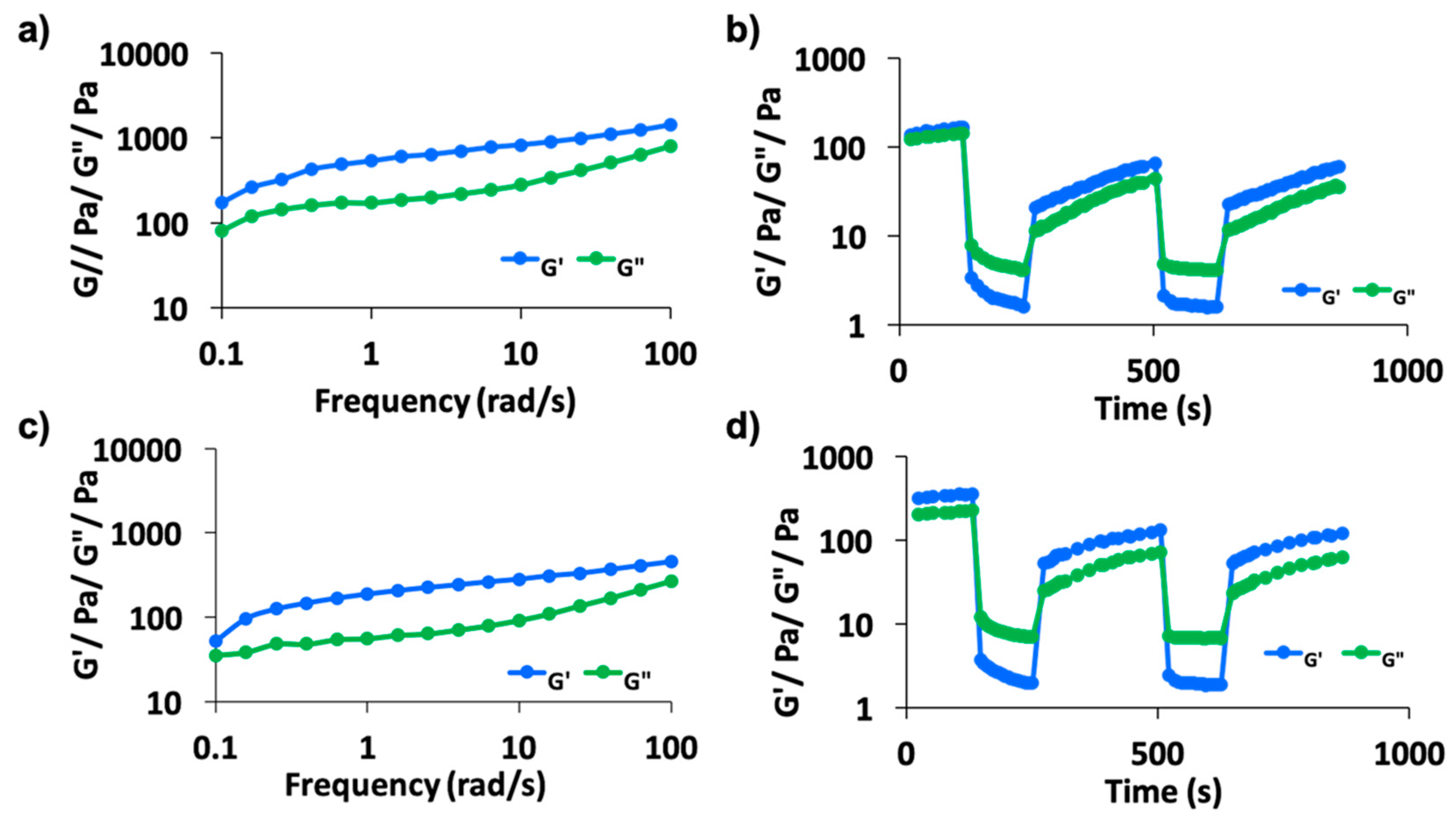
2.2. Rheology Studies
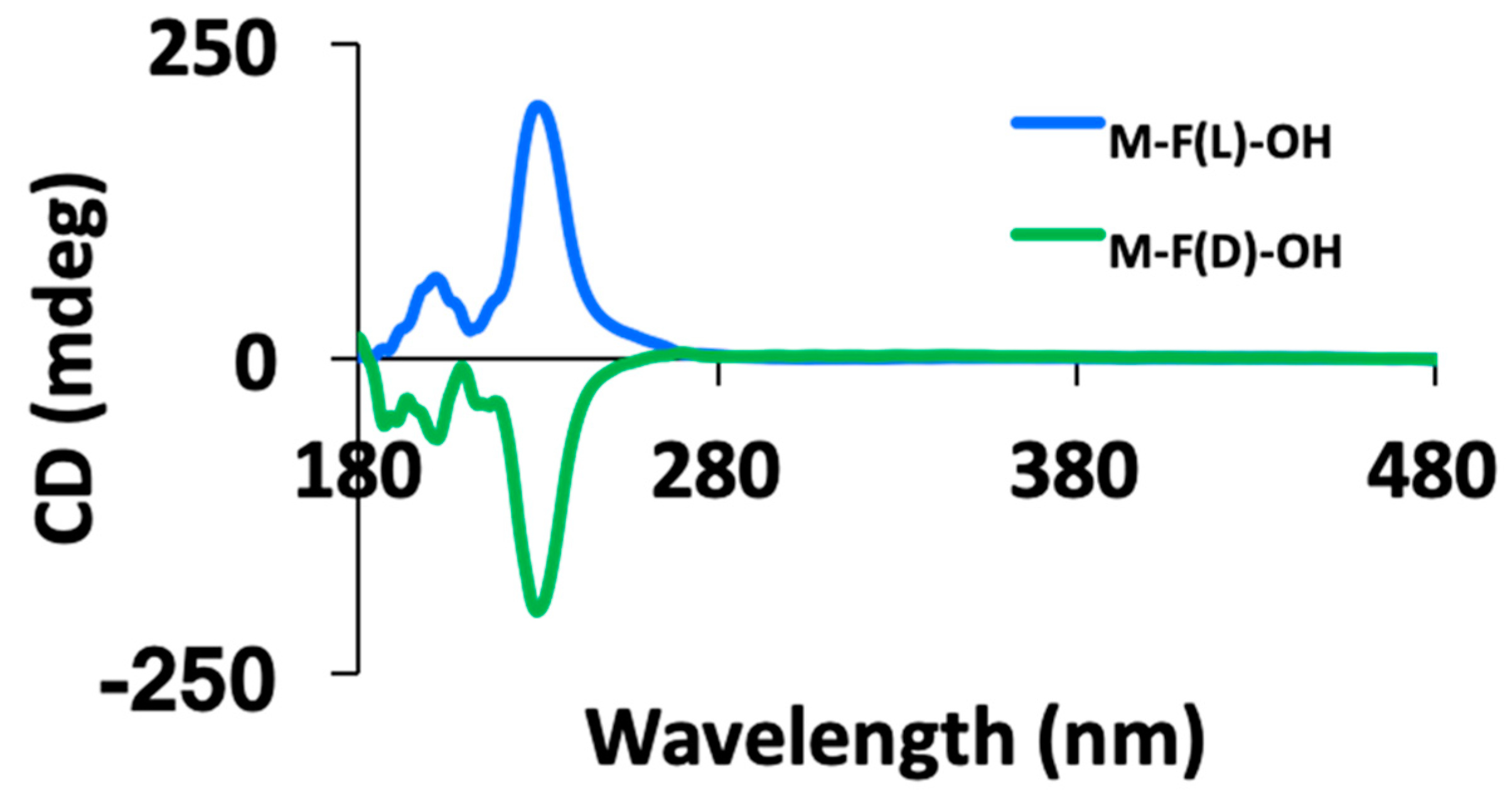
2.3. Circular Dichroism
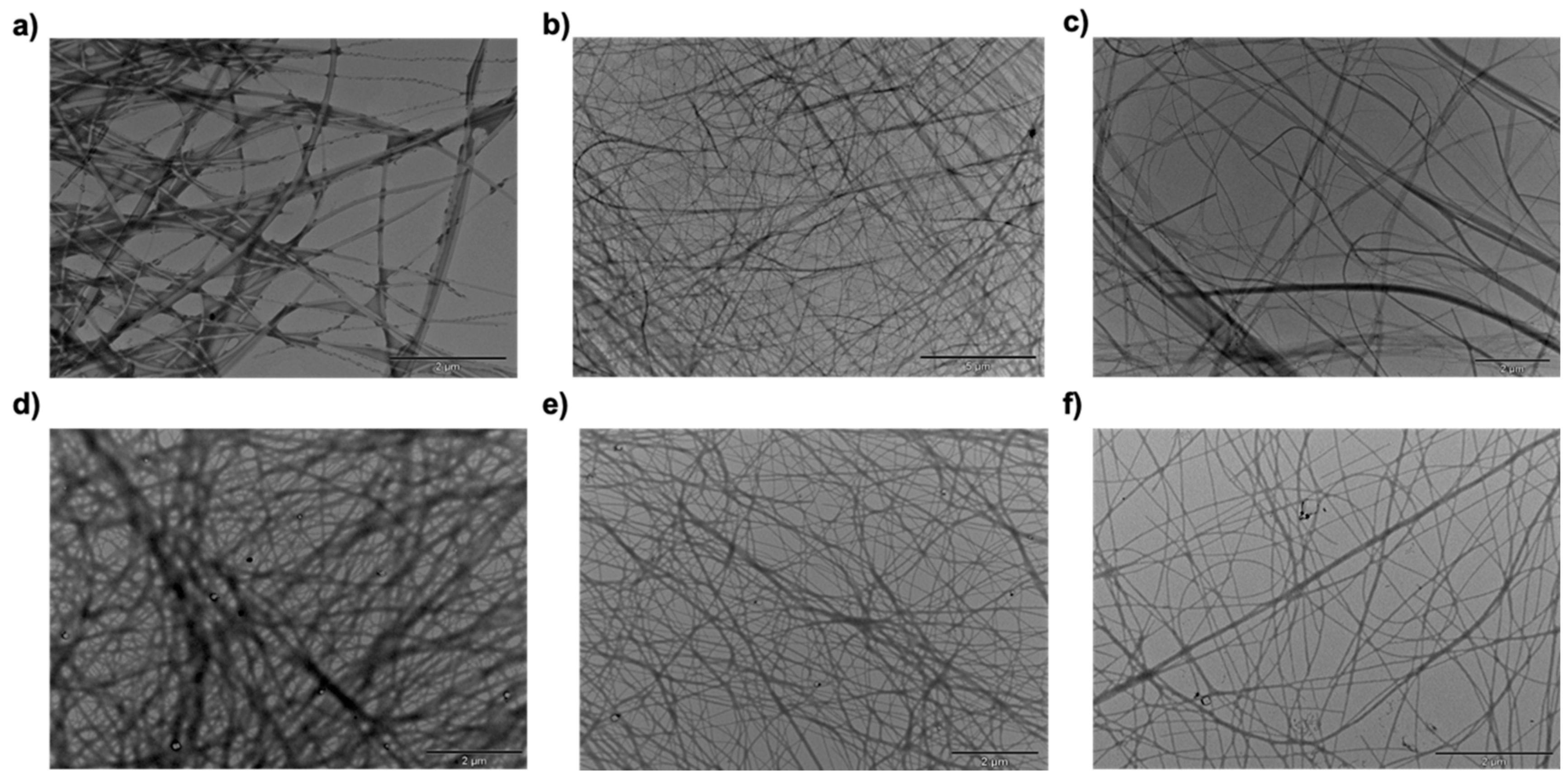
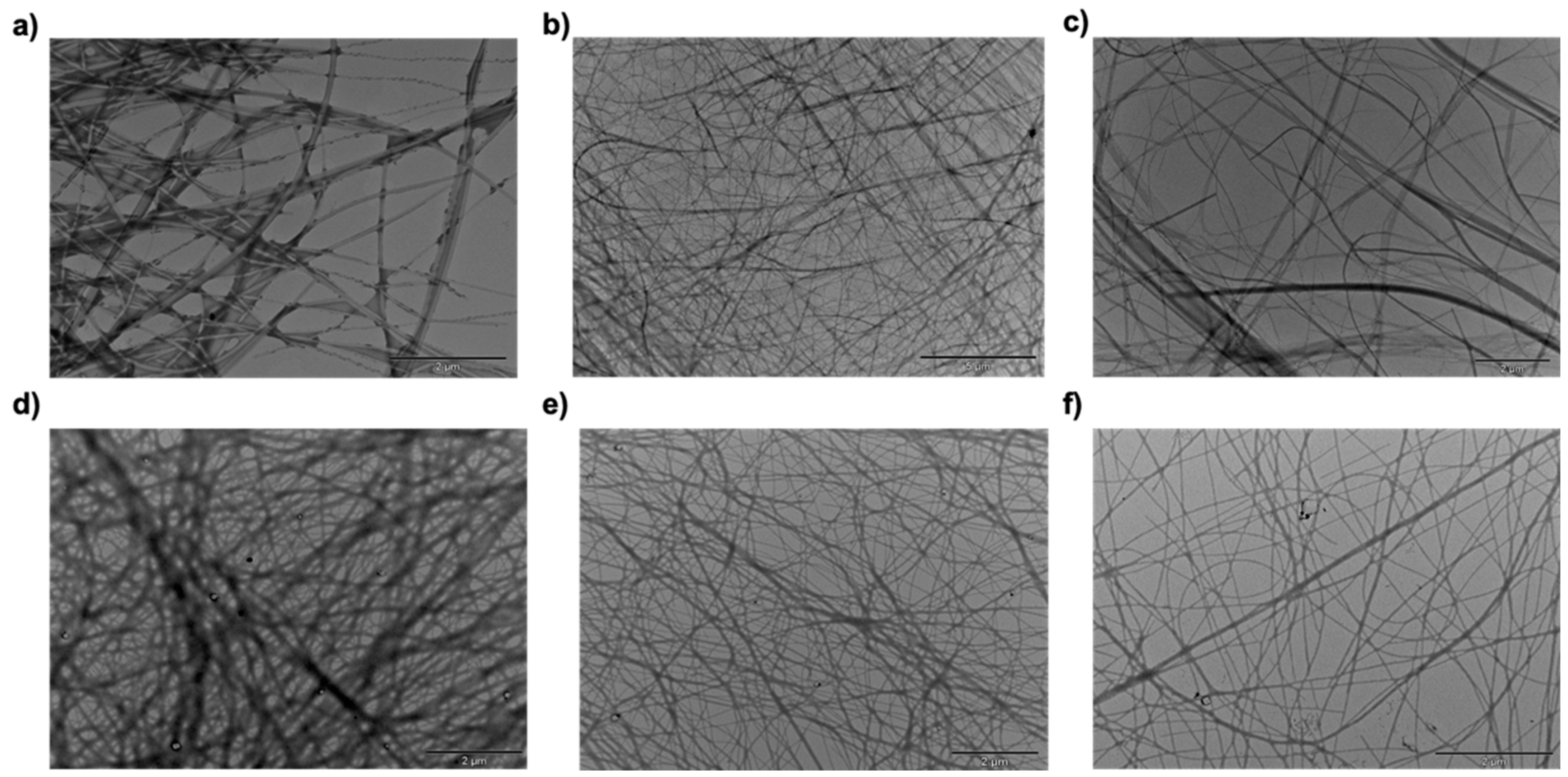
2.4. Morphological Investigation
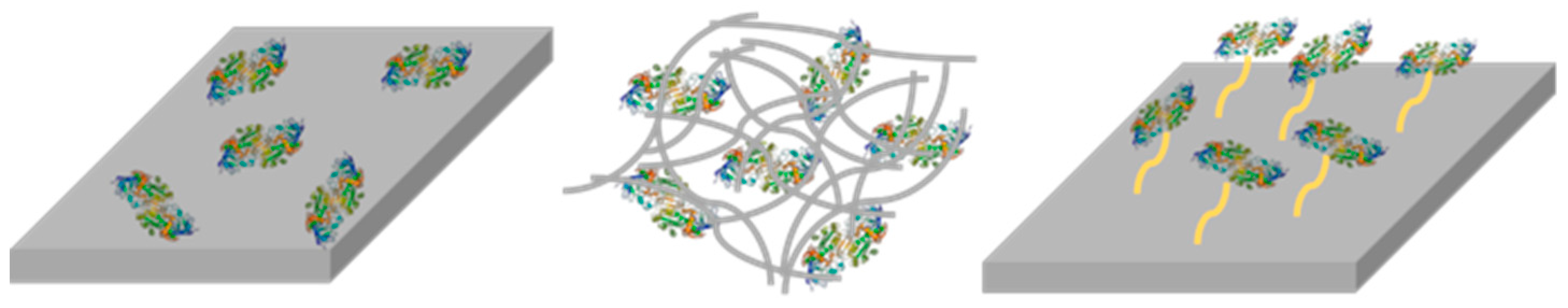
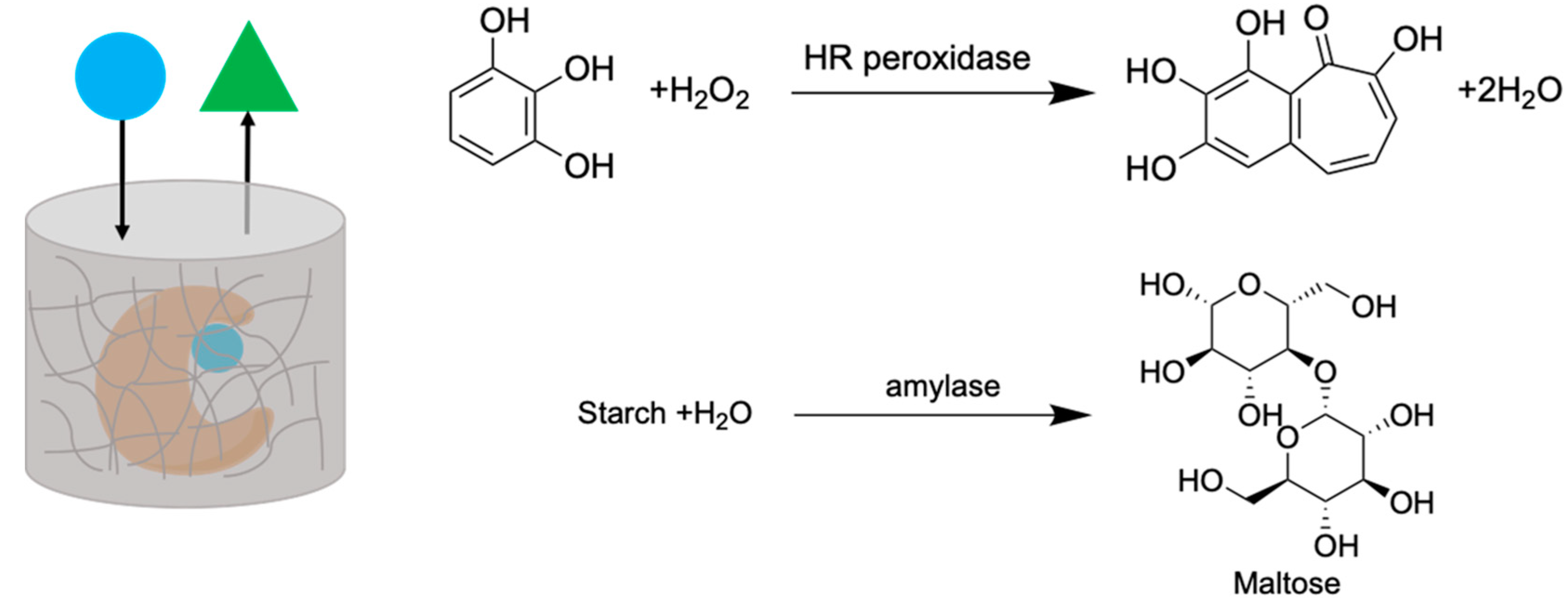
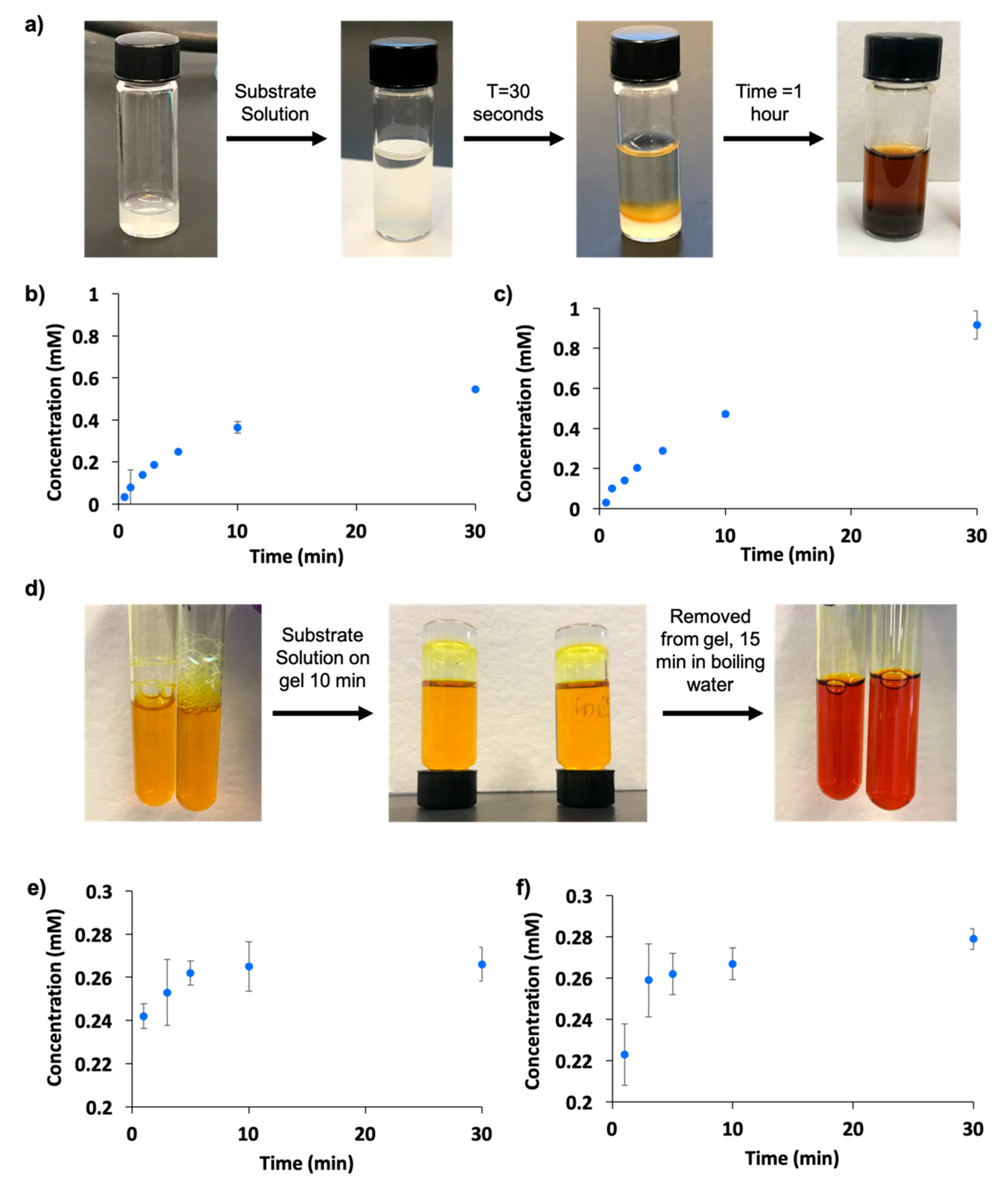
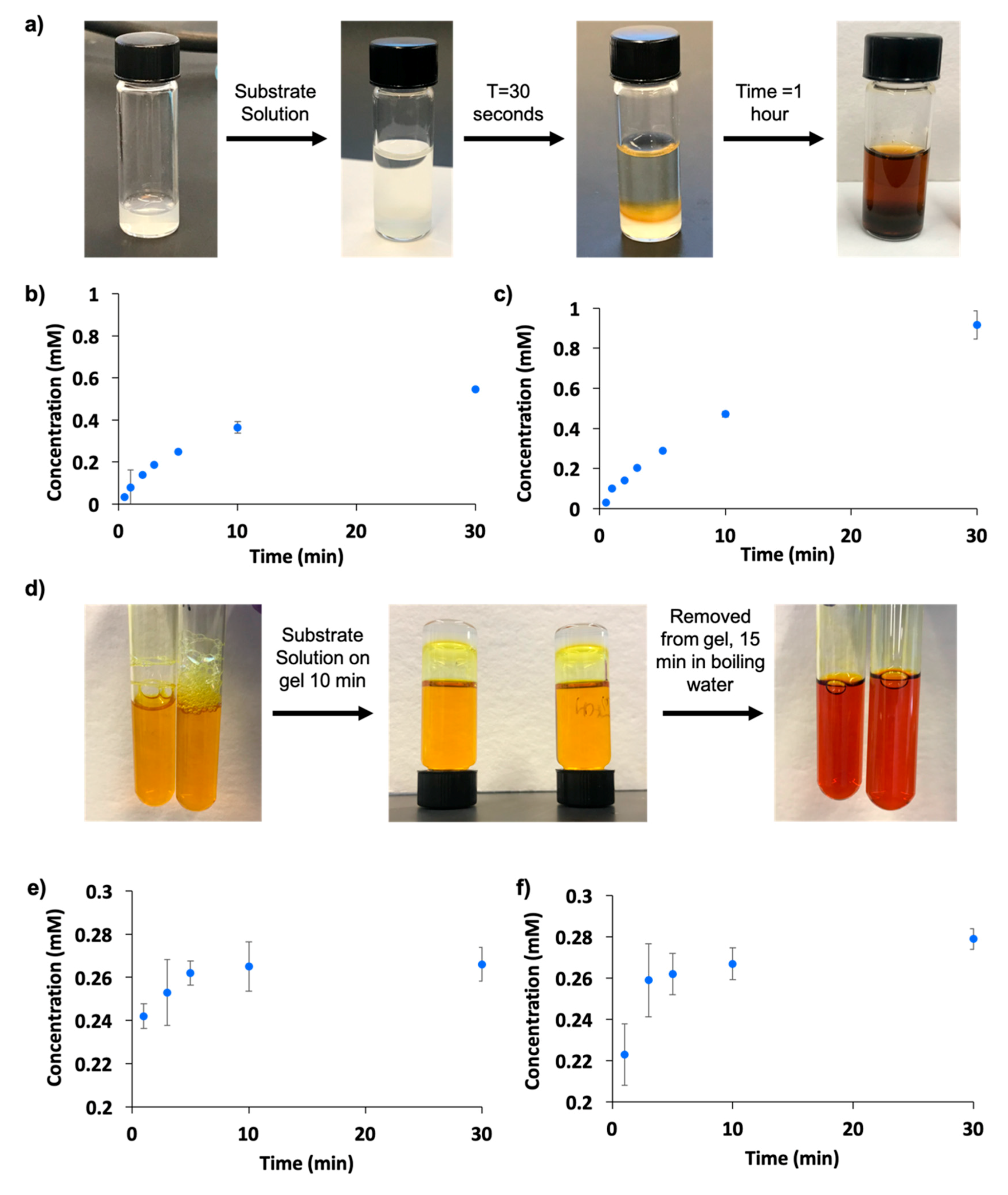
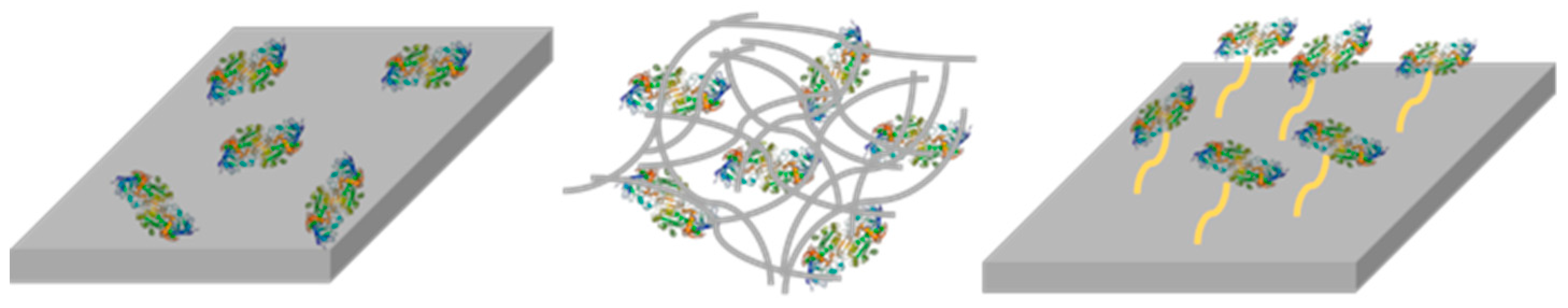
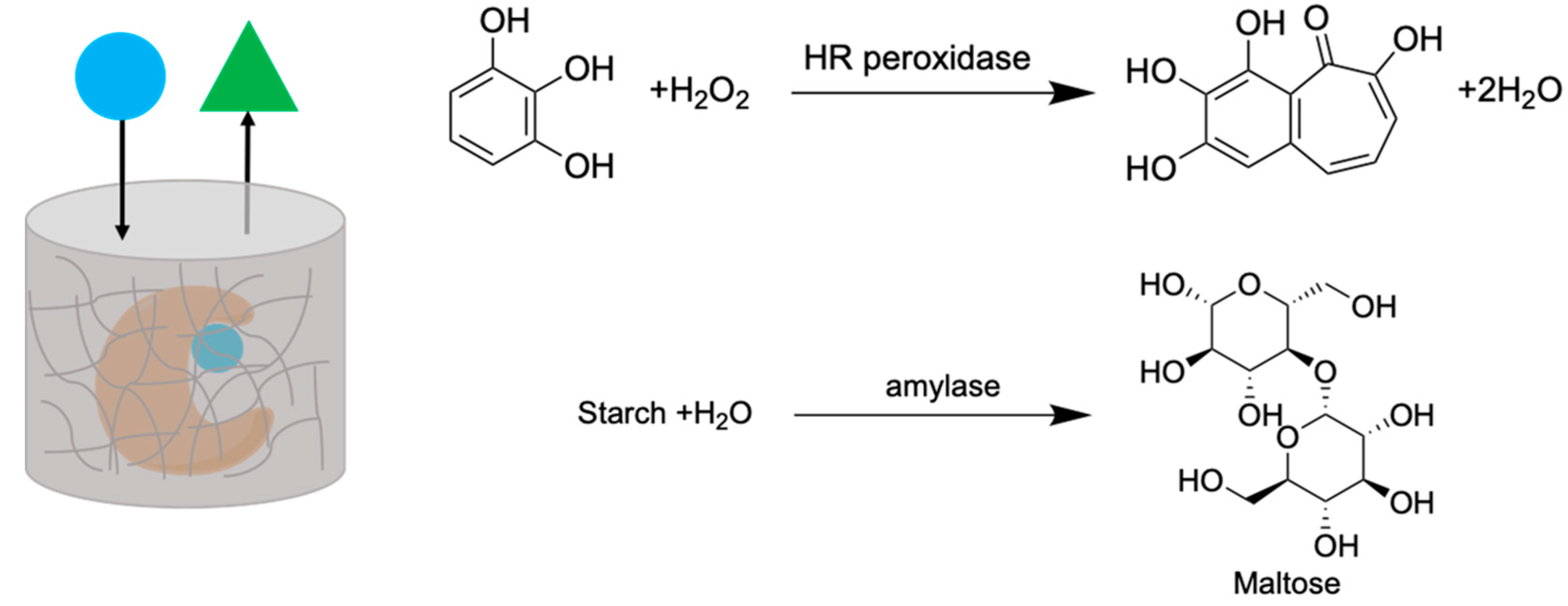
2.5. Enzyme Entrapment
2.6. Enzyme Leaching
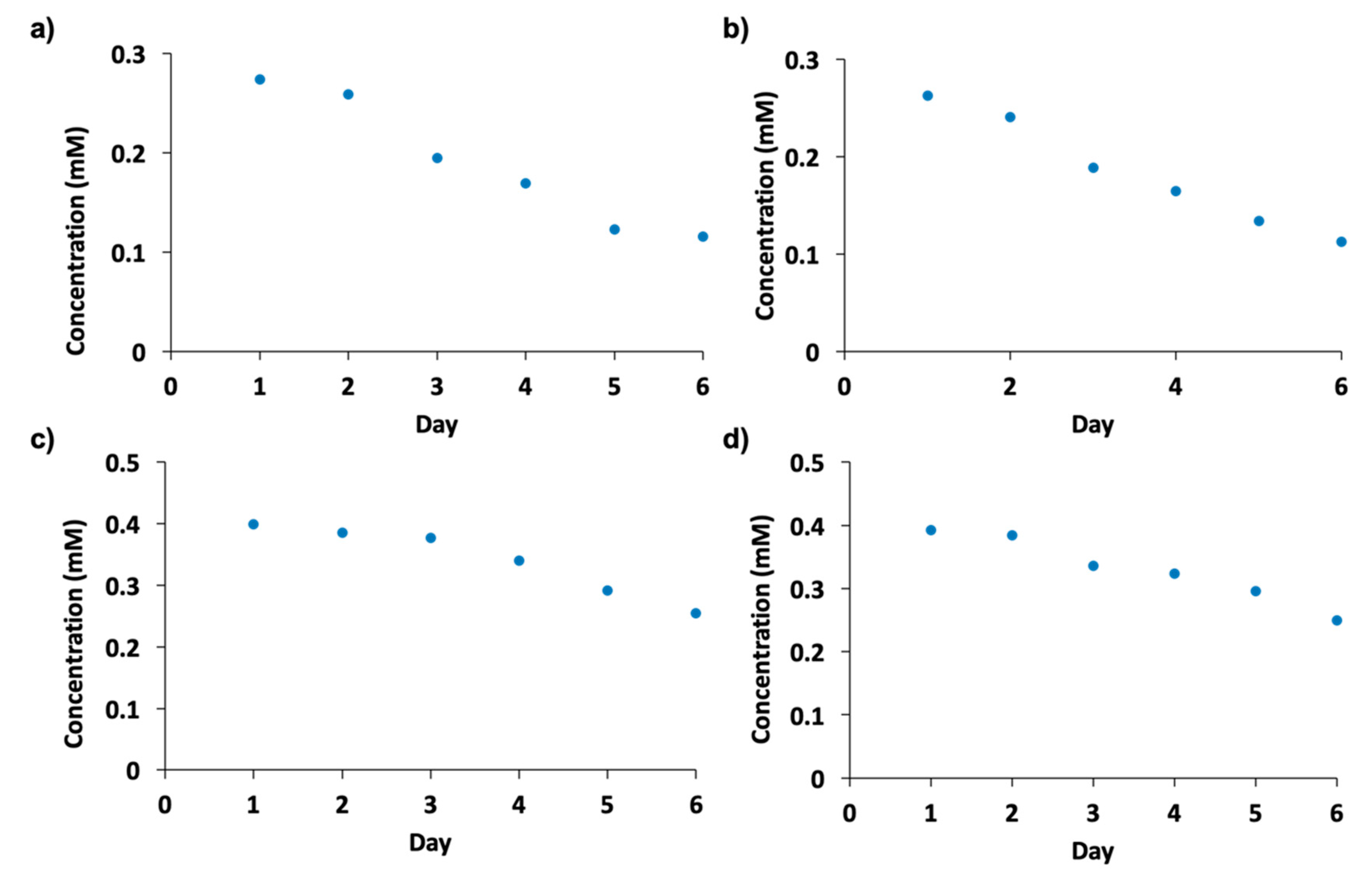
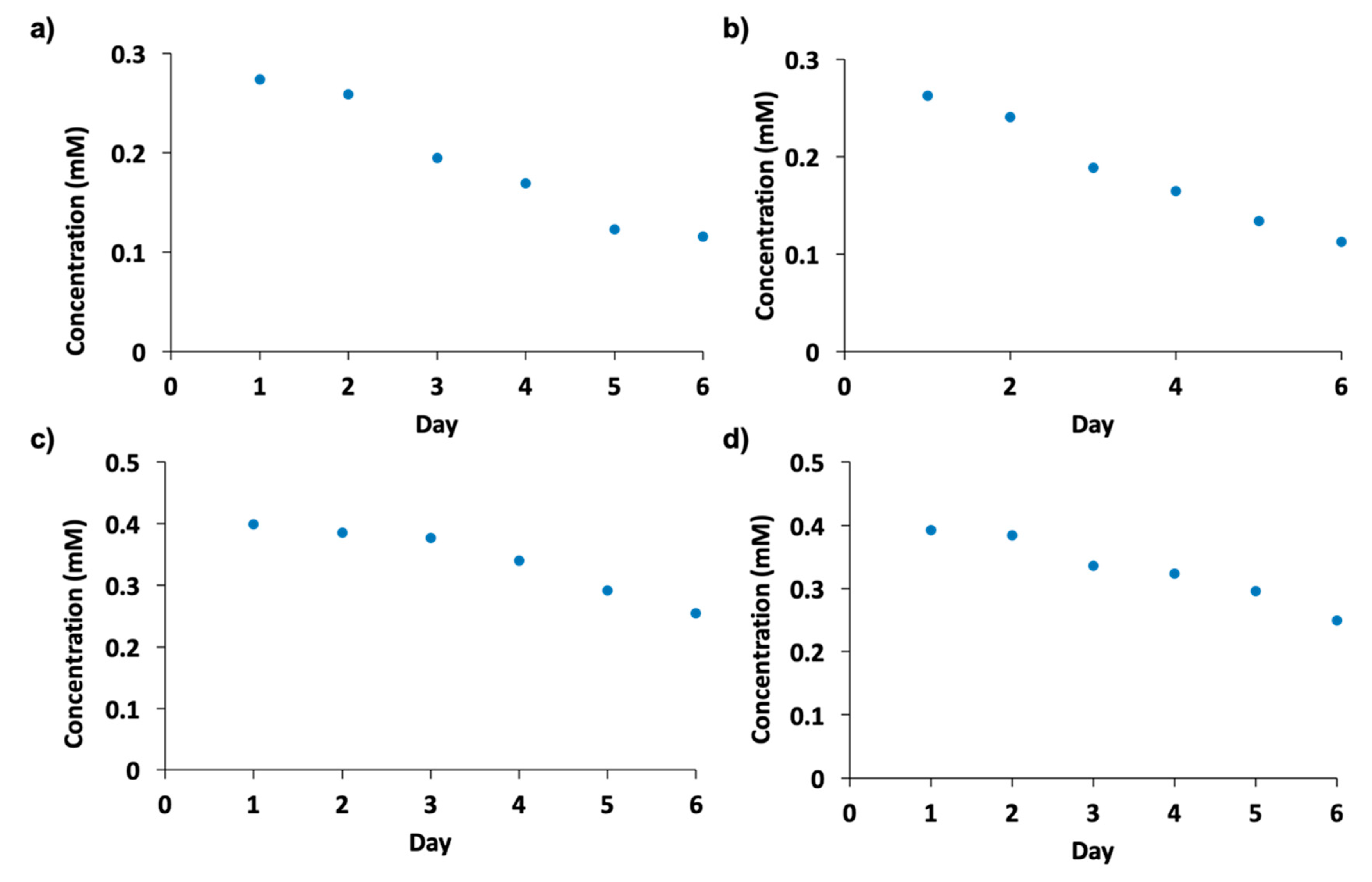
2.7. Enzyme Recycling and Stability
3. Discussion
4. Materials and Methods
4.1. Nuclear Magnetic Resonance (NMR) Spectroscoopy
4.2. Fourier Transform—Infrrared Spectroscopy
4.3. Transmission Electron Miscroscopy (TEM)
4.4. Circular Dichroism
4.5. Rheology Studies
4.6. Enzyme Entrapment, Activity, Leaching, and Quantification Using Ultraviolet (UV) Scpectroscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Falcone, N.; Kraatz, H.-N. Supramolecular Assembly of Peptide and Metallopeptide Gelators and Their Stimuli-Responsive Properties in Biomedical Applications. Chem. Eur. J. 2018, 24, 14316–14328. [Google Scholar] [CrossRef] [PubMed]
- Panda, J.J.; Mishra, A.; Basu, A.; Chauhan, V.S. Stimuli-responsive self-assembled hydrogel of low molecular weight free dipeptide with potential for tunable drug delivery. Biomacromolecules 2008, 9, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Falcone, N.; Shao, T.; Sun, X.; Kraatz, H.-B. Systematic exploration of the pH dependence of a peptide hydrogel. Can. J. Chem. 2018. [Google Scholar] [CrossRef]
- Jayawarna, V.; Richardson, S.M.; Hodson, A.R.; Saiani, A.; Gough, J.E.; Ulijn, R.V. Introducing chemical functionality in Fmoc-peptide gels for cell culture. Acta Biomater. 2009, 5, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Jayawarna, V.; Richardson, S.M.; Gough, J.E.; Ulijn, R.V. Self-assembling peptide hydrogels: Directing cell behaviour by chemical composition. Tissue Eng. Part A 2008, 14, 1937–3341. [Google Scholar]
- Basak, S.; Nanda, J.; Banerjee, A. Formation of hybrid hydrogels consisting of tripeptide and different solver nanoparticle-capped ligands: Modulation of the mechanical strength of gel phase materials. Soft Matter 2009, 5, 3452–3460. [Google Scholar]
- Adhikari, B.; Paluri, G.; Banerjee, A. A new aromatic amino acid based organogel for oil spill recovery. J. Mater. Chem. 2012, 22, 11658–11664. [Google Scholar]
- Salick, D.A.; Kretsinger, J.K.; Pochan, D.J.; Schreider, J.P. Inherent antibacterial activity of a peptide-based β-hairpin hydrogel. J. Am. Chem. Soc. 2007, 129, 14793–14799. [Google Scholar] [CrossRef]
- Laverty, G.; McClosky, A.P.; Gilmore, B.F.; Jones, D.S.; Zhou, J.; Xu, B. Ultrashort cationic naphthalene-derived self-assembled peptides as antimicrobial nanomaterials. Biomacromolecules 2014, 15, 3429–3439. [Google Scholar] [CrossRef]
- Altunbas, A.; Lee, S.J.; Rajasekaran, S.A.; Schreider, J.P.; Pochan, D.J. Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles. Biomaterials 2011, 32, 5906–5914. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A.; Pereria, C. Biocatalysis engineering: The big picture. Chem. Soc. Rev. 2017, 46, 2678–2691. [Google Scholar] [CrossRef]
- Huang, X.J.; Chen, P.C.; Huang, F.; Ou, Y.; Chen, M.R.; Xu, Z.K. Immobilization of Candida rugose lipase on electrospun cellulose nanofiber membrane. J. Mol. Catal. B Enzym. 2011, 70, 95–100. [Google Scholar] [CrossRef]
- Shen, Q.; Yang, R.; Hua, X.; Ye, F.; Zhang, W.; Zhao, W. Gelatin-templated biomimetic calcification for-galactosidase immobilization. Process Biochem. 2011, 46, 1565–1571. [Google Scholar] [CrossRef]
- Cunha, A.G.; Fernandez-Lorente, G.; Bevilaqua, J.V.; Destain, S.; Pavia, L.M.; Freire, D.M.; Fernandes-Lafuente, R.; Guisan, J.M. Immobilization of Yarrowia liplytica Lipase—A Comparison of Stability of Physical Adsorption and Covalent Attachement Techniques. Appl. Biochem. Biotechnol. 2008, 146, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ispas, C.; Sokolov, I.; Andreesu, S. Enzyme-functionalized mesoporous silica for bioanalytical applications. Anal. Bioanal. Chem. 2009, 393, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.C.; Kuan, I.C.; Hang, J.R.; Tsai, B.H.; Lee, S.L.; Yu, C.Y. Activity enhancement and stabilization of lipase from Pseudomonas cepacia in polyallylamine-mediated biomimetic silica. Biotechnol. Lett. 2011, 33, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.G.; Wan, L.S.; Liu, Z.M.; Huang, X.J.; Xu, Z.K. Enzyme immobilization on electrospun nanofibers: An overview. J. Mol. Catal. B Enzym. 2009, 56, 189–195. [Google Scholar] [CrossRef]
- Koch-Schmidt, A.-C. Gel-entrapment of Enzymes. In Biomedical Applications of Immobilized Enzymes and Proteins; Springer: Boston, MA, USA, 1977; pp. 47–67. [Google Scholar]
- Reetz, M.T. Entrapment of biocatalysts in hydrophobic sol-gel materials for use in organic chemistry. Adv. Mater. 1997, 9, 943–954. [Google Scholar] [CrossRef]
- Frenkel-Mullerad, H.; Avnir, D. Sel-gel materials as efficient enzyme protectors: Preserving the activity of phosphatases under extreme pH conditions. J. Am. Chem. Soc. 2005, 127, 8077–8081. [Google Scholar] [CrossRef]
- Hinberg, I.; Kapoulas, A.; Korus, R.; O’Driscoll, K. Gel entrapment of enzymes: Kinetic studies of immobilized glucose oxidase. Biotechnol. Bioeng. 1974, 16, 159–168. [Google Scholar] [CrossRef]
- Wang, Z.; Etienne, M.; Kohring, G.W.; Walcarius, A. Critical effect of polyelectrolytes on the electrochemical response of dehydrogenase entrapped in sol-gel thin films. Electroanalysis 2010, 22, 2092–2100. [Google Scholar] [CrossRef]
- Li, Y.Z.; He, N.; Wang, X.; Chang, W.B.; Ci, Y.X. Mimicry of peroxidase by immobilization of hemin on N-isopropylacrylamide-based hydrogel. Analyst 1998, 123, 359–364. [Google Scholar] [CrossRef]
- Mahajan, R.; Gupta, V.K.; Sharma, J. Comparison and suitability of gel matrix for entrapping higher content of enzymes for commercial applications. Indian J. Pharm. Sci. 2010, 2, 223–228. [Google Scholar]
- Konsoula, Z.; Liakopoulou-Kyriakides, M. Thermostable α-amylase production by Bacillus subtilis entrapped in calcium alginate gel capsules. Enzym. Microb. Technol. 2006, 39, 690–696. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, F.; Wang, Q.; Zhang, Y.; Xu, B. Small peptide nanofibers as the matricies of molecular hydrogels for mimicking enzymes and enhancing the activity of enzymes. Chem. Soc. Rev. 2010, 39, 3425–3433. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lim, S.Y.; Nam, D.H.; Ryu, J.; Ku, S.H.; Park, C.B. Self-assembled photoluminescent peptide hydrogel as a versatile platform for enzyme-based optical biosensors. Biosens. Bioelectron. 2011, 26, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, Z.; Wang, L.; Ma, M.; Xu, B. Molecular hydrogel-immobilized enzymes exhibit superactivity and high stability in organic solvents. Chem. Commun. 2007, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, Z.; Gao, Y.; Ge, W.; Wang, L.; Xu, B. Enzymatic hydrogelation to immobilize an enzyme for high activity and stability. Soft Matter 2008, 4, 550–553. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, Z.; Zhang, X.; Xiao, X.; Chang, C.K.; Xu, B. A supramolecular-hydrogel-encapsulated Hemin as an Artifical Enzyme to Mimic Peroxidase. Angew. Chem. Int. Ed. 2007, 46, 4285–4289. [Google Scholar] [CrossRef] [PubMed]
- Escuder, B.; Rodriguez-Llansola, F.; Miravet, J.F. Supramolecular gels as active media for organic reactions and catalysis. New J. Chem. 2010, 34, 1044–1054. [Google Scholar] [CrossRef]
- Hickling, C.; Toogood, H.S.; Saiani, A.; Scrutton, N.S.; Miller, A.F. Nanofibrillar Peptide Hydrogels for the Immobilization of Biocatalysts for Chemical Transformations. Macromol. Rapid Commun. 2014, 35, 868–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, S.; Singh, I.; Banerjee, A.; Kraatz, H.-B. Amino-Acid Based Amphiphilic Hydrogels: Metal-Ion Induced Tuning of Mechanical and Thermal Stability. RSC Adv. 2017, 7, 14461–14465. [Google Scholar] [CrossRef]
- Benoiton, N.L.; Kuroda, K.; Chen, F.M.F. Racemization in Peptide Synthesis: A Laboratory Experiment for Senior Undergraduates. Int. J. Pept. Protein Res. 1980, 15, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Pochan, D.J. Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 2010, 39, 3528–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orbach, R.; Mironi-Harpaz, I.; Adler-Abramovich, L.; Mossou, E.; Mitchell, E.P.; Forsyth, V.T.; Gazit, E.; Seliktar, D. The rheological and structural properties of Fmoc-peptide-based-hydrogels: The effect of aromatic molecular architecture on self-assembly and physical characteristics. Langmuir 2012, 28, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Keung, A.J.; Irwin, E.F.; Li, Y.; Little, L.; Schaffer, D.V.; Healy, K.E. Substrate Modulus Directs Neural Stem Cell Behavior. Biophys. J. 2008, 95, 4426–4438. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Yang, J.H.; Zhou, J.; Xu, F.; Zrinyi, M.; Dussault, B.H.; Osada, Y.; Chen, Y.M. Self-healing gels based on constitutional dynamic chemistry and their potential applications. Chem. Soc. Rev. 2014, 43, 8114–8131. [Google Scholar] [CrossRef]
- Nanda, J.; Biswas, A.; Banerjee, A. Single Amino Acid-Based Thixotropic Hydrogel Formation and pH-Dependent Morphological Change of Gel Nanofibers. Soft Matter 2013, 9, 4198–4208. [Google Scholar] [CrossRef]
- Amdursky, N.; Stevens, M.M. Circular Dichroism of Amino Acids: Following the Structural Formation of Phenylalanine. ChemPhysChem 2015, 16, 2768–2774. [Google Scholar] [CrossRef]
- Kirin, S.I.; Kraatz, H.B.; Metzler-Nolte, N. Systematizing structural motifs and nomenclature in 1, n’-disubstituted ferrocene peptides. Chem. Soc. Rev. 2006, 35, 348–354. [Google Scholar] [CrossRef]
- Adhikari, B.; Afraisibi, R.; Kraatz, H.B. Ferrocene-Tryptophan Conjugate: An Example of a Redox-Controlled Reversible Supramolecular Nanofiber Network. Organometallics 2013, 32, 5899–5905. [Google Scholar] [CrossRef]
- Ramos-Sasselli, I.; Halling, P.J.; Ulijn, R.V.; Tuttle, T. Supramolecular Fibers in Gels Can Be at Thermodynamic EquilibriumL ASimple Packing Model Reveals Preferential Fibril Formation versus Crystallization. ACS Nano 2016, 10, 2661–26608. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Singh, I.; Kraatz, H.-B. Ion-Dependent Modulation of Self-Healing Hydrogels. ChemistrySelect 2017, 2, 451–457. [Google Scholar] [CrossRef]
- Cho, H.; Balaji, S.; Sheikh, A.Q.; Hurley, J.R.; Tian, Y.F.; Collier, J.H.; Cromblehome, T.M.; Narmoneva, D.A. Regulation of Endothelial Cell Activation and Angiogenesis by Injectable Peptide Nanofibers. Acta Biomater. 2012, 8, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Falcone, N.; Basak, S.; Dong, B.; Syed, J.; Ferranco, A.; Lough, A.; She, Z.; Kraatz, H.B. A Ferrocene-Tryptophan Conjugate: The Role of the Indolic Nitrogen in Supramolecular Assembly. ChemPlusChem 2017, 82, 1282–1289. [Google Scholar] [CrossRef]
- Miller, G.L. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- Saqib, A.A.N.; Whitney, P.J. Differential Behavior if the dinitrosalicylic acid (DNS) reagent towards mono- and di-saccharide sugars. Biomass Bioenergy 2011, 35, 4748–4750. [Google Scholar] [CrossRef]
- Avnir, D.; Coradin, T.; Lev, O.; Livage, J. Recent bio-applications of sol-gel materials. J. Mater. Chem. 2006, 16, 1013–1030. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds; MA-F(L/D)-OH are available from the authors upon request. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MGC (w/v) | Tgel (°C) (MGC/1%) | Tan δ (Gel-) | Tan δ (Gel + HRP) | Tan δ (Gel + Amylase) |
|---|---|---|---|---|---|
| MA-l-Phe-OH | 0.25% | 37/51 | 0.41 ± 0.03 | 0.43 ± 0.07 | 0.42 ± 0.12 |
| MA-d-Phe-OH | 0.25% | 37/51 | 0.41 ± 0.11 | 0.43 ± 0.02 | 0.42 ± 0.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcone, N.; Shao, T.; Rashid, R.; Kraatz, H.-B. Enzyme Entrapment in Amphiphilic Myristyl-Phenylalanine Hydrogels. Molecules 2019, 24, 2884. https://doi.org/10.3390/molecules24162884
Falcone N, Shao T, Rashid R, Kraatz H-B. Enzyme Entrapment in Amphiphilic Myristyl-Phenylalanine Hydrogels. Molecules. 2019; 24(16):2884. https://doi.org/10.3390/molecules24162884
Chicago/Turabian StyleFalcone, Natashya, Tsuimy Shao, Roomina Rashid, and Heinz-Bernhard Kraatz. 2019. "Enzyme Entrapment in Amphiphilic Myristyl-Phenylalanine Hydrogels" Molecules 24, no. 16: 2884. https://doi.org/10.3390/molecules24162884
APA StyleFalcone, N., Shao, T., Rashid, R., & Kraatz, H.-B. (2019). Enzyme Entrapment in Amphiphilic Myristyl-Phenylalanine Hydrogels. Molecules, 24(16), 2884. https://doi.org/10.3390/molecules24162884