Genetically Engineered Proteins to Improve Biomass Conversion: New Advances and Challenges for Tailoring Biocatalysts

 , , ,
, , ,
Abstract
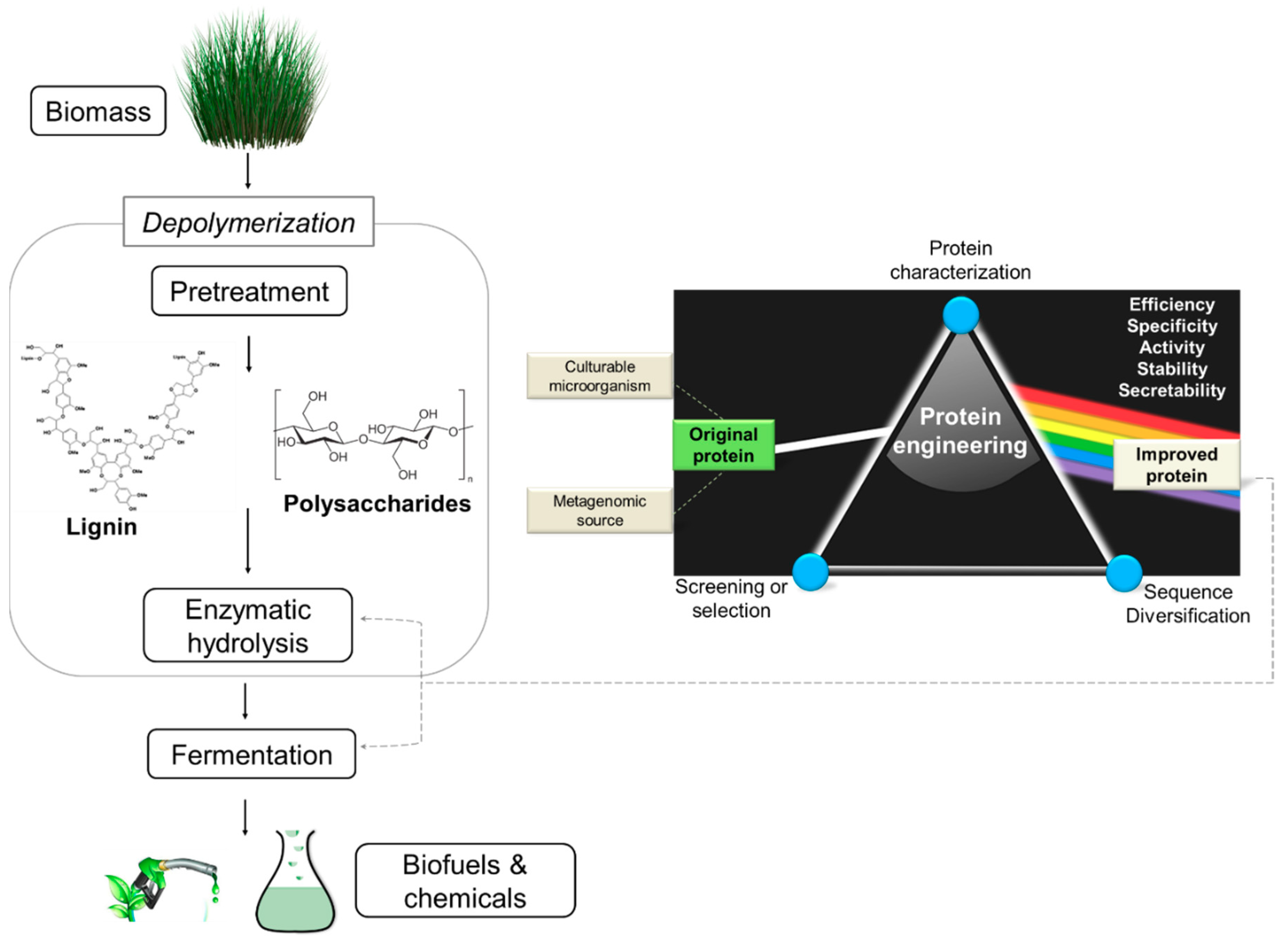
:1. Introduction
2. Protein Engineering
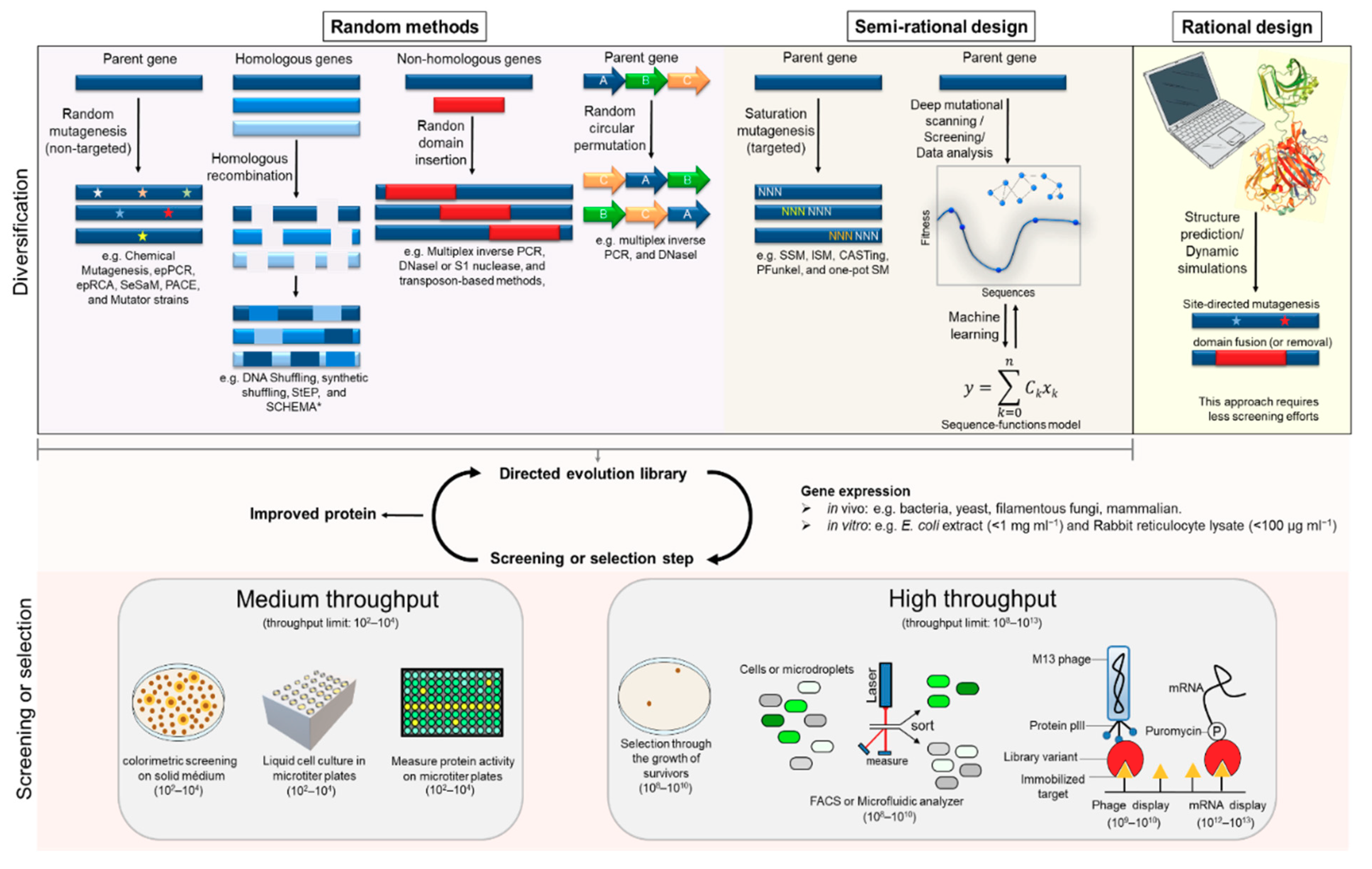
2.1. General Tools
2.1.1. Directed Evolution
2.1.2. Rational Design
2.2. Strategies to Improve Biomass Conversion
2.2.1. Engineering Protein Activity
Engineering Cellulosomes and Protein Scaffolds to Improve Biomass Deconstruction
Engineering Multifunctional Enzymes: One Enzyme with Multiple Activities
2.2.2. Engineering Protein Stability
Engineering Thermostability
Engineering Ionic Liquid Stable Variants
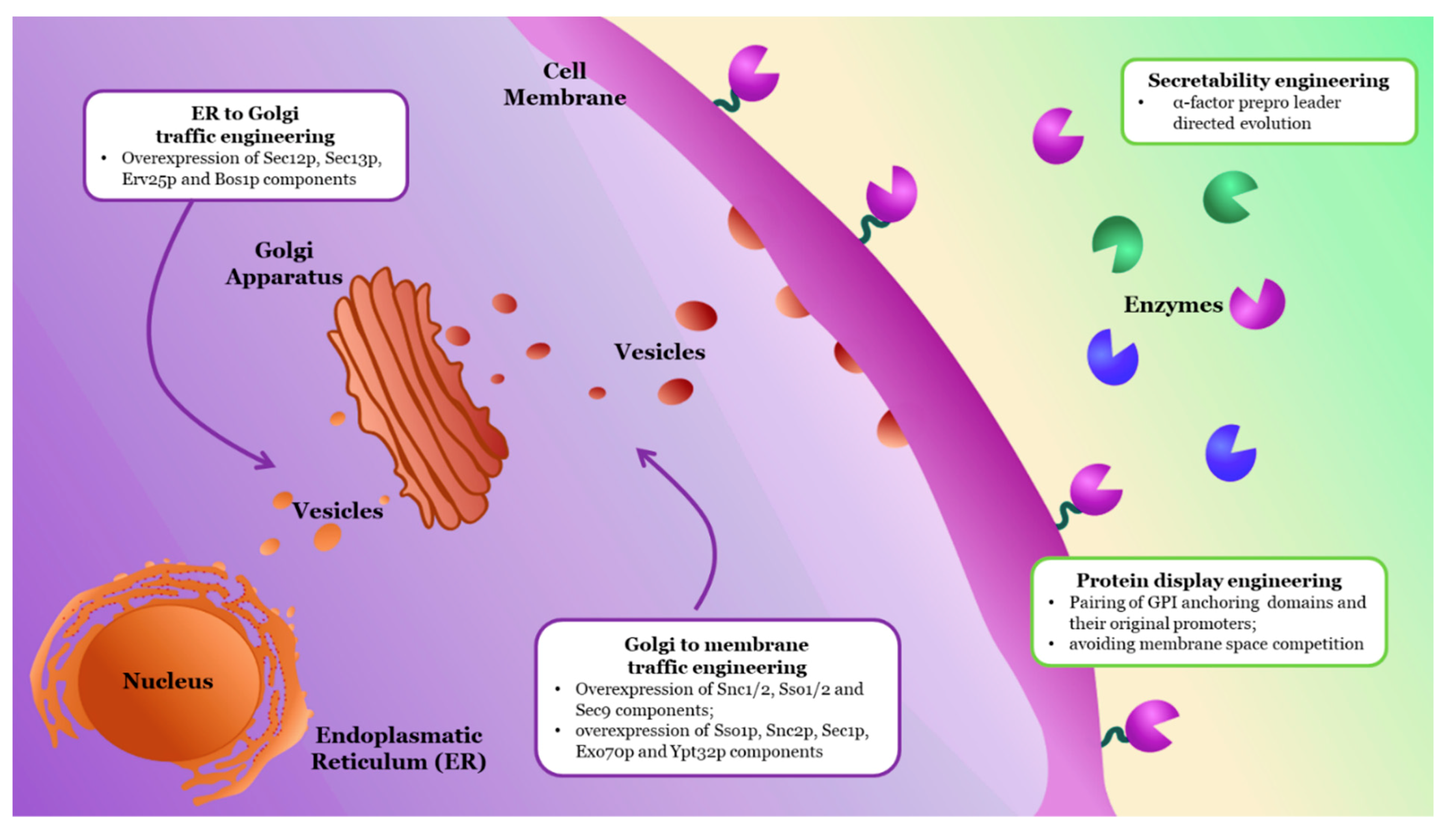
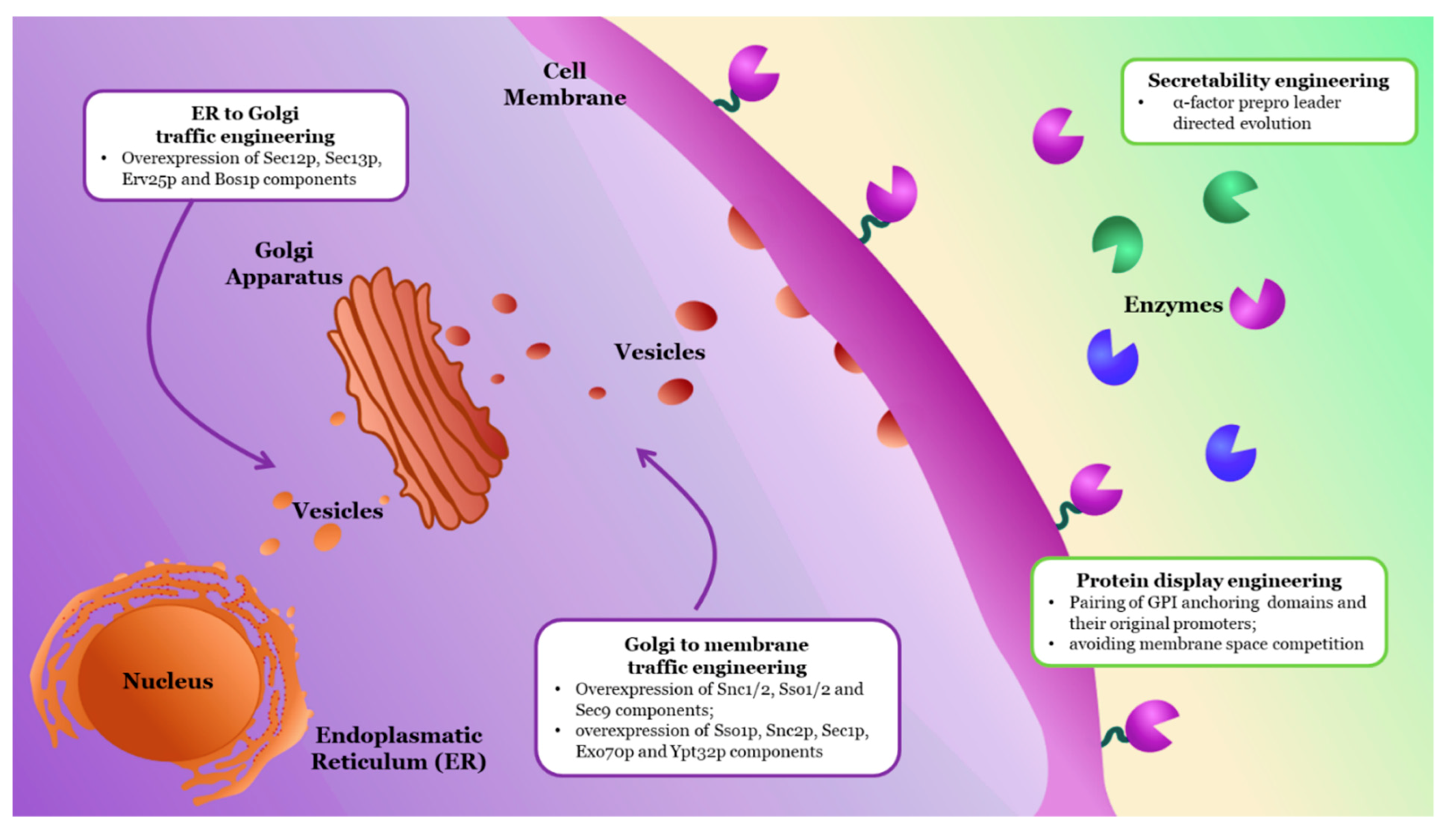
2.2.3. Engineering Functional Expression and Cellular Localization
2.2.4. Protein Engineering Guided by Deep Mutational Scanning
3. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saini, J.K.; Saini, R.; Tewari, L. Lignocellulosic agriculture wastes as biomass feedstocks for second-generation bioethanol production: Concepts and recent developments. 3 Biotech 2015, 5, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M. Sustainability of biofuels and renewable chemicals production from biomass. Curr. Opin. Chem. Biol. 2015, 29, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Jullesson, D.; David, F.; Pfleger, B.; Nielsen, J. Impact of synthetic biology and metabolic engineering on industrial production of fine chemicals. Biotechnol. Adv. 2015, 33, 1395–1402. [Google Scholar] [CrossRef]
- Lorenz, P.; Eck, J. Metagenomics and industrial applications. Nat. Rev. Microbiol. 2005, 3, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Handelsman, J. Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 2003, 14, 303–310. [Google Scholar] [CrossRef]
- Guazzaroni, M.E.; Silva-Rocha, R.; Ward, R.J. Synthetic biology approaches to improve biocatalyst identification in metagenomic library screening. Microb. Biotechnol. 2015, 8, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, F.; Hess, M. A perspective: Metatranscriptomics as a tool for the discovery of novel biocatalysts. J. Biotechnol. 2009, 142, 91–95. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Hu, J.; Davies, J.; Mok, Y.K.; Arato, C.; Saddler, J.N. The potential of using immobilized xylanases to enhance the hydrolysis of soluble, biomass derived xylooligomers. Materials 2018, 11, 2005. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, C.T.; Chiu, Y.T.; Wu, K.C.W. An effective cellulose-to-glucose-to-fructose conversion sequence by using enzyme immobilized Fe3O4-loaded mesoporous silica nanoparticles as recyclable biocatalysts. ChemCatChem 2013, 5, 2153–2157. [Google Scholar] [CrossRef]
- Dutta, S.; Wu, K.C.W. Enzymatic breakdown of biomass: Enzyme active sites, immobilization, and biofuel production. Green Chem. 2014, 16, 4615–4626. [Google Scholar] [CrossRef]
- Poluri, K.M.; Gulati, K. Protein Engineering Techniques: Gateways to Synthetic Protein Universe; Springer: Berlin, Germany, 2017; ISBN 9789811027314. [Google Scholar]
- Engqvist, M.K.M.; Rabe, K.S. Applications of Protein Engineering and Directed Evolution in Plant Research. Plant Physiol. 2019, 179, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Cobb, R.E.; Si, T.; Zhao, H. Directed evolution: An evolving and enabling synthetic biology tool. Curr. Opin. Chem. Biol. 2012, 16, 285–291. [Google Scholar] [CrossRef]
- Tiwari, M.K.; Singh, R.; Singh, R.K.; Kim, I.; Lee, J. Computational approaches for rational design of proteins with novel functionalities structural, catalytic, sensory, and regulatory functions. Comput. Struct. Biotechnol. J. 2012, 2, e201204002. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Tuning the activity of an enzyme for unusual environments: Sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc. Natl. Acad. Sci. USA 1993, 90, 5618–5622. [Google Scholar] [CrossRef]
- Arnold, F.H. Design by Directed Evolution. Acc. Chem. Res. 1998, 31, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379. [Google Scholar] [CrossRef]
- Currin, A.; Swainston, N.; Day, P.J.; Kell, D.B. Synthetic biology for the directed evolution of protein biocatalysts: Navigating sequence space intelligently. Chem. Soc. Rev. 2015, 44, 1172–1239. [Google Scholar] [CrossRef]
- Ribeiro, L.F.; Ribeiro, L.F.C. Improving fungal enzyme properties through protein engineering. In Fungal Enzymes; Polizeli, M., de Lourdes, T.M., Rai, M., Eds.; CRC Press: Boca Raton, FL, USA, 2013; Volume 1, pp. 1–425. [Google Scholar]
- Ribeiro, L.F.; Xiong, T.; Hauk, P.; Ribeiro, L.F.C. Protein engineering strategies to improve efficiency in biomass degradation. In Fungal Biotechnology for Biofuel Production; Bentham Science Publishers: Sharjah, UAE, 2015; pp. 202–221. [Google Scholar]
- Ribeiro, L.F.; Warren, T.D.; Ostermeier, M. Construction of protein switches by domain insertion and directed evolution. In Synthetic Protein Switches; Humana Press: New York, NY, USA, 2017; Volume 1596. [Google Scholar]
- Leung, D.W.; Chen, E.; Goeddel, D.V. A method for random mutagenesis of defined DNA segment using modified Polymerase chain reaction. Tech. A J. Methods Cell Mol. Biol. 1989, 1, 11–15. [Google Scholar]
- Cadwell, R.C.; Joyce, G.F. Randomization of genes by PCR mutagenesis. Genome Res. 1992, 2, 28–33. [Google Scholar] [CrossRef]
- Fujii, R.; Kitaoka, M.; Hayashi, K. Error-prone rolling circle amplification: The simplest random mutagenesis protocol. Nat. Protoc. 2006, 1, 2493. [Google Scholar] [CrossRef]
- Wong, T.S. Sequence saturation mutagenesis (SeSaM): A novel method for directed evolution. Nucleic Acids Res. 2004, 32, e26. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A system for the continuous directed evolution of biomolecules. Nature 2011, 472, 499. [Google Scholar] [CrossRef]
- Greener, A.; Callahan, M.; Jerpseth, B. An Efficient Random Mutagenesis Technique Using an E. coli Mutator Strain. Appl. Biochem. Biotechnol. Part B Mol. Biotechnol. 1997, 7, 189–195. [Google Scholar]
- Stemmer, W.P.C. Rapid Evolution of a Protein in-Vitro by DNA Shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Ness, J.E.; Kim, S.; Gottman, A.; Pak, R.; Krebber, A.; Borchert, T.V.; Govindarajan, S.; Mundorff, E.C.; Minshull, J. Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently. Nat. Biotechnol. 2002, 20, 1251. [Google Scholar] [CrossRef]
- Zhao, H.M.; Giver, L.; Shao, Z.X.; Affholter, J.A.; Arnold, F.H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 1998, 16, 258–261. [Google Scholar] [CrossRef]
- Voigt, C.A.; Martinez, C.; Wang, Z.G.; Mayo, S.L.; Arnold, F.H. Protein building blocks preserved by recombination. Nat. Struct. Biol. 2002, 9, 553. [Google Scholar] [CrossRef]
- Ribeiro, L.F.; Amarelle, V.; Ribeiro, L.F.C.; Guazzaroni, M.-E. Converting a Periplasmic Binding Protein into a Synthetic Biosensing Switch through Domain Insertion. Biomed Res. Int. 2019. [Google Scholar] [CrossRef]
- Ribeiro, L.F.; Nicholes, N.; Tullman, J.; Ribeiro, L.F.C.; Fuzo, C.A.; Vieira, D.S.; Furtado, G.P.; Ostermeier, M.; Ward, R.J. Insertion of a xylanase in xylose binding protein results in a xylose-stimulated xylanase. Biotechnol. Biofuels 2015, 8, 118. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, L.F.; Tullman, J.; Nicholes, N.; Silva, S.R.B.; Vieira, D.S.; Ostermeier, M.; Ward, R.J. A xylose-stimulated xylanase-xylose binding protein chimera created by random nonhomologous recombination. Biotechnol. Biofuels 2016, 9, 119. [Google Scholar] [CrossRef]
- Tullman, J.; Guntas, G.; Dumont, M.; Ostermeier, M. Protein Switches Identified from Diverse Insertion Libraries Created Using S1 Nuclease Digestion of Supercoiled-Form Plasmid DNA. Biotechnol. Bioeng. 2011, 108, 2535–2543. [Google Scholar] [CrossRef]
- Tullman, J.; Nicholes, N.; Dumont, M.R.; Ribeiro, L.F.; Ostermeier, M. Enzymatic protein switches built from paralogous input domains. Biotechnol. Bioeng. 2016, 113, 852–858. [Google Scholar] [CrossRef]
- Edwards, W.R.; Busse, K.; Allemann, R.K.; Jones, D.D. Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res. 2008, 36, e78. [Google Scholar] [CrossRef]
- Graf, R.; Schachman, H.K. Random circular permutation of genes and expressed polypeptide chains: Application of the method to the catalytic chains of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 1996, 93, 11591–11596. [Google Scholar] [CrossRef]
- Reitinger, S.; Yu, Y.; Wicki, J.; Ludwiczek, M.; D’Angelo, I.; Baturin, S.; Okon, M.; Strynadka, N.C.J.; Lutz, S.; Withers, S.G.; et al. Circular Permutation of Bacillus circulam Xylanase: A Kinetic and Structural Study. Biochemistry 2010, 49, 2464–2474. [Google Scholar] [CrossRef]
- Reidhaar-Olson, J.F.; Sauer, R.T. Combinatorial cassette mutagenesis as a probe of the informational content of protein sequences. Science 1988, 241, 53–57. [Google Scholar] [CrossRef]
- Reetz, M.T.; Carballeira, J.D. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2007, 2, 891. [Google Scholar] [CrossRef]
- Reetz, M.T.; Bocola, M.; Carballeira, J.D.; Zha, D.; Vogel, A. Expanding the range of substrate acceptance of enzymes: Combinatorial active-site saturation test. Angewandte Chemie. Int. Ed. 2005, 44, 4192–4196. [Google Scholar] [CrossRef]
- Firnberg, E.; Ostermeier, M. PFunkel: Efficient, Expansive, User-Defined Mutagenesis. PLoS ONE 2012, 7, e52031. [Google Scholar] [CrossRef]
- Wrenbeck, E.E.; Klesmith, J.R.; Stapleton, J.A.; Adeniran, A.; Tyo, K.E.J.; Whitehead, T.A. Plasmid-based one-pot saturation mutagenesis. Nat. Methods 2016, 13, 928. [Google Scholar] [CrossRef]
- Dixon, H.B.F.; Bielka, H.; Cantor, C.R. Nomenclature for incompletely specified bases in nucleic acid sequences. Recommendations 1984. J. Biol. Chem. 1986, 261, 13–17. [Google Scholar]
- Nov, Y. When Second Best Is Good Enough: Another Probabilistic Look at Saturation Mutagenesis. Appl. Environ. Microbiol. 2012, 78, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Damborsky, J.; Brezovsky, J. Computational tools for designing and engineering enzymes. Curr. Opin. Chem. Biol. 2014, 19, 8–16. [Google Scholar] [CrossRef]
- Hiraga, K.; Arnold, F.H. General method for sequence-independent site-directed chimeragenesis. J. Mol. Biol. 2003, 330, 287–296. [Google Scholar] [CrossRef]
- Wu, Z.; Kan, S.B.J.; Lewis, R.D.; Wittmann, B.J.; Arnold, F.H. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. USA 2019, 116, 8852–8858. [Google Scholar] [CrossRef] [Green Version]
- Steiner, K.; Schwab, H. Recent advances in rational approaches for enzyme engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209010. [Google Scholar] [CrossRef]
- Steffens, D.L.; Williams, J.G.K. Efficient site-directed saturation mutagenesis using degenerate oligonucleotides. J. Biomol. Tech. 2007, 18, 147. [Google Scholar]
- Cecchini, D.A.; Pepe, O.; Pennacchio, A.; Fagnano, M.; Faraco, V. Directed evolution of the bacterial endo-β-1,4-glucanase from Streptomyces sp. G12 towards improved catalysts for lignocellulose conversion. AMB Express 2018, 8, 74. [Google Scholar] [CrossRef]
- Zheng, F.; Tu, T.; Wang, X.; Wang, Y.; Ma, R.; Su, X.; Xie, X.; Yao, B.; Luo, H. Enhancing the catalytic activity of a novel GH5 cellulase GtCel5 from Gloeophyllum trabeum CBS 900.73 by site-directed mutagenesis on loop 6. Biotechnol. Biofuels 2018, 11, 1–13. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Z.; Zhou, S.; Mou, H.; Zhang, R. Cloning and expression of a β-mannanase gene from Bacillus sp. MK-2 and its directed evolution by random mutagenesis. Enzym. Microb. Technol. 2019, 124, 70–78. [Google Scholar] [CrossRef]
- Kim, S.-K.; Chung, D.; Himmel, M.E.; Bomble, Y.J.; Westpheling, J. Engineering the N -terminal end of CelA results in improved performance and growth of Caldicellulosiruptor bescii on crystalline cellulose. Biotechnol. Bioeng. 2017, 114, 945–950. [Google Scholar] [CrossRef]
- Wu, X.; Tian, Z.; Jiang, X.; Zhang, Q.; Wang, L. Biotechnologically relevant enzymes and proteins Enhancement in catalytic activity of Aspergillus niger XynB by selective site-directed mutagenesis of active site amino acids. Appl. Microbiol. Biotechnol. 2018, 102, 249–260. [Google Scholar] [CrossRef]
- Cheng, Y.-S.; Chen, C.-C.; Huang, J.-W.; Ko, T.-P.; Huang, Z.; Guo, R.-T. Improving the catalytic performance of a GH11 xylanase by rational protein engineering. Appl. Microbiol. Biotechnol. 2015, 99, 9503–9510. [Google Scholar] [CrossRef]
- Larue, K.; Melgar, M.; Martin, V.J.J. Directed evolution of a fungal β-glucosidase in Saccharomyces cerevisiae. Biotechnol. Biofuels 2016, 9, 1–15. [Google Scholar] [CrossRef]
- Saavedra, J.M.; Azócar, M.A.; Rodríguez, V.; Ramírez-Sarmiento, C.A.; Andrews, B.A.; Asenjo, J.A.; Parra, L.P. Relevance of Local Flexibility Near the Active Site for Enzymatic Catalysis: Biochemical Characterization and Engineering of Cellulase Cel5A From Bacillus agaradherans. Biotechnol. J. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Pourzardosht, N.; Dehnavi, E.; Ranaei Siadat, S.O.; Zamani, M.R.; Motallebi, M.; Nikzad Jamnani, F.; Aghaeepoor, M.; Barshan Tashnizi, M. Disulfide bonds elimination of endoglucanase II from Trichoderma reesei by site-directed mutagenesis to improve enzyme activity and thermal stability: An experimental and theoretical approach. Int. J. Biol. Macromol. 2018, 120, 1572–1580. [Google Scholar] [CrossRef]
- Torktaz, I.; Karkhane, A.A.; Hemmat, J. Rational engineering of Cel5E from Clostridium thermocellum to improve its thermal stability and catalytic activity. Appl. Microbiol. Biotechnol. 2018, 102, 8389–8402. [Google Scholar] [CrossRef]
- Bai, W.; Cao, Y.; Liu, J.; Wang, Q.; Jia, Z. Improvement of alkalophilicity of an alkaline xylanase Xyn11A-LC from Bacillus sp. SN5 by random mutation and Glu135 saturation mutagenesis. BMC Biotechnol. 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Smith, M.A.; Bedbrook, C.N.; Wu, T.; Arnold, F.H. Hypocrea jecorina cellobiohydrolase i stabilizing mutations identified using noncontiguous recombination. ACS Synth. Biol. 2013, 2, 690–696. [Google Scholar] [CrossRef]
- Goedegebuur, F.; Dankmeyer, L.; Gualfetti, P.; Karkehabadi, S.; Hansson, H.; Jana, S.; Huynh, V.; Kelemen, B.R.; Kruithof, P.; Larenas, E.A.; et al. Improving the thermal stability of cellobiohydrolase Cel7A from Hypocrea jecorina by directed evolution. J. Biol. Chem. 2017, 292, 17418–17430. [Google Scholar] [CrossRef]
- Heinzelman, P.; Snow, C.D.; Wu, I.; Nguyen, C.; Villalobos, A.; Govindarajan, S.; Minshull, J.; Arnold, F.H. A family of thermostable fungal cellulases created by structure-guided recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 5610–5615. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.; Arnold, F.H. Engineered thermostable fungal Cel6A and Cel7A cellobiohydrolases hydrolyze cellulose efficiently at elevated temperatures. Biotechnol. Bioeng. 2013, 110, 1874–1883. [Google Scholar] [CrossRef]
- Chokhawala, H.A.; Roche, C.M.; Kim, T.-W.; Atreya, M.E.; Vegesna, N.; Dana, C.M.; Blanch, H.W.; Clark, D.S. Mutagenesis of Trichoderma reesei endoglucanase I: Impact of expression host on activity and stability at elevated temperatures. BMC Biotechnol. 2015, 15, 11. [Google Scholar] [CrossRef]
- Akcapinar, G.B.; Venturini, A.; Martelli, P.L.; Casadio, R.; Sezerman, U.O. Modulating the thermostability of Endoglucanase I from Trichoderma reesei using computational approaches. Protein Eng. Des. Sel. 2015, 28, 127–135. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Song, X.; Hong, J.; Zhang, Y.; Yao, L. Improving Trichoderma reesei Cel7B Thermostability by Targeting the Weak Spots. J. Chem. Inf. Model. 2014, 54, 2826–2833. [Google Scholar] [CrossRef]
- Trudeau, D.L.; Lee, T.M.; Arnold, F.H. Engineered thermostable fungal cellulases exhibit efficient synergistic cellulose hydrolysis at elevated temperatures. Biotechnol. Bioeng. 2014, 111, 2390–2397. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, T.; Watanabe, M.; Yaoi, K. Improved thermostability of a metagenomic glucose-tolerant β-glycosidase based on its X-ray crystal structure. Appl. Microbiol. Biotechnol. 2017, 101, 8353–8363. [Google Scholar] [CrossRef]
- Tanghe, M.; Danneels, B.; Last, M.; Beerens, K.; Stals, I.; Desmet, T. Disulfide bridges as essential elements for the thermostability of lytic polysaccharide monooxygenase LPMO10C from Streptomyces coelicolor. Protein Eng. Des. Sel. 2017, 30, 401–408. [Google Scholar] [CrossRef]
- Niu, C.; Zhu, L.; Xu, X.; Li, Q. Rational design of thermostability in bacterial 1,3-1,4-β-glucanases through spatial compartmentalization of mutational hotspots. Appl. Microbiol. Biotechnol. 2017, 101, 1085–1097. [Google Scholar] [CrossRef]
- Khodakarami, A.; Goodarzi, N.; Hoseinzadehdehkordi, M.; Amani, F.; Khodaverdian, S.; Khajeh, K.; Ghazi, F.; Ranjbar, B.; Amanlou, M.; Dabirmanesh, B. Rational design toward developing a more efficient laccase: Catalytic efficiency and selectivity. Int. J. Biol. Macromol. 2018, 112, 775–779. [Google Scholar] [CrossRef]
- Ventorim, R.Z.; de Oliveira Mendes, T.A.; Trevizano, L.M.; dos Santos Camargos, A.M.; Guimarães, V.M. Impact of the removal of N-terminal non-structured amino acids on activity and stability of xylanases from Orpinomyces sp. PC-2. Int. J. Biol. Macromol. 2018, 106, 312–319. [Google Scholar] [CrossRef]
- Wang, K.; Luo, H.; Tian, J.; Turunen, O.; Huang, H.; Shi, P.; Hua, H.; Wang, C.; Wang, S.; Yao, B. Thermostability Improvement of a Streptomyces Xylanase by Introducing Proline and Glutamic Acid Residues. Appl. Environ. Microbiol. 2014, 80, 2158–2165. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Liu, Y.; Sun, M.; Han, Y.; Wang, J.; Sun, J.; Lu, F. Improvement of alkali stability and thermostability of Paenibacillus campinasensis Family-11 xylanase by directed evolution and site-directed mutagenesis. J. Ind. Microbiol. Biotechnol. 2014, 41, 153–162. [Google Scholar] [CrossRef]
- Shoseyov, O.; Shani, Z.; Levy, I. Carbohydrate Binding Modules: Biochemical Properties and Novel Applications. Microbiol. Mol. Biol. Rev. 2006, 70, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.J.; Huang, M.Y.; Pang, H.; Zhao, J.; Wu, C.X.; Feng, J.X. Characterization of a novel theme C glycoside hydrolase family 9 cellulase and its CBM-chimeric enzymes. Appl. Microbiol. Biotechnol. 2017, 101, 5723–5737. [Google Scholar] [CrossRef]
- Walker, J.A.; Takasuka, T.E.; Deng, K.; Bianchetti, C.M.; Udell, H.S.; Prom, B.M.; Kim, H.; Adams, P.D.; Northen, T.R.; Fox, B.G. Multifunctional cellulase catalysis targeted by fusion to different carbohydrate-binding modules. Biotechnol. Biofuels 2015, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Maldonado, R.; Meleiro, L.P.; Mendes, L.F.S.; Alves, L.F.; Carli, S.; Morero, L.D.; Basso, L.G.M.; Costa-Filho, A.J.; Ward, R.J. Lignocellulose binding of a Cel5A-RtCBM11 chimera with enhanced β-glucanase activity monitored by electron paramagnetic resonance. Biotechnol. Biofuels 2017, 10, 269. [Google Scholar] [CrossRef]
- Cicortas Gunnarsson, L.; Ohlin, M.; Albrekt, A.-S.; Holst, O.; Nordberg Karlsson, E.; Andersson, M. A carbohydrate binding module as a diversity-carrying scaffold. Protein Eng. Des. Sel. 2004, 17, 213–221. [Google Scholar] [CrossRef]
- Strobel, K.L.; Pfeiffer, K.A.; Blanch, H.W.; Clark, D.S. Structural insights into the affinity of Cel7A carbohydratebinding module for lignin. J. Biol. Chem. 2015, 290, 22818–22826. [Google Scholar] [CrossRef]
- Furtado, G.P.; Lourenzoni, M.R.; Fuzo, C.A.; Fonseca-Maldonado, R.; Guazzaroni, M.E.; Ribeiro, L.F.; Ward, R.J. Engineering the affinity of a family 11 carbohydrate binding module to improve binding of branched over unbranched polysaccharides. Int. J. Biol. Macromol. 2018, 120, 2509–2516. [Google Scholar] [CrossRef]
- Artzi, L.; Bayer, E.A.; Moraïs, S. Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef]
- Rentmeister, A. Current developments in cellulase engineering. Chemie Ingenieur Technik 2013, 85, 818–825. [Google Scholar] [CrossRef]
- Chen, C.; Cui, Z.; Song, X.; Liu, Y.J.; Cui, Q.; Feng, Y. Integration of bacterial expansin-like proteins into cellulosome promotes the cellulose degradation. Appl. Microbiol. Biotechnol. 2016, 100, 2203–2212. [Google Scholar] [CrossRef]
- Shamshoum, M.; Peleg, Y.; Bayer, E.A.; Arfi, Y.; Rogachev, I. Integration of bacterial lytic polysaccharide monooxygenases into designer cellulosomes promotes enhanced cellulose degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 9109–9114. [Google Scholar] [Green Version]
- Davidi, L.; Moraïs, S.; Artzi, L.; Knop, D.; Hadar, Y.; Arfi, Y.; Bayer, E.A. Toward combined delignification and saccharification of wheat straw by a laccase-containing designer cellulosome. Proc. Natl. Acad. Sci. USA 2016, 113, 10854–10859. [Google Scholar] [CrossRef] [Green Version]
- Dueber, J.E.; Wu, G.C.; Malmirchegini, G.R.; Moon, T.S.; Petzold, C.J.; Ullal, A.V.; Prather, K.L.J.; Keasling, J.D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. [Google Scholar] [CrossRef]
- Fierobe, H.P.; Mingardon, F.; Mechaly, A.; Bélaïch, A.; Rincon, M.T.; Pagès, S.; Lamed, R.; Tardif, C.; Bélaïch, J.P.; Bayer, E.A. Action of designer cellulosomes on homogeneous Versus complex substrates: Controlled incorporation of three distinct enzymes into a defined trifunctional scaffoldin. J. Biol. Chem. 2005, 280, 16325–16334. [Google Scholar] [CrossRef]
- Stern, J.; Moraïs, S.; Lamed, R.; Bayer, E.A. Adaptor scaffoldins: An original strategy for extended designer cellulosomes, inspired from nature. MBio 2016. [Google Scholar] [CrossRef]
- Mori, Y.; Ozasa, S.; Kitaoka, M.; Noda, S.; Tanaka, T.; Ichinose, H.; Kamiya, N. Aligning an endoglucanase Cel5A from Thermobifida fusca on a DNA scaffold: Potent design of an artificial cellulosome. Chem. Commun. 2013, 49, 6971–6973. [Google Scholar] [CrossRef]
- Moraïs, S.; Heyman, A.; Barak, Y.; Caspi, J.; Wilson, D.B.; Lamed, R.; Shoseyov, O.; Bayer, E.A. Enhanced cellulose degradation by nano-complexed enzymes: Synergism between a scaffold-linked exoglucanase and a free endoglucanase. J. Biotechnol. 2010, 147, 205–211. [Google Scholar] [CrossRef]
- Mitsuzawa, S.; Kagawa, H.; Li, Y.; Chan, S.L.; Paavola, C.D.; Trent, J.D. The rosettazyme: A synthetic cellulosome. J. Biotechnol. 2009, 143, 139–144. [Google Scholar] [CrossRef]
- Kim, D.M.; Nakazawa, H.; Umetsu, M.; Matsuyama, T.; Ishida, N.; Ikeuchi, A.; Takahashi, H.; Asano, R.; Kumagai, I. A nanocluster design for the construction of artificial cellulosomes. Catal. Sci. Technol. 2012, 2, 499–503. [Google Scholar] [CrossRef]
- Jeon, E.Y.; Baek, A.H.; Bornscheuer, U.T.; Park, J.B. Enzyme fusion for whole-cell biotransformation of long-chain sec-alcohols into esters. Appl. Microbiol. Biotechnol. 2015, 99, 6267–6275. [Google Scholar] [CrossRef]
- Blankley, R.T.; Tee, K.L.; Parker, D.A.; Matthews, S.; McLean, K.J.; Leys, D.; Munro, A.W.; Rattray, N.J. Production of alkenes and novel secondary products by P450 OleT JE using novel H2O2-generating fusion protein systems. FEBS Lett. 2017, 591, 737–750. [Google Scholar]
- Corrado, M.L.; Knaus, T.; Mutti, F.G. A Chimeric Styrene Monooxygenase with Increased Efficiency in Asymmetric Biocatalytic Epoxidation. ChemBioChem 2018, 19, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Neidle, E.L.; Johnson, C.W.; Beckham, G.T.; Linger, J.G.; Eiteman, M.A.; Tumen-Velasquez, M.; Ahmed, A.A.M. Chimeric Enzymes for Conversion of Lignin-Derived Chemicals. US Patent US10087425B2, 25 July 2017. [Google Scholar]
- Xu, Q.; Baker, J.O.; Himmel, M.E. Chimeric Enzymes with Improved Cellulase Activity. US Patent 8,993,276, 31 March 2015. [Google Scholar]
- Beck, Z.Q.; Fujdala, M.K.; Hansson, H.; Kaper, T.; Kralj, S.; Liu, A.D.; Mikkelsen, N.E.; Sandgren, M. Engineered Multifunctional Enzymes and Methods of Use. EP3234119A1, 25 October 2017. [Google Scholar]
- Arnold, F.H.; Heinzelman, P. Stable, functional chimeric cellobo hydrolase class enzymes. US20170037441A1, 9 February 2017. [Google Scholar]
- Taylor, L.E.; Knott, B.C.; Baker, J.O.; Alahuhta, P.M.; Hobdey, S.E.; Linger, J.G.; Lunin, V.V.; Amore, A.; Subramanian, V.; Podkaminer, K.; et al. Engineering enhanced cellobiohydrolase activity. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rizk, M.; Elleuche, S.; Antranikian, G. Generating bifunctional fusion enzymes composed of heat-active endoglucanase (Cel5A) and endoxylanase (XylT). Biotechnol. Lett. 2015, 37, 139–145. [Google Scholar] [CrossRef]
- Ribeiro, L.F.; Furtado, G.P.; Lourenzoni, M.R.; Costa-Filho, A.J.; Santos, C.R.; Peixoto Nogueira, S.C.; Betini, J.A.; Polizeli, M.D.L.T.M.; Murakami, M.T.; Ward, R.J. Engineering bifunctional laccase-xylanase chimeras for improved catalytic performance. J. Biol. Chem. 2011, 286, 43026–43038. [Google Scholar] [CrossRef]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72. [Google Scholar] [CrossRef]
- Kazlauskas, R. Engineering more stable proteins. Chem. Soc. Rev. 2018, 47, 9026–9045. [Google Scholar] [CrossRef]
- Khare, S.K.; Larroche, C. Current perspectives in enzymatic saccharification of lignocellulosic biomass. Biochem. Eng. J. 2015, 102, 38–44. [Google Scholar] [CrossRef]
- Hosseini Koupaie, E.; Dahadha, S.; Bazyar Lakeh, A.A.; Azizi, A.; Elbeshbishy, E. Enzymatic pretreatment of lignocellulosic biomass for enhanced biomethane production—A review. J. Environ. Manage. 2019, 233, 774–784. [Google Scholar] [CrossRef]
- Hendriks, A.T.W.M.; Zeeman, G. Pretreatments to enhance the digestibility of lignocellulosic biomass. Bioresour. Technol. 2009, 100, 10–18. [Google Scholar] [CrossRef]
- Druzhinina, I.S.; Kubicek, C.P. Genetic engineering of Trichoderma reesei cellulases and their production. Microb. Biotechnol. 2017, 10, 1485–1499. [Google Scholar] [CrossRef]
- Gupta, V.K.; O’Donovan, A.; Tuohy, M.G.; Sharma, G.D. Trichoderma in bioenergy research. In Biotechnology and Biology of Trichoderma; Elsevier: Amsterdam, The Netherlands, 2014; pp. 325–336. [Google Scholar]
- Smith, M.A.; Romero, P.A.; Wu, T.; Brustad, E.M.; Arnold, F.H. Chimeragenesis of distantly-related proteins by noncontiguous recombination. Protein Sci. 2013, 22, 231–238. [Google Scholar] [CrossRef]
- Lantz, S.E.; Goedegebuur, F.; Hommes, R.; Kaper, T.; Kelemen, B.R.; Mitchinson, C.; Wallace, L.; Ståhlberg, J.; Larenas, E.A. Hypocrea jecorina CEL6A protein engineering. Biotechnol. Biofuels 2010, 3, 20. [Google Scholar] [CrossRef]
- Ghandi, K. A Review of Ionic Liquids, Their Limits and Applications. Green Sustain. Chem. 2014, 4, 44. [Google Scholar] [CrossRef]
- Xu, J.; Xiong, P.; He, B. Advances in improving the performance of cellulase in ionic liquids for lignocellulose biorefinery. Bioresour. Technol. 2016, 200, 961–970. [Google Scholar] [CrossRef]
- Sadaf, A.; Grewal, J.; Khare, S.K. Ionic liquid stable cellulases and hemicellulases: Application in biobased production of biofuels. In Waste Biorefinery; Elsevier B.V.: Amsterdam, The Netherlands, 2018; ISBN 9780444639929. [Google Scholar]
- Wolski, P.W.; Dana, C.M.; Clark, D.S.; Blanch, H.W. Engineering ionic liquid-tolerant cellulases for biofuels production. Protein Eng. Des. Sel. 2016, 29, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Wolski, P.W.; Clark, D.S.; Blanch, H.W. Green fluorescent protein as a screen for enzymatic activity in ionic liquid–aqueous systems for in situhydrolysis of lignocellulose. Green Chem. 2011, 13, 3107–3110. [Google Scholar] [CrossRef]
- Pottkämper, J.; Barthen, P.; Ilmberger, N.; Schwaneberg, U.; Schenk, A.; Schulte, M.; Ignatiev, N.; Streit, W.R. Applying metagenomics for the identification of bacterial cellulases that are stable in ionic liquids. Green Chem. 2009, 11, 957–965. [Google Scholar] [CrossRef]
- Chen, Z.; Pereira, J.H.; Liu, H.; Tran, H.M.; Hsu, N.S.Y.; Dibble, D.; Singh, S.; Adams, P.D.; Sapra, R.; Hadi, M.Z.; et al. Improved activity of a thermophilic cellulase, Cel5A, from Thermotoga maritima on ionic liquid pretreated switchgrass. PLoS ONE 2013, 8, 1–9. [Google Scholar] [CrossRef]
- Lynd, L.R.; Van Zyl, W.H.; McBride, J.E.; Laser, M. Consolidated bioprocessing of cellulosic biomass: An update. Curr. Opin. Biotechnol. 2005, 16, 577–583. [Google Scholar] [CrossRef]
- Lambertz, C.; Garvey, M.; Klinger, J.; Heesel, D.; Klose, H.; Fischer, R.; Commandeur, U. Challenges and advances in the heterologous expression of cellulolytic enzymes: A review. Biotechnol. Biofuels 2014, 7, 135. [Google Scholar] [CrossRef]
- Zsebo, K.; Lu, H.; Fieschko, J.; Goldstein, L.; Davis, J.; Duker, K.; Suggs, S.; Lai, P.; Bitter, G. Protein secretion from Saccharomyces cerevisiae directed by the prepro-alpha-factor leader region. J. Biol. Chem. 1986, 261, 5858–5865. [Google Scholar]
- Camarero, S.; Pardo, I.; Cañas, A.I.; Molina, P.; Record, E.; Martínez, A.T.; Martínez, M.J.; Alcalde, M. Engineering Platforms for Directed Evolution of Laccase from Pycnoporus cinnabarinus. Appl. Environ. Microbiol. 2012, 78, 1370–1384. [Google Scholar] [CrossRef]
- Mateljak, I.; Tron, T.; Alcalde, M. Evolved α-factor prepro-leaders for directed laccase evolution in Saccharomyces cerevisiae. Microb. Biotechnol. 2017, 10, 1830–1836. [Google Scholar] [CrossRef]
- Avelar, M.; Olvera, C.; Aceves-Zamudio, D.; Folch, J.L.; Ayala, M. Recombinant expression of a laccase from Coriolopsis gallica in Pichia pastoris using a modified α-factor preproleader. Protein Expr. Purif. 2017, 136, 14–19. [Google Scholar] [CrossRef]
- Van Zyl, J.H.D.; Den Haan, R.; Van Zyl, W.H. Over-expression of native Saccharomyces cerevisiae exocytic SNARE genes increased heterologous cellulase secretion. Appl. Microbiol. Biotechnol. 2014, 98, 5567–5578. [Google Scholar] [CrossRef]
- Tang, H.; Song, M.; He, Y.; Wang, J.; Wang, S.; Shen, Y.; Hou, J.; Bao, X. Engineering vesicle trafficking improves the extracellular activity and surface display efficiency of cellulases in Saccharomyces cerevisiae. Biotechnol. Biofuels 2017, 10, 1–13. [Google Scholar] [CrossRef]
- Ueda, M.; Tanaka, A. Cell surface engineering of yeast: Construction of arming yeast with biocatalyst. J. Biosci. Bioeng. 2000, 90, 125–136. [Google Scholar] [CrossRef]
- Inokuma, K.; Bamba, T.; Ishii, J.; Ito, Y.; Hasunuma, T.; Kondo, A. Enhanced cell-surface display and secretory production of cellulolytic enzymes with Saccharomyces cerevisiae Sed1 signal peptide. Biotechnol. Bioeng. 2016, 113, 2358–2366. [Google Scholar] [CrossRef]
- Kondo, A.; Ueda, M. Yeast cell-surface display—Applications of molecular display. Appl. Microbiol. Biotechnol. 2004, 64, 28–40. [Google Scholar] [CrossRef]
- Inokuma, K.; Hasunuma, T.; Kondo, A. Efficient yeast cell-surface display of exo-and endo-cellulase using the SED1 anchoring region and its original promoter. Biotechnol. Biofuels 2014, 7, 1–11. [Google Scholar] [CrossRef]
- Bamba, T.; Inokuma, K.; Hasunuma, T.; Kondo, A. Enhanced cell-surface display of a heterologous protein using SED1 anchoring system in SED1-disrupted Saccharomyces cerevisiae strain. J. Biosci. Bioeng. 2018, 125, 306–310. [Google Scholar] [CrossRef]
- Yıldırım, Z.; Çelik, E. Periplasmic and extracellular production of cellulase from recombinant Escherichia coli cells. J. Chem. Technol. Biotechnol. 2017, 92, 319–324. [Google Scholar] [CrossRef]
- Huang, G.L.; Gosschalk, J.E.; Kim, Y.S.; Ogorzalek Loo, R.R.; Clubb, R.T. Stabilizing displayed proteins on vegetative Bacillus subtilis cells. Appl. Microbiol. Biotechnol. 2018, 102, 6547–6565. [Google Scholar] [CrossRef]
- Araya, C.L.; Fowler, D.M. Deep mutational scanning: Assessing protein function on a massive scale. Trends Biotechnol. 2011, 29, 435–442. [Google Scholar] [CrossRef]
- Fowler, D.M.; Stephany, J.J.; Fields, S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc. 2014, 9, 2267–2284. [Google Scholar] [CrossRef]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef]
- Wrenbeck, E.E.; Bedewitz, M.A.; Klesmith, J.R.; Noshin, S.; Barry, C.S.; Whitehead, T.A. An Automated Data-Driven Pipeline for Improving Heterologous Enzyme Expression. ACS Synth. Biol. 2019, 8, 474–481. [Google Scholar] [CrossRef]
- Klesmith, J.R.; Bacik, J.-P.; Wrenbeck, E.E.; Michalczyk, R.; Whitehead, T.A. Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proc. Natl. Acad. Sci. USA 2017, 114, 2265–2270. [Google Scholar] [CrossRef] [Green Version]
- Elazar, A.; Weinstein, J.; Biran, I.; Fridman, Y.; Bibi, E.; Fleishman, S.J. Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. Elife 2016, 5, 1–19. [Google Scholar] [CrossRef]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef] [Green Version]
- Klesmith, J.R.; Bacik, J.P.; Michalczyk, R.; Whitehead, T.A. Comprehensive Sequence-Flux Mapping of a Levoglucosan Utilization Pathway in E. coli. ACS Synth. Biol. 2015, 4, 1235–1243. [Google Scholar] [CrossRef]
- Lapidoth, G.; Khersonsky, O.; Lipsh, R.; Dym, O.; Albeck, S.; Rogotner, S.; Fleishman, S.J. Highly active enzymes by automated combinatorial backbone assembly and sequence design. Nat. Commun. 2018, 9, 2780. [Google Scholar] [CrossRef]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320. [Google Scholar] [CrossRef]
- Ribeiro, L.F.; Ribeiro, L.F.; Barreto, M.Q.; Ward, R.J. Protein engineering strategies to expand CRISPR-Cas9 applications. Int. J. Genom. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Jakočiūnas, T.; Pedersen, L.E.; Lis, A.V.; Jensen, M.K.; Keasling, J.D. CasPER, a method for directed evolution in genomic contexts using mutagenesis and CRISPR/Cas9. Metab. Eng. 2018, 48, 288–296. [Google Scholar] [CrossRef]
- Price, M.A.; Cruz, R.; Baxter, S.; Escalettes, F.; Rosser, S.J. CRISPR-Cas9 In Situ engineering of subtilisin E in Bacillus subtilis. PLoS ONE 2019, 14, e0210121. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Degenerate Codon a | Number of Codons | Number of Amino Acids | Number of Stop Codons | Encoded Amino Acid | Library Size for 2 Positions b | Library Size for 3 Positions b |
|---|---|---|---|---|---|---|
| NNN | 64 | 20 | 3 | All | 995 | 25585 |
| NNK | 20 | 20 | 1 | All | 875 | 21051 |
| NNS | 32 | 20 | 1 | All | 875 | 21051 |
| DBK | 18 | 12 | 0 | ARCGILMFSTWV | 279 | 3812 |
| NDT | 12 | 12 | 0 | RNDCGHILFSYV | 215 | 2587 |
| NRT | 8 | 8 | 0 | RNDCGHSY | 95 | 766 |
| NAN | 16 | 7 | 2 | YHNQKDE | 95 | 766 |
| NTN | 16 | 5 | 0 | MFLIV | 62 | 409 |
| NCN | 16 | 4 | 0 | SPTA | 23 | 95 |
| RST | 4 | 4 | 0 | AGST | 23 | 95 |
| Enzyme | Organism Source | Methods a | Variant | Improved Characteristic b | Refs | ||
|---|---|---|---|---|---|---|---|
| Activity | Thermostability | pH | |||||
| endo-β-1,4-glucanase (GH12) | Streptomyces sp. G12 | epPCR | 1) epCS_2 (G145D/ N207K) 2) epCS_1 (G263C/ R307H) 3) epCS_4 (T67N/ D142E/ S218N/ V242D/ D330E) | ↑ ~1.4-fold, ~1.4-fold, ~1.7-fold in activity at 60 °C for epCS_2, epCS_1 and epCS_4, respectively, in AZO-CMC | 34% remaining activity after 72 h at 60 °C for epCS_1 (17% for WT) | epCS_4 activity ↑ ~2 fold at pH 5 and ~2.6 fold at pH 6 | [54] |
| GH5 cellulase Cel5 | Gloeophyllum trabeum CBS 900.73 | SDM | 1) N233A 2) N233G | ↑ kcat/KM: 45% and 52% for N233A and N233G, respectively, using CMC as substrate | NSI | N233G retained ~75% activity after 1h at 37 °C in pH 2 (~30% for WT) | [55] |
| β-mannanase | Bacillus sp. MK-2 | epPCR | 1) Q112R 2) L211I 3) K291E | ↑ kcat/KM: 2.8-, 1.7- and 4.2-fold for Q112R, L211I, and K291E, respectively, using konjac glucomannan as substrate | NSI | ND | [56] |
| CelA GH9 and CelA GH48 | Caldicellulosiruptor bescii | rational design | 1) D5-GH48 2) D5-GH9 | D5-GH48 30%↑ activity in Avicel D5-GH9 82%↑activity in CMC | ND | ND | [57] |
| XynB xylanase | Aspergillus niger ATCC1015 | SDM | 1) T43E 2) S41N/T43E | ↑ kcat/KM: 20% and 65% for T43E and S41N/T43E, respectively | S41N/T43E: 60% and 35% remaining activity after 60 min and 120 min at 60 °C, respectively (~12% for WT at both times) | ND | [58] |
| XynCDBFV xylanase | Neocallimastix patriciarum | SDM | 1) W125F 2) F163W 3) W125F/F163W | ↑ enzyme activity: 10% for single mutants and 20% for double mutant using xylan beechwood as substrate | W125F/F163W slightly more residual activity (~6-8%) at 80 °C and 90 °C for 120 and 30 min, respectively. | ND | [59] |
| β-glucosidase GH3 (BGL1) | A. niger | directed evolution | 1) Y305C 2) Q140L | Y305C reduced transglucosylation activity Q140L reduced the inhibitory effect of glucose | ND | ND | [60] |
| Cel5A | Bacillus agaradherans | SDM | 1) N141L 2) A137Y 3) I102A/A137Y | 3-, 1.5-, and 3.4-fold ↑ activity for N141L, A137Y and I102A/A137Y, respectively | ND | ND | [61] |
| Glucanase II (Cel5A) | Trichoderma reesei | SDM | 1) C99V 2) C323H | ↑ kcat/KM 1.87-fold and 1.3-fold for C99V and C323H, respectively, using CMC as substrate | ↑T1/2 2.4-fold (80 °C), 2.01-fold (70 °C), 1.8-fold (60 °C) for C99V ↑T1/2 2.34-fold (80 °C), 1.81 -fold (70 °C), 1.6-fold (60 °C) for C232H | ND | [62] |
| GH5 Cel5E | Clostridium thermocellum | rational engineering using overlapping PCR | 1) N94W 2) N94F 3) E133F 4) N94A | N94W, N94F, E133F, and N94A mutants showed 1.92-, 1.29-, 1.1-, and 1.15-fold ↑ CMCase activity and 1.46-, 1.29-, 1.11- and 1.12-fold ↑β-glucanase activity on barley β-glucan | N94W, N94F, E133F, and N94A retained 92%, 91%, 93%, and 90% of residual CMCase activity (WT ~86%) and 91%, 89%, 91%, and 88% of residual β-glucanase activity (WT 82%), during 4 h at 62 °C | N94W ~2-fold and 1.5-fold ↑activity at pH 6–8 in CMC and barley β-glucan, respectively | [63] |
| cellulase cocktail from T. reesei | T. reesei | succinylation | change enzyme surface charges | 2-fold ↑ in cellulose conversion in 15% (v/v) 1-butyl-3-methylimidazolium chloride and further reduction in apparent KM of the enzyme cocktail for Avicel by 2.7-fold due to reducing lignin inhibition | ND | ND | |
| family 11 alkaline xylanase Xyn11A-LC | Bacillus sp. SN5 | epPCR | 1) V116A/ E135V 2) E135V 3) E135R | 1.1-, 1.5-, and 1.3-fold ↑ in catalytic efficiencies for V116A/ E135V , E135V, and E135R, respectively | E135V and E135R, 73.1% and 77.8% remaining activity, respectively, at 60 °C for 30 min (33.5% for WT) | E135V ↑relative activity regarding the WT: 17.5% at pH 8.5, 18.9% at pH 9 and 14.3% at pH 10 E135V and E135R retain over 80% of activity in pH 4.5–10 (WT in pH 8–10) | [64] |
| Cel7A | Hypocrea jecorina | SCHEMA | F362M | ↑ activity by ~3-fold at 90 min and 49 °C on MUL | ↑ stability by 3 °C | ND | [65] |
| Cel7A | H. jecorina | directed evolution | FCA398 (S8P/T41I/N49S/A68T/N89D/S92T/S113N/S196T/P227L/D249K/T255P/S278P/E295K/T296P/T332Y/V403D/S411F/T462I) | ↑ activity by ~2-fold at 70 h and 65 °C on PASC | ↑Tm in 10.4 °C and ↑T1/2 by 44-fold, 34-fold, and 9-fold at 62 °C, 66 °C, and 69 °C, respectively. | ND | [66] |
| Cel6A | H. jecorina | SCHEMA, random mutagenesis and directed evolution | 1) HJPlus (not specify) 2) 1G6 (S317P respect to HJPlus) 3) 2B3 (Q277L/S317P respect to HJPlus) 4) 3C6P (S30F/V128A/M135L/Q277L/S317P/S406P/S413P respect to HJPlus) | ↑in activity on Avicel after 2h: ~1.25-fold, ~1.8-fold, and ~6.5-fold at 60 °C, 65 °C, and 70 °C, respectively, for both HJPlus and 3C6P ~9-fold and ~15-fold at 75 °C, 8-fold and 28-fold at 80 °C for HJPlus and 3C6P, respectively ~16-fold at 85 °C for 3C6P | 3.5-fold, 5.6-fold, 14.8-fold and 112-fold ↑ in T1/2 for HJPlus, 1G6, 2B3 and 3C6P, respectively. 6.7 °C, 8 °C, 10.5 °C and 14.9 °C ↑ in T50 for HJPlus, 1G6, 2B3 and 3C6P, respectively. | ~3-fold and ~5-fold ↑activity at pH 7 and pH 8 for HJPlus | [67,68] |
| Cel7B | T. reesei | structure-guided evolution | G230A/D113S/D115T | ↑ in specific activity after 15 h: ~4-fold at 50 °C on CMC ~1.4 and ~2-fold at 60 °C and 65 °C, respectively, on Avicel ~1.6 and ~3-fold at 60 °C and 65 °C, respectively, on MUC | ↑Tm in 3 °C and ↑ T1/2 by ~2-fold at 60 °C | ND | [69] |
| Cel7B | T. reesei | SDM | Q274V | NSI | ~10 °C ↑ in optimal temperature ~80% and ~40% remaining activity at 45 °C and 65 °C, respectively, for 8 h (WT ~40% and ~20%) | Retained 70% activity at pH 4 (WT 20%) | [70] |
| Cel7B | T. reesei | SDM to introduce disulfide bonds | G4C -F71C/N160C-G183C/S168T | ↑ in specific activity ∼1.3-fold and ∼2.5-fold in Avicel and FP, respectively, after 24 h at 50 °C | 8.2 °C ↑ in T50 and ∼10 °C ↑in optimal temperature | ND | [71] |
| Cel5A | H. jecorina | SDM | OptCel5A (F191V/T233V/V265T/S318P/D271Y/S79P/E53D/T57N/G293A/V101I/N155E/T80E/S133R/G239E/S309W/G189S) | ↑~1.5-fold in activity on Avicel after 60h at 70 °C in comparison to the WT at 60 °C | 17 °C ↑ in optimal temperature | ND | [72] |
| β-glucosidase MeBglD2 | Isolated in a function-driven metagenomic screening | SDM | 1)N59C/A295G 2)H8L/N59C/A295G | NSI | ∼10 °C ↑in optimal temperature for both variants 7 °C and 9 °C ↑ in Tm for N59C/A295G and H8L/N59C/A295G, respectively. 50% remaining activity at 62 °C for 30 min for both variants (WT inactive already at 56 °C) | ND | [73] |
| LPMO | Streptomyces coelicolor LPMO10C | SDM | A143C-P183C/ S73C-A115C | ND | 12 °C ↑ in Tm and 60% remaining activity after 2 h at 80 °C (WT 30%) | ND | [74] |
| BglT | Bacillus terquilensis | ISM | E46P/S43E/ H205P/S40E | ↑ specific activity 64.4% after 10min at 40 °C on AZO- barley β-glucan and ∼1.5-fold ↑ in kcat/KM | ∼20 °C ↑ in optimal temperature and 13.8 °C ↑ in Tm ↑T1/2 by 3.86-fold and 7.13-fold at 60 °C and 70 °C, respectively | Optimal pH shifted from pH 6.5 to pH 6.0 Retained 63.1% and 80.7% activity at pH 4.5 and pH 5.5, respectively (WT 7.9% and 59.9%) | [75] |
| laccase | Bacillus HR03 | SDM | 1) T415G 2) T415I 3) T418I | ↑kcat/KM 1.5-, 3.7-, and 1.5-fold on ABTS for T415G, T415I and T418I, respectively | ↑T1/2 by 2-fold for T415I at 80 °C | ND | [76] |
| xylanase | Orpinomyces sp. PC-2 | directed evolution guided by molecular dynamics | SM2 (V135A/A226and T and N-terminal region removed) | 30,250-fold increase in kcat/KM in beechwood xylan (2.9 × 1010 mL min−1 mg−1 vs. 8.0 × 106 mL min−1) | ↑T1/2 by 38-fold at 50 °C | Retained 45% activity at pH 3 (WT ~10%) | [77] |
| xylanase XynAS9 | Streptomyces sp. strain S9 | SDM | 1)V81P/G82E 2)V81P/G82E/D185P/S186E | NSI | ↑T1/2 by 16- and 14-fold at 65 °C, and 3.7- and 4.5-fold at 80 °C, for V81P/G82E and V81P/G82E/D185P/S186E, respectively 6.9 °C and 11.2 °C ↑in Tm for V81P/G82E and V81P/G82E/D185P/S186E, respectively 17 °C ↑ in optimal temperature for both variants | ND | [78] |
| Family-11 xylanase | Paenibacillus campinasensis | directed evolution and SDM | 1) XynG1-1B43(V90R/P172H) 2) XynG1-1B43cc16(V90R/P172H/T84C-T182C/D16Y) | 1.2- and 1.4-fold↑in kcat/KM for XynG1-1B43 and XynG1-1B43cc16, respectively, on beechwood xylan | 10 °C ↑ in optimal temperature for both variants 24.5% remaining activity at 90 °C for 120 min for XynG1-1B43cc16 (WT inactive) | ~3.5-fold ↑activity at pH 11 for XynG1-1B43cc16. | [79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, L.F.; Amarelle, V.; Alves, L.d.F.; Viana de Siqueira, G.M.; Lovate, G.L.; Borelli, T.C.; Guazzaroni, M.-E. Genetically Engineered Proteins to Improve Biomass Conversion: New Advances and Challenges for Tailoring Biocatalysts. Molecules 2019, 24, 2879. https://doi.org/10.3390/molecules24162879
Ribeiro LF, Amarelle V, Alves LdF, Viana de Siqueira GM, Lovate GL, Borelli TC, Guazzaroni M-E. Genetically Engineered Proteins to Improve Biomass Conversion: New Advances and Challenges for Tailoring Biocatalysts. Molecules. 2019; 24(16):2879. https://doi.org/10.3390/molecules24162879
Chicago/Turabian StyleRibeiro, Lucas Ferreira, Vanesa Amarelle, Luana de Fátima Alves, Guilherme Marcelino Viana de Siqueira, Gabriel Lencioni Lovate, Tiago Cabral Borelli, and María-Eugenia Guazzaroni. 2019. "Genetically Engineered Proteins to Improve Biomass Conversion: New Advances and Challenges for Tailoring Biocatalysts" Molecules 24, no. 16: 2879. https://doi.org/10.3390/molecules24162879