Site Selective and Efficient Spin Labeling of Proteins with a Maleimide-Functionalized Trityl Radical for Pulsed Dipolar EPR Spectroscopy

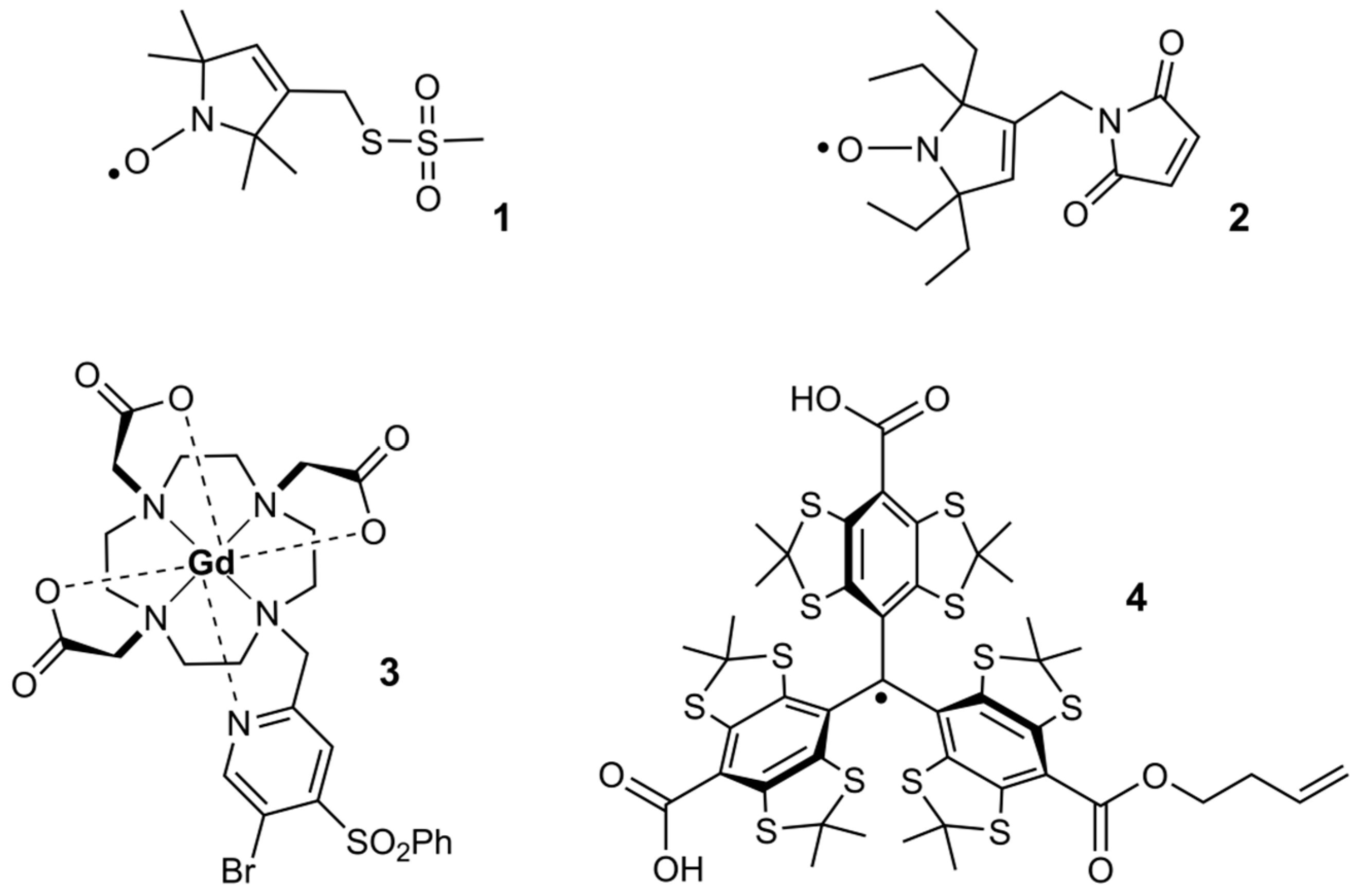{kind=link}
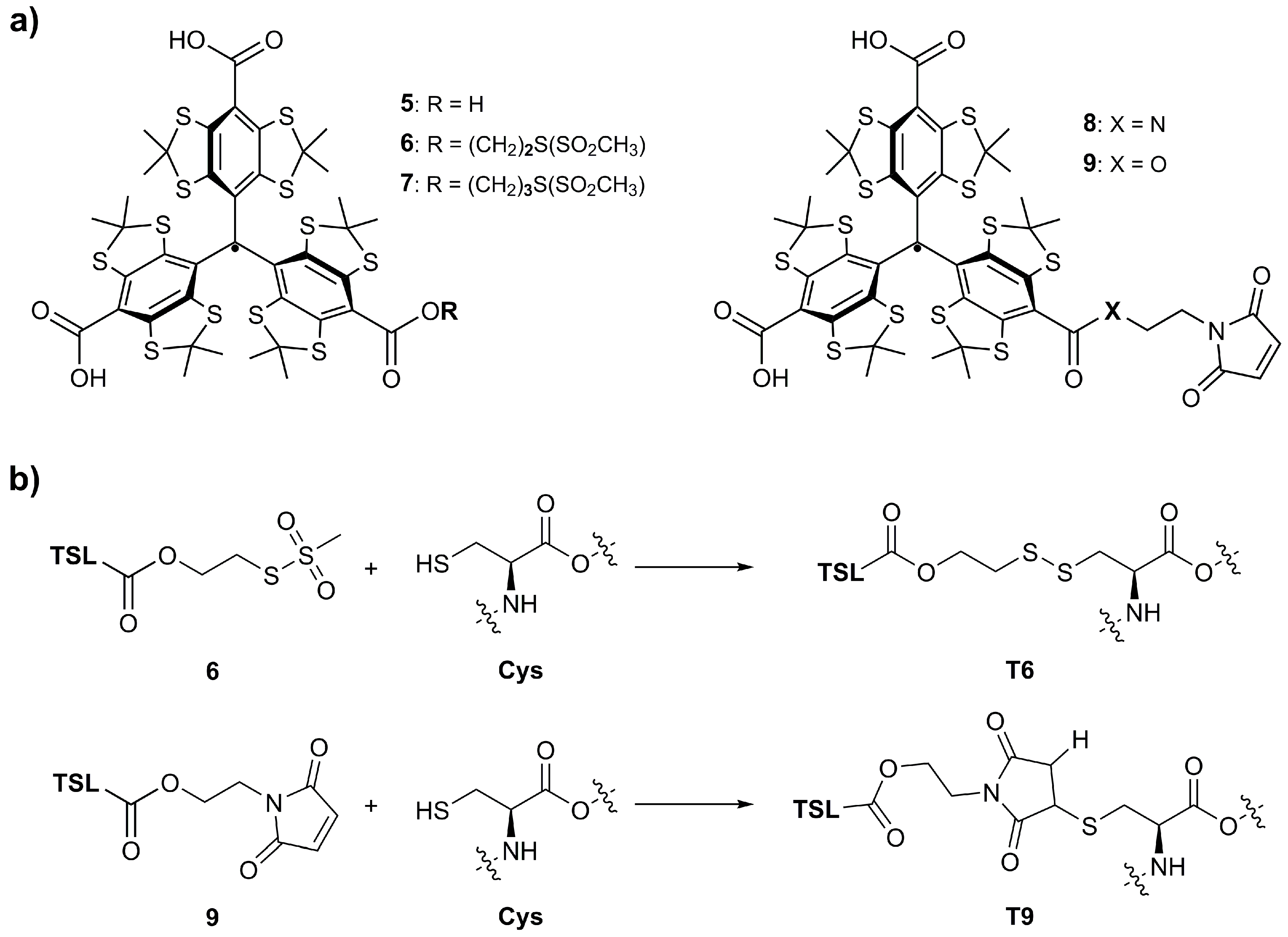{kind=link}
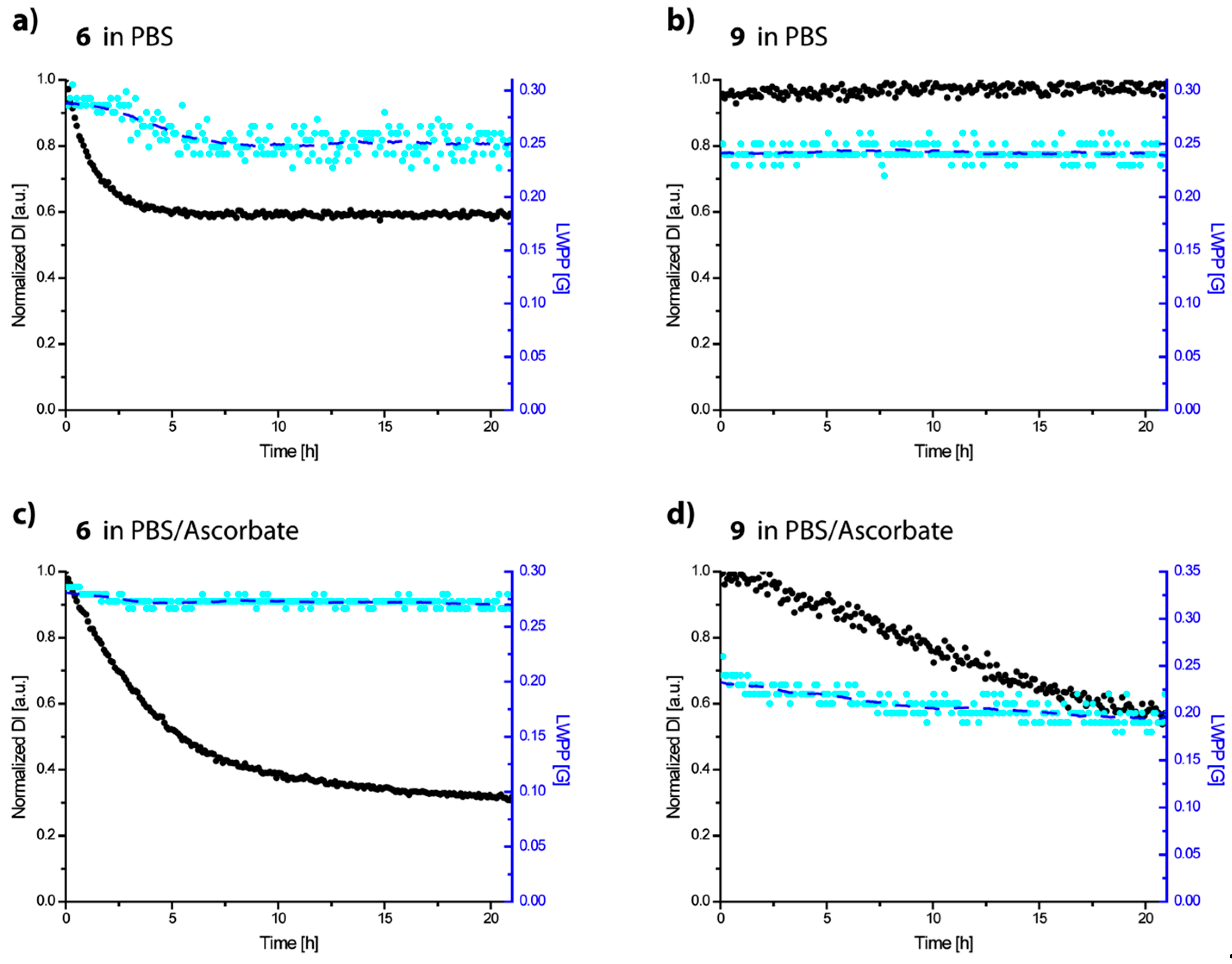{kind=link}
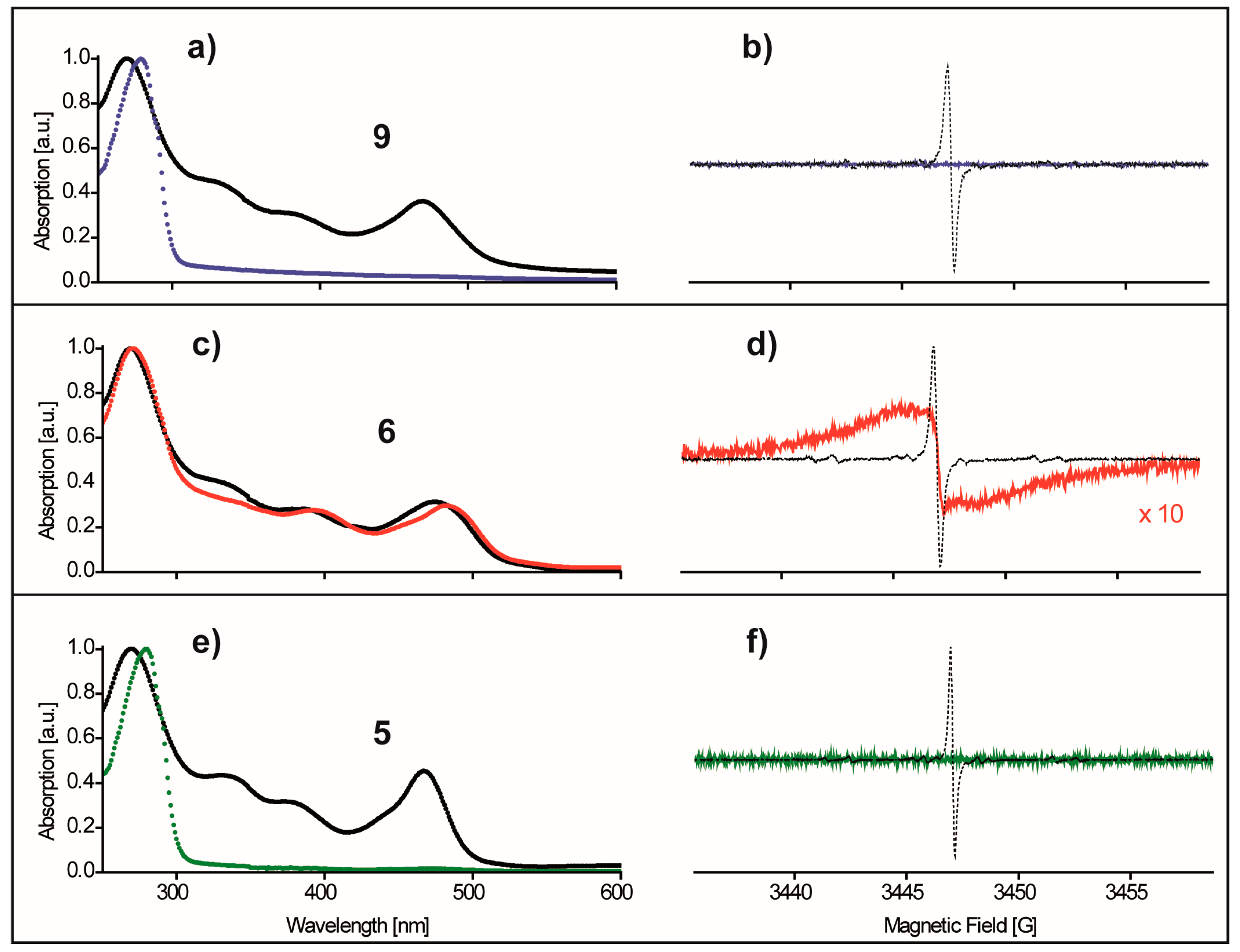{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
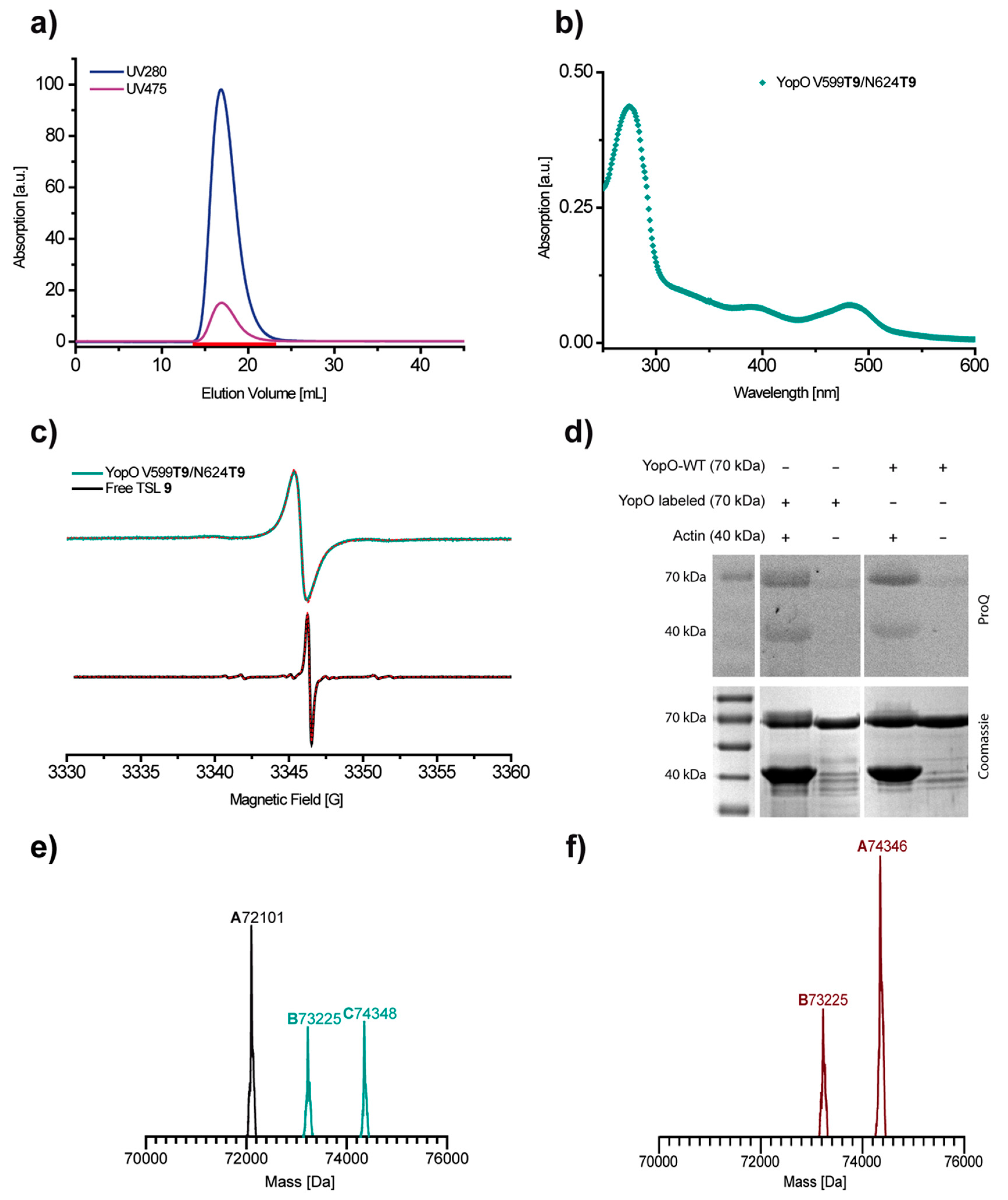
2.1. Synthesis
2.2. Redox-Stability of Trityls
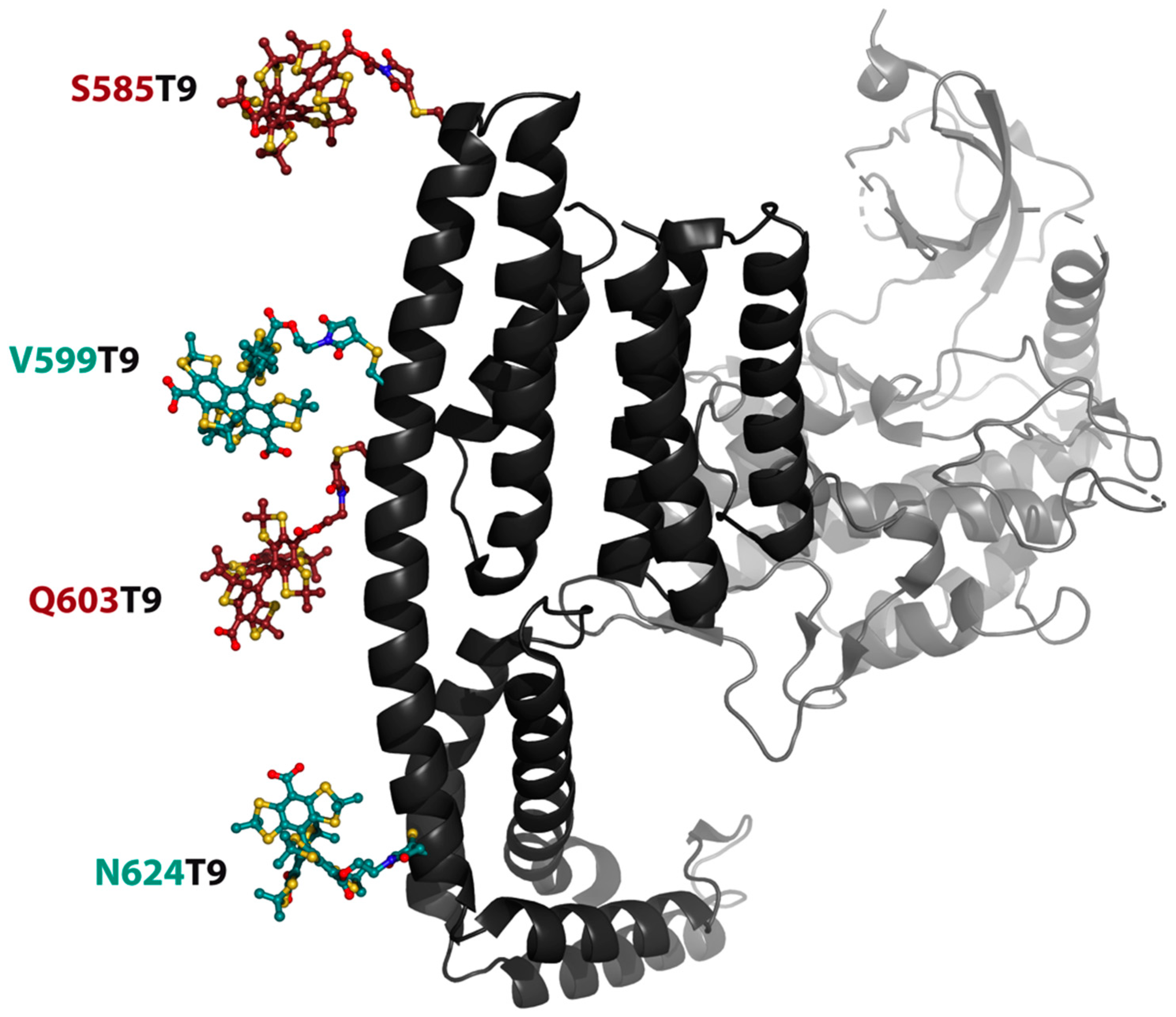
2.3. Labeling
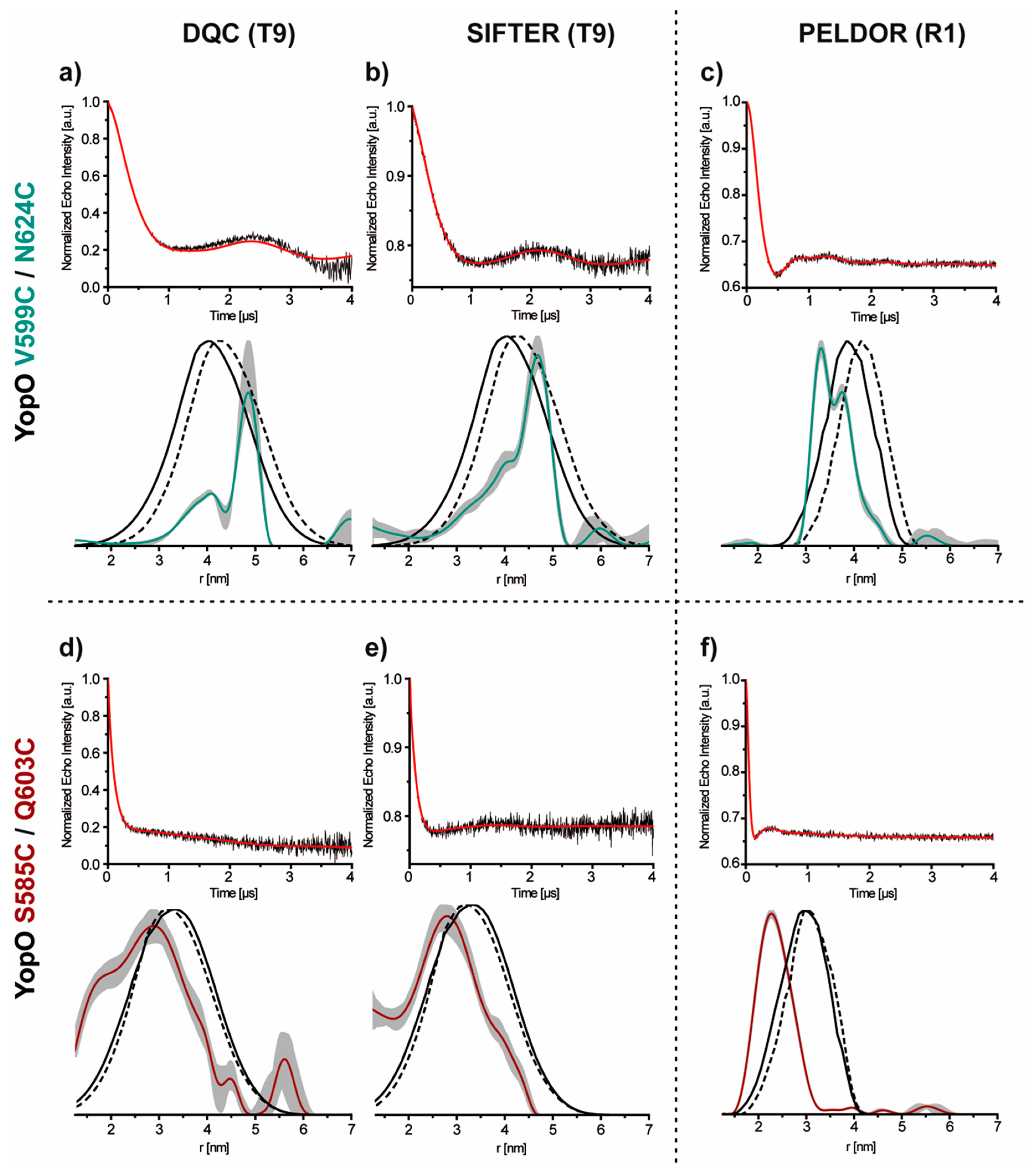
2.4. PDS Measurements
3. Conclusions
4. Materials and Methods
4.1. Synthesis of 9
4.2. Protocol for Labeling YopO with 9
4.3. UV-Vis Setup
4.4. Mass Spectrometry (MS) Setup
4.5. EPR Sample Preparation
4.6. EPR Setup
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schiemann, O.; Prisner, T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q. Rev. Biophys. 2007, 40, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef] [Green Version]
- Timmel, C.R.; Harmer, J.R. Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences. Struct. Bond. 2014, 152, 1–332. [Google Scholar]
- Milov, A.D.; Salikhov, K.M.; Schirov, M.D. Application of ELDOR in electron-spin echo for paramagnetic center space distribution in solids. Fiz. Tverd. Tela (Leningrad) 1981, 23, 975–982. [Google Scholar]
- Milov, A.; Ponomarev, A.; Tsvetkov, Y. Electron-electron double resonance in electron spin echo: Model biradical systems and the sensitized photolysis of decalin. Chem. Phys. Lett. 1984, 110, 67–72. [Google Scholar] [CrossRef]
- Saxena, S.; Freed, J.H. Double quantum two-dimensional Fourier transform electron spin resonance: Distance measurements. Chem. Phys. Lett. 1996, 251, 102–110. [Google Scholar] [CrossRef]
- Saxena, S.; Freed, J.H. Theory of double quantum two-dimensional electron spin resonance with application to distance measurements. J. Chem. Phys. 1997, 107, 1317–1340. [Google Scholar] [CrossRef] [Green Version]
- Borbat, P.P.; Freed, J.H. Multiple-quantum ESR and distance measurements. Chem. Phys. Lett. 1999, 313, 145–154. [Google Scholar] [CrossRef]
- Jeschke, G.; Pannier, M.; Godt, A.; Spiess, H. Dipolar spectroscopy and spin alignment in electron paramagnetic resonance. Chem. Phys. Lett. 2000, 331, 243–252. [Google Scholar] [CrossRef]
- Kulik, L.; Dzuba, S.; Grigoryev, I.; Tsvetkov, Y. Electron dipole–dipole interaction in ESEEM of nitroxide biradicals. Chem. Phys. Lett. 2001, 343, 315–324. [Google Scholar] [CrossRef]
- Milikisyants, S.; Scarpelli, F.; Finiguerra, M.G.; Ubbink, M.; Huber, M. A pulsed EPR method to determine distances between paramagnetic centers with strong spectral anisotropy and radicals: The dead-time free RIDME sequence. J. Magn. Reson. 2009, 201, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Borbat, P.P.; Gonzalez-Bonet, G.; Bhatnagar, J.; Pollard, A.M.; Freed, J.H.; Bilwes, A.M.; Crane, B.R. Reconstruction of the chemotaxis receptor–kinase assembly. Nat. Struct. Mol. Boil. 2006, 13, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Banham, J.E.; Timmel, C.R.; Abbott, R.J.M.; Lea, S.M.; Jeschke, G. The Characterization of Weak Protein–Protein Interactions: Evidence from DEER for the Trimerization of a von Willebrand Factor a Domain in Solution. Angew. Chem. Int. Ed. 2006, 45, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Denysenkov, V.P.; Prisner, T.F.; Stubbe, J.; Bennati, M. High-field pulsed electron–electron double resonance spectroscopy to determine the orientation of the tyrosyl radicals in ribonucleotide reductase. Proc. Natl. Acad. Sci. USA 2006, 103, 13386–13390. [Google Scholar] [CrossRef] [PubMed]
- Pilotas, C.; Ward, R.; Branigan, E.; Rasmussen, A.; Hagelueken, G.; Huang, H.; Black, S.S.; Booth, I.R.; Schiemann, O.; Naismith, J.H. Conformational state of the MscS mechanoselective channel in solution revealed by pulsed electron-electron double resonance (PELDOR) spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, E2675–E2682. [Google Scholar] [CrossRef] [PubMed]
- Herget, M.; Baldauf, C.; Schölz, C.; Parcej, D.; Wiesmüller, K.-H.; Tampé, R.; Abele, R.; Bordignon, E. Conformation of peptides bound to the transporter associated with antigen processing (TAP). Proc. Natl. Acad. Sci. USA 2011, 108, 1349–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaenzer, J.; Peter, M.F.; Thomas, G.H.; Hagelueken, G. PELDOR Spectroscopy Reveals Two Defined States of a Sialic Acid TRAP Transporter SBP in Solution. Biophys. J. 2017, 112, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullin, D.; Florin, N.; Hagelueken, G.; Schiemann, O. EPR-Based Approach for the Localization of Paramagnetic Metal Ions in Biomolecules. Angew. Chem. Int. Ed. 2015, 54, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Schiemann, O.; Cekan, P.; Margraf, D.; Prisner, T.F.; Sigurdsson, S.T. Relative Orientation of Rigid Nitroxides by PELDOR: Beyond Distance Measurements in Nucleic Acids. Angew. Chem. 2009, 121, 3342–3345. [Google Scholar] [CrossRef]
- Marko, A.; Denysenkov, V.P.; Margraf, D.; Cekan, P.; Schiemann, O.; Sigurdsson, S.T.H.; Prisner, T.F. Conformational Flexibility of DNA. J. Am. Chem. Soc. 2011, 133, 13375–13379. [Google Scholar] [CrossRef]
- Stelzl, L.S.; Erlenbach, N.; Heinz, M.; Prisner, T.F.; Hummer, G. Resolving the Conformational Dynamics of DNA with Ångstrom Resolution by Pulsed Electron–Electron Double Resonance and Molecular Dynamics. J. Am. Chem. Soc. 2017, 139, 11674–11677. [Google Scholar] [CrossRef] [PubMed]
- Krstić, I.; Frolow, O.; Sezer, D.; Endeward, B.; Weigand, J.E.; Suess, B.; Engels, J.W.; Prisner, T.F. PELDOR Spectroscopy Reveals Preorganization of the Neomycin-Responsive Riboswitch Tertiary Structure. J. Am. Chem. Soc. 2010, 132, 1454–1455. [Google Scholar] [CrossRef] [PubMed]
- Kerzhner, M.; Matsuoka, H.; Wuebben, C.; Famulok, M.; Schiemann, O. High-Yield Spin Labeling of Long RNAs for Electron Paramagnetic Resonance Spectroscopy. Biochemistry 2018, 57, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Duss, O.; Michel, E.; Yulikov, M.; Schubert, M.; Jeschke, G.; Allain, F.H.-T. Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature 2014, 509, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Feintuch, A.; Collauto, A.; Adams, L.A.; Aurelio, L.; Graham, B.; Otting, G.; Goldfarb, D. Selective Distance Measurements Using Triple Spin Labeling with Gd3+, Mn2+, and a Nitroxide. J. Phys. Chem. Lett. 2017, 8, 5277–5282. [Google Scholar] [CrossRef]
- Berliner, L.J.; Grunwald, J.; Hankovszky, H.; Hideg, K. A novel reversible thiol-specific spin label: Papain active site labeling and inhibition. Anal. Biochem. 1982, 119, 450–455. [Google Scholar] [CrossRef]
- Likhtenshtein, G.I.; Yamauchi, J.; Nakatsuji, S.; Smirnov, A.I.; Tamura, R. Nitroxides: Applications in Chemistry, Biomedicine, and Materials Science, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; ISBN 978-3-527-31889-6. [Google Scholar]
- Kuzhelev, A.A.; Krumkacheva, O.A.; Shevelev, G.Y.; Yulikov, M.; Fedin, M.V.; Bagryanskaya, E.G.; Shevelev, G.Y. Room-temperature distance measurements using RIDME and the orthogonal spin labels trityl/nitroxide. Phys. Chem. Chem. Phys. 2018, 20, 10224–10230. [Google Scholar] [CrossRef]
- Azarkh, M.; Okle, O.; Eyring, P.; Dietrich, D.R.; Drescher, M. Evaluation of spin labels for in-cell EPR by analysis of nitroxide reduction in cell extract of Xenopus laevis oocytes. J. Magn. Reson. 2011, 212, 450–454. [Google Scholar] [CrossRef]
- Lawless, M.J.; Shimshi, A.; Cunningham, T.F.; Kinde, M.N.; Tang, P.; Saxena, S. Analysis of Nitroxide-Based Distance Measurements in Cell Extracts and in Cells by Pulsed ESR Spectroscopy. ChemPhysChem 2017, 18, 1653–1660. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed. 2018, 57, 1366–1370. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, F.; Gong, Y.-J.; Bahrenberg, T.; Feintuch, A.; Su, X.-C.; Goldfarb, D. High Sensitivity In-Cell EPR Distance Measurements on Proteins Using an Optimized Gd(III) Spin Label. J. Phys. Chem. Lett. 2018, 9, 6119–6123. [Google Scholar] [CrossRef] [PubMed]
- Jassoy, J.J.; Berndhaeuser, A.; Duthie, F.; Kuehn, S.P.; Hagelueken, G.; Schiemann, O. Versatile Trityl Spin Labels for Nanometer Distance Measurements on Biomolecules In Vitro and within Cells. Angew. Chem. Int. Ed. 2017, 56, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T.H. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Radical Res. 2015, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bridges, M.D.; López, C.J.; Rogozhnikova, O.Y.; Trukhin, D.V.; Brooks, E.K.; Tormyshev, V.; Halpern, H.J.; Hubbell, W.L. A Triarylmethyl Spin Label for Long-Range Distance Measurement at Physiological Temperatures Using T1 Relaxation Enhancement. J. Magn. Reson. 2016, 269, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Tormyshev, V.M.; Rogozhnikova, O.Y.; Akhmetzyanov, D.; Bagryanskaya, E.G.; Prisner, T.F. Selective High-Resolution Detection of Membrane Protein-Ligand Interaction in Native Membranes Using Trityl-Nitroxide PELDOR. Angew. Chem. 2016, 128, 11710–11714. [Google Scholar] [CrossRef]
- Gmeiner, C.; Klose, D.; Mileo, E.; Belle, V.; Marque, S.R.A.; Dorn, G.; Allain, F.H.T.; Guigliarelli, B.; Jeschke, G.; Yulikov, M. Orthogonal Tyrosine and Cysteine Site-Directed Spin Labeling for Dipolar Pulse EPR Spectroscopy on Proteins. J. Phys. Chem. Lett. 2017, 8, 4852–4857. [Google Scholar] [CrossRef] [PubMed]
- Reginsson, G.W.; Kunjir, N.C.; Sigurdsson, S.T.; Schiemann, O. Trityl Radicals: Spin Labels for Nanometer-Distance Measurements. Chem. A Eur. J. 2012, 18, 13580–13584. [Google Scholar] [CrossRef]
- Kunjir, N.C.; Reginsson, G.W.; Schiemann, O.; Sigurdsson, S.T. Measurements of short distances between trityl spin labels with cw EPR, DQC and PELDOR. Phys. Chem. Chem. Phys. 2013, 15, 19673. [Google Scholar] [CrossRef]
- Akhmetzyanov, D.; Schöps, P.; Kunjir, N.C.; Sigurdsson, S.T.; Marko, A.; Prisner, T.F. Pulsed EPR dipolar spectroscopy at Q- and G-band on a trityl biradical. Phys. Chem. Chem. Phys. 2015, 17, 24446–24451. [Google Scholar] [CrossRef]
- Owenius, R.; Eaton, G.R.; Eaton, S.S. Frequency (250 MHz to 9.2 GHz) and viscosity dependence of electron spin relaxation of triarylmethyl radicals at room temperature. J. Magn. Reson. 2005, 172, 168–175. [Google Scholar] [CrossRef]
- Kuzhelev, A.A.; Trukhin, D.V.; Krumkacheva, O.A.; Strizhakov, R.K.; Rogozhnikova, O.Y.; Troitskaya, T.I.; Fedin, M.V.; Tormyshev, V.M.; Bagryanskaya, E.G. Room—Temperature Electron Spin Relaxation of Triarylmethyl Radicals at the X- and Q-Bands. J. Phys. Chem. B 2015, 119, 13630–13640. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Y.; Borbat, P.; Zweier, J.L.; Freed, J.H.; Hubbell, W.L. Pulsed ESR dipolar spectroscopy for distance measurements in immobilized spin labeled proteins in liquid solution. J. Am. Chem. Soc. 2012, 134, 9950–9952. [Google Scholar] [CrossRef] [PubMed]
- Shevelev, G.Y.; Krumkacheva, O.A.; Lomzov, A.A.; Kuzhelev, A.A.; Rogozhnikova, O.Y.; Trukhin, D.V.; Troitskaya, T.I.; Tormyshev, V.M.; Fedin, M.V.; Pyshnyi, D.V.; et al. Physiological-Temperature Distance Measurement in Nucleic Acid using Triarylmethyl-Based Spin Labels and Pulsed Dipolar EPR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 9874–9877. [Google Scholar] [CrossRef] [PubMed]
- Krumkacheva, O.; Bagryanskaya, E. EPR-based distance measurements at ambient temperature. J. Magn. Reson. 2017, 280, 117–126. [Google Scholar] [CrossRef]
- Andersson, S.; Rydbeck, A.; Mahno, R.S. Free Radicals. US Patent 5728370, 17 March 1998. [Google Scholar]
- Reddy, T.J.; Iwama, T.; Halpern, H.J.; Rawal, V.H. General Synthesis of Persistent Trityl Radicals for EPR Imaging of Biological Systems. J. Org. Chem. 2002, 67, 4635–4639. [Google Scholar] [CrossRef]
- Dhimitruka, I.; Velayutham, M.; Bobko, A.A.; Khramtsov, V.V.; Villamena, F.A.; Hadad, C.M.; Zweier, J.L. Large Scale Synthesis of a Persistent Trityl Radical for Use in Biomedical EPR Applications and Imaging. Bioorganic Med. Chem. Lett. 2007, 17, 6801–6805. [Google Scholar] [CrossRef]
- Rogozhnikova, O.Y.; Vasiliev, V.G.; Troitskaya, T.I.; Trukhin, D.V.; Mikhalina, T.V.; Halpern, H.J.; Tormyshev, V.M. Generation of Trityl Radicals by Nucleophilic Quenching of Tris(2,3,5,6-tetrathiaaryl)methyl Cations and Practical and Convenient Large-Scale Synthesis of Persistent Tris(4-carboxy-2,3,5,6-tetrathiaaryl)methyl Radical. Eur. J. Org. Chem. 2013, 2013, 3347–3355. [Google Scholar] [CrossRef]
- Hintz, H.; Vanas, A.; Klose, D.; Jeschke, G.; Godt, A. Trityl Radicals with a Combination of the Orthogonal Functional Groups Ethyne and Carboxyl: Synthesis without a Statistical Step and EPR Characterization. J. Org. Chem. 2019, 84, 3304–3320. [Google Scholar] [CrossRef]
- Liu, Y.; Villamena, F.A.; Sun, J.; Xu, Y.; Dhimitruka, I.; Zweier, J.L. Synthesis and Characterization of Ester-Derivatized Tetrathiaarylmethyl Radicals as Intracellular Oxygen Probes. J. Org. Chem. 2008, 73, 1490–1497. [Google Scholar] [CrossRef]
- Dhimitruka, I.; Bobko, A.A.; Hadad, C.M.; Zweier, J.L.; Khramtsov, V.V. Synthesis and Characterization of Amino Derivatives of Persistent Trityl Radicals as Dual Function pH and Oxygen Paramagnetic Probes. J. Am. Chem. Soc. 2008, 130, 10780–10787. [Google Scholar] [CrossRef] [Green Version]
- Decroos, C.; Prangé, T.; Mansuy, D.; Boucher, J.-L.; Li, Y. Unprecedented ipso aromatic nucleophilic substitution upon oxidative decarboxylation of tris(pcarboxyltetrathiaaryl) methyl (TAM) radicals: A new access to diversely substituted TAM radicals. Chem. Commun. 2011, 47, 4805–4807. [Google Scholar] [CrossRef] [PubMed]
- Driesschaert, B.; Marchand, V.; Leveque, P.; Gallez, B.; Marchand-Brynaert, J. A phosphonated triarylmethyl radical as a probe for measurement of pH by EPR. Chem. Commun. 2012, 48, 4049. [Google Scholar] [CrossRef] [PubMed]
- Tormyshev, V.M.; Rogozhnikova, O.Y.; Bowman, M.K.; Trukhin, D.V.; Troitskaya, T.I.; Vasiliev, V.G.; Shundrin, L.A.; Halpern, H.J. Preparation of Diversely Substituted Triarylmethyl Radicals by the Quenching of Tris(2,3,5,6-tetrathiaaryl)methyl Cations with C-, N-, P-, and S-Nucleophiles. Eur. J. Org. Chem. 2014, 2014, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Driesschaert, B.; Bobko, A.A.; Khramtsov, V.V.; Zweier, J.L. Nitro-Triarylmethyl Radical as Dual Oxygen and Superoxide Probe. Cell Biochem. Biophys. 2017, 75, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Tao, S.; Liu, W.; Rockenbauer, A.; Villamena, F.A.; Zweier, J.L.; Song, Y.; Liu, Y. Synthesis and Characterization of the Perthiatriarylmethyl Radical and Its Dendritic Derivatives with High Sensitivity and Selectivity to Superoxide Radical. Chem. A Eur. J. 2018, 24, 6958–6967. [Google Scholar] [CrossRef] [PubMed]
- Fleck, N.; Hett, T.; Brode, J.; Meyer, A.; Richert, S.; Schiemann, O. C–C Cross Coupling of Trityl Radicals: Spin Density Delocalization, Exchange Coupling, and a Spin Label. J. Org. Chem. 2019, 84, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Li, Y.; Tan, X.; Zhai, W.; Han, G.; Hou, J.; Liu, G.; Song, Y.; Liu, Y. Synthesis and Characterization of Hydrophilic Trityl Radical TFO for Biomedical and Biophysical Applications. Chem. A Eur. J. 2019, 25, 7888–7895. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Elewa, M.; Said, M.M.; El Shihawy, H.A.; El-Sadek, M.; Muller, D.; Meister, A.; Hause, G.; Drescher, S.; Metz, H.; et al. Synthesis, Characterization, and Nanoencapsulation of Tetrathiatriarylmethyl and Tetrachlorotriarylmethyl (Trityl) Radical Derivatives—A Study To Advance Their Applicability as in Vivo EPR Oxygen Sensors. J. Org. Chem. 2015, 80, 6754–6766. [Google Scholar] [CrossRef]
- Marchand, V.; Levêque, P.; Driesschaert, B.; Marchand-Brynaert, J.; Gallez, B.; Marchand-Brynaert, J. In vivo EPR extracellular pH-metry in tumors using a triphosphonated trityl radical. Magn. Reson. Med. 2016, 77, 2438–2443. [Google Scholar] [CrossRef]
- Khramtsov, V.V.; Bobko, A.A.; Tseytlin, M.; Driesschaert, B. Exchange Phenomena in the Electron Paramagnetic Resonance Spectra of the Nitroxyl and Trityl Radicals: Multifunctional Spectroscopy and Imaging of Local Chemical Microenvironment. Anal. Chem. 2017, 89, 4758–4771. [Google Scholar] [CrossRef]
- Kishimoto, S.; Krishna, M.C.; Khramtsov, V.V.; Utsumi, H.; Lurie, D.J. In Vivo Application of Proton-Electron Double-Resonance Imaging. Antioxid. Redox Signal. 2018, 28, 1345–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peman, A.; Vilaseca, M.; Vitalla, F.L.; Van Doorslaer, S.; Marin-Montesinos, I.; Paniagua, J.C.; Pons, M. Paramagnetic spherical nanoparticles by the self-assembly of persistent trityl radicals. Phys. Chem. Chem. Phys. 2016, 18, 3151–3158. [Google Scholar]
- Giannoulis, A.; Yang, Y.; Gong, Y.-J.; Tan, X.; Feintuch, A.; Carmieli, R.; Bahrenberg, T.; Liu, Y.; Su, X.-C.; Goldfarb, D. DEER distance measurements on trityl/trityl and Gd(iii)/trityl labelled proteins. Phys. Chem. Chem. Phys. 2019, 21, 10217–10227. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, Y.; Liu, W.; Villamena, F.A.; Zweier, J.L. Characterization of the Binding of the Finland Trityl Radical with Bovine Serum Albumin. RSC Adv. 2014, 4, 47649–47656. [Google Scholar] [CrossRef] [PubMed]
- Galyov, E.E.; Håkansson, S.; Forsberg, A.; Wolf-Watz, H. A secreted protein kinase of Yersinia pseudotuberculosis is an indispensable virulence determinant. Nature 1993, 361, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Grimes, J.M.; Robinson, R.C. Yersinia effector YopO uses actin as bait to phosphorylate proteins that regulate actin polymerization. Nat. Struct. Mol. Boil. 2015, 22, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Chen, L.; Song, Y.; Rockenbauer, A.; Villamena, F.A.; Zweier, J.L.; Liu, Y. Thiol-Dependent Reduction of the Triester and Triamide Derivatives of Finland Trityl Radical Triggers O2-Dependent Superoxide Production. Chem. Res. Toxicol. 2017, 30, 1664–1672. [Google Scholar] [CrossRef]
- Jassoy, J.J.; Meyer, A.; Spicher, S.; Wuebben, C.; Schiemann, O. Synthesis of Nanometer Sized Bis- and Tris-trityl Model Compounds with Different Extent of Spin–Spin Coupling. Molecules 2018, 23, 682. [Google Scholar] [CrossRef]
- Brewer, C.F.; Riehm, J.P. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal. Biochem. 1967, 18, 248–255. [Google Scholar] [CrossRef]
- Machida, M.; Machida, M.I.; Kanaoka, Y. Hydrolysis of N-substituted maleimides: Stability of fluorescence thiol reagents in aqueous media. Chem. Pharm. Bull. 1977, 25, 2739–2743. [Google Scholar] [CrossRef]
- Denes, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. ChemInform Abstract: The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 45, 724–744. [Google Scholar] [CrossRef]
- Hagelueken, G.; Ward, R.; Naismith, J.H.; Schiemann, O. MtsslWizard: In Silico Spin-Labeling and Generation of Distance Distributions in PyMOL. Appl. Magn. Reson. 2012, 42, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.D.; Kiick, K.L. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjugate Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis2006—A comprehensive software package for analyzing pulsed ELDOR data. Appl. Magn. Reson. 2006, 30, 473–498. [Google Scholar] [CrossRef]
- Meyer, A.; Jassoy, J.J.; Spicher, S.; Berndhäuser, A.; Schiemann, O. Performance of PELDOR, RIDME, SIFTER, and DQC in measuring distances in trityl based bi- and triradicals: Exchange coupling, pseudosecular coupling and multi-spin effects. Phys. Chem. Chem. Phys. 2018, 20, 13858–13869. [Google Scholar] [CrossRef]
- Borbat, P.P.; Freed, J.H. EPR Spectroscopy: Fundamentals and Methods; Goldfarb, D., Stoll, S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2018; Chapter 20; pp. 425–462. ISBN 9781119162988. [Google Scholar]
- Shevelev, G.Y.; Gulyak, E.L.; Lomzov, A.A.; Kuzhelev, A.A.; Krumkacheva, O.A.; Kupryushkin, M.S.; Tormyshev, V.M.; Fedin, M.V.; Bagryanskaya, E.G.; Pyshnyi, D.V. A Versatile Approach to Attachment of Triarylmethyl Labels to DNA for Nanoscale Structural EPR Studies at Physiological Temperatures. J. Phys. Chem. B 2018, 122, 137–143. [Google Scholar] [CrossRef]
- Bahrenberg, T.; Yang, Y.; Goldfarb, D.; Feintuch, A. rDEER: A Modified DEER Sequence for Distance Measurements Using Shaped Pulses. Magnetochemistry 2019, 5, 20. [Google Scholar] [CrossRef]
- Hagelueken, G.; Ingledew, W.J.; Huang, H.; Petrovic-Stojanovska, B.; Whitfield, C.; ElMkami, H.; Schiemann, O.; Naismith, J.H. PELDOR Spectroscopy Distance Fingerprinting of the Octameric Outer-Membrane Protein Wza fromEscherichia coli. Angew. Chem. 2009, 121, 2948–2950. [Google Scholar] [CrossRef]
- Jeschke, G. Interpretation of Dipolar EPR Data in Terms of Protein Structure. Struct. Bond. 2014, 152, 83–120. [Google Scholar]
- Abdullin, D.; Hagelueken, G.; Schiemann, O. Determination of nitroxide spin label conformations via PELDOR and X-ray crystallography. Phys. Chem. Chem. Phys. 2016, 18, 10428–10437. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.F.; Tuukkanen, A.T.; Heubach, C.A.; Selsam, A.; Duthie, F.G.; Svergun, D.I.; Schiemann, O.; Hagelueken, G. Studying Conformational Changes of the Yersinia Type-III-Secretion Effector YopO in Solution by Integrative Structural Biology. Structure 2019, 27, 1–11. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jassoy, J.J.; Heubach, C.A.; Hett, T.; Bernhard, F.; Haege, F.R.; Hagelueken, G.; Schiemann, O. Site Selective and Efficient Spin Labeling of Proteins with a Maleimide-Functionalized Trityl Radical for Pulsed Dipolar EPR Spectroscopy. Molecules 2019, 24, 2735. https://doi.org/10.3390/molecules24152735
Jassoy JJ, Heubach CA, Hett T, Bernhard F, Haege FR, Hagelueken G, Schiemann O. Site Selective and Efficient Spin Labeling of Proteins with a Maleimide-Functionalized Trityl Radical for Pulsed Dipolar EPR Spectroscopy. Molecules. 2019; 24(15):2735. https://doi.org/10.3390/molecules24152735
Chicago/Turabian StyleJassoy, J. Jacques, Caspar A. Heubach, Tobias Hett, Frédéric Bernhard, Florian R. Haege, Gregor Hagelueken, and Olav Schiemann. 2019. "Site Selective and Efficient Spin Labeling of Proteins with a Maleimide-Functionalized Trityl Radical for Pulsed Dipolar EPR Spectroscopy" Molecules 24, no. 15: 2735. https://doi.org/10.3390/molecules24152735