Abstract
Three new γ-hydroxyl butenolides (1–3), a pair of new enantiomeric spiro-butenolides (4a and 4b), a pair of enantiomeric cyclopentenones (5a new and 5b new natural), and six known compounds (6–11), were isolated from Aspergillus sclerotiorum. Their structures were established by spectroscopic data and electronic circular dichroism (ECD) spectra. Two pairs of enantiomers [(+)/(–)-6c and (+)/(–)-6d] obtained from the reaction of 6 with acetyl chloride (AcCl) confirmed that 6 was a mixture of two pairs of enantiomers. In addition, the X-ray data confirmed that 7 was also a racemate. The new metabolites (1−5) were evaluated for their inhibitory activity against cancer and non-cancer cell lines. As a result, compound 1 exhibited moderate cytotoxicity to HL60 and A549 with IC50 values of 6.5 and 8.9 µM, respectively, and weak potency to HL-7702 with IC50 values of 17.6 µM. Furthermore, compounds 1−9 were screened for their antimicrobial activity using the micro-broth dilution method. MIC values of 200 μg/mL were obtained for compounds 2 and 3 towards Staphylococcus aureus and Escherichia coli, while compound 8 exhibited a MIC of 50 μ/mL towards Candida albicans.
1. Introduction
Aspergillus sclerotiorum widely distributes in various environments such as marine samples, soil, sea-salt field, and rotting apples, and produces a series of bioactive metabolites. Representatives include asterriquinones [], aspochracin derivative [], lovastatin analogues [], butenolides [], cyclopeptides [,,], sclerotiamide [], hydroxamic acids [], and ochratoxin []. In addition, co-incubation of this strain with Penicillium citrinum resulted in the production of furanone derivatives and alkaloids [].
The Yellow River Delta is formed mainly by the deposition of sand and mud carried by the Yellow River, which is the world’s youngest wetland ecosystem []. High evaporation and tidal intrusion heavily salinizes and alkalizes the soil, half of which is barren and not suitable for the growth of crops [,]. The saline soil is typically characterized by poor nutrient and high salinity, which endows the microorganism special biosynthetic pathways during the evolutionary process to produce structurally novel and biologically active secondary metabolites. Up to now, only a few research papers have been carried out on the fungi from this unique environment []. In our continuation of investigation on the saline soil-derived fungi from the Yellow River Delta, dozens of natural products (NPs) with multifarious structural features and a wide range of biological activities were obtained [,]. As part of our ongoing efforts in seeking for new bioactive NPs, A. sclerotiorum JH42 with antimicrobial activity was isolated from saline soil, and subjected to chemical exploration, which led to the achievement of six new (1–3, 4a, 4b, and 5a), a new natural (5b), and six known (6–11) compounds (Figure 1). Additionally, the isolated γ-hydroxyl butenolides in the current work were all proved to undergo tautomerism at C-4 based on the analyses of physicochemical properties, calculated ECD data, and X-ray diffraction. Herein, details of the isolation, structure elucidation, acetylation, cytotoxic, and antimicrobial activities of these compounds are reported.
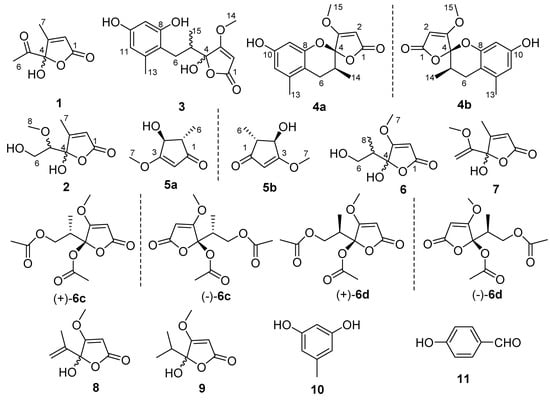
Figure 1.
Structures of Compounds 1−3, 4a, 4b, 5a, 5b, 6−11, (+)/(−)-6c and (+)/(−)-6d.
2. Results
Compound 1 was obtained as an optically inactive colorless oil with the molecular formula of C7H8O4 as determined by the deprotonated-ion HRESIMS at m/z 155.0339 [M − H]− (calcd for C7H7O4, 155.0344). Its IR spectrum indicated the presence of hydroxyl (3374 cm−1), keto carbonyl (1755 cm−1), α,β-unsaturated lactone groups (1731, 1652 cm−1) []. The 1H NMR spectrum (Table 1) in DMSO-d6 showed two singlet methyls at δH 2.25 and 1.96, a singlet olefinic proton at δH 6.18, and a hydroxyl group at δH 8.50. Its 13C NMR spectrum revealed seven carbon resonances, corresponding to a keto carbonyl (δC 201.5), four diagnostic carbon signals (δC 170.2, 165.7, 118.9 and 105.7) for a γ-hydroxyl butenolide moiety [], and two methyl groups (δC 24.7 and 12.7) (Table 1). The gross structure of 1 was unambiguously established by the HMBC correlations from H-2 to C-1, C-3 and C-4, from H-6 to C-4 and C-5, and from H-7 to C-1, C-3 and C-4 (Figure 2). It was a mixture of inseparable enantiomers, which mutually transformed through the γ-keto-acid form as existence in penicillic acid [].

Table 1.
1H (400 MHz) and 13C (100 MHz) NMR data for 1, 2 and 5 [δH, mult (J in Hz)].
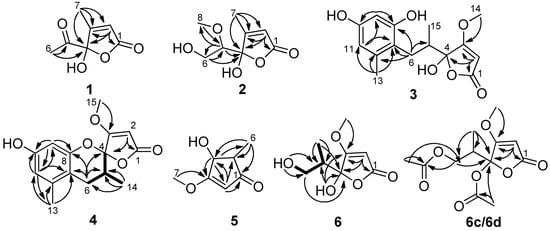
Figure 2.
Key COSY and HMBC correlations of 1–6, 6c and 6d.
Compound 2 was also isolated as an optically inactive colorless oil with the molecular formula of C8H12O5 as elucidated by the HRESIMS m/z 187.0610 [M − H]− (calcd for C8H11O5, 187.0601). The IR absorption bands at 3373, 1739 and 1652 cm−1 suggested the presence of hydroxyl, α,β-unsaturated lactone groups. The 1H NMR spectrum in MeOH-d4 displayed signals for a singlet olefinic proton at δH 5.86, an oxymethine at δH 3.56 (dd, J = 7.0, 3.2 Hz), a diastereotopic methylene at δH 3.90 (m) and 3.66 (dd, J = 11.7, 7.0 Hz), a methoxyl at δH 3.50 (s) and a methyl at δH 2.09 (Table 1). The 13C NMR spectrum (Table 1), with the help of DEPT and HSQC data, showed the presence of two methyls (one oxygenated), an oxymethylene, an olefinic and an oxymethine, a hemiketal carbon (δC 110.1), and two quaternary carbons (δC 172.9 and 168.7). The above data indicated 2 was also a γ-hydroxyl butenolide as 1. The main differences were the appearances of an oxygenated methyl, an oxymethylene and an oxymethine, together with the disappearances of a carbonyl and a methyl. The planar structure and the assignments of the NMR data were completed by the HMBC correlations (Figure 2). Since no optical rotation and no CD absorption exhibited, compound 2 was also an inseparable racemic mixture.
Compound 3 was a colorless solid with zero optical rotation. Its HRESIMS at m/z 293.1021 [M − H]− (calcd for C15H17O6, 293.1020) gave the molecular formula of C15H18O6. The 1H NMR spectrum displayed signals for two meta-coupled aromatic protons at δH 6.38 and 6.20 (d, J = 1.6 Hz, each), one isolated olefinic proton at δH 5.20, a set of nonequivalent methylene protons at δH 2.58 and 2.08, one methine at δH 2.35 (m), one methoxyl at δH 3.94 and two methyl protons at δH 2.19 and 0.86 (Table 2). The 13C NMR spectrum displayed fifteen resonance signals (Table 2). The carbon signals at δC 180.9, 170.7, 106.3, 90.2 and 60.0 showed the representative resonances for a γ-hydroxyl-β-methoxyl butenolide moiety as in dihydropenicillic acid [], which was also isolated in the current report. Obviously, two aromatic methines and four aromatic quaternary carbons constructed a tetrasubstituted phenyl ring, and the linkages of substituents were completed by the HMBC correlations (Figure 2). The splitting behaviors of methylene at δH 2.58 (dd, J = 13.6, 11.2 Hz) and 2.08 (disturbed by the signals of solvent), methine at δH 2.35 (m) and methyl at δH 0.86 (br s) completed the structure of 3. The weak carbon signals of C-4, C-6, C-7, and C-14, together with the broad singlet of H-14 (should be doublet) (Figure S15 and S16), implied that the C-4 anomers mutually transformed in solution. Owing to no optical rotation and no CD absorption displayed, 3 existed as an inseparable racemic mixture.
Table 2.
1H (400 MHz) and 13C (100 MHz) NMR data for 3 and 4 in acetone-d6.
Compound 4 was afforded as an optically inactive colorless solid. The HRESIMS m/z 277.1071 [M + H]+ gave a molecular formula of C15H16O5, which was one H2O unit less than that of 3. Its 1H and 13C NMR data were similar to those of 3, except for the chemical shifts of C-4, C/H-5, C/H-6, C-7 and C-8 (Table 2). The above information, especially the chemical shift of C-8 (δC 153.2), implied that 8-OH and 4-OH in 3 should be dehydrated into an ether linkage in 4. The 2D NMR spectra (Figure 2) established its planar structure, which was different from aspergispiroketal in the locations of the substituents at benzyl moiety [], as proved by the HMBC correlations from H-6 to C-7 and C-8, from H-9 to C-8, C-10 and C-11, from H-11 to C-7, C-9, C-10 and C-13, and from H-13 to C-7, C-11 and C-12. In view of no optical activity and ECD absorption, 4 was a racemate and separated into (+)-4 and (–)-4 using high performance liquid chromatography (HPLC) on a chiral column. Their absolute configurations were determined according to the experimental and calculated ECD data (Figure 3). Based on the optimized structures, the ECD calculation was conducted using time-dependent density functional theory (TD-DFT) at BP86/6-311G (d,p) for four isomers of 4 (Attachment S1). Then the absolute configurations of (+)-4 and (–)-4 were determined to be (4S,5R)-4b and (4R,5S)-4a, respectively.
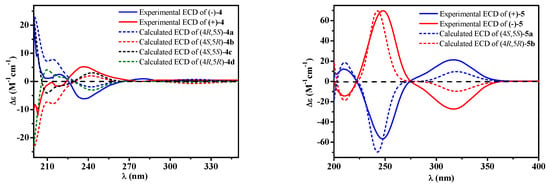
Figure 3.
Experimental [(+)/(–)-4, (+)/(–)-5] and calculated [4a, 4b, 4c, 4d, 5a and 5b] electronic circular dichroism (ECD) spectra.
Compound 5, a colorless solid with zero optical rotation, was assigned the molecular formula of C7H10O3 on the basis of its positive HRESIMS at m/z 143.0704 [M + H]+ (calcd for C7H11O3, 143.0703). The 1H NMR spectrum showed an olefinic proton at δH 6.37 (d, J = 2.6 Hz), a oxymethine at δH 4.37 (dd J = 2.6, 1.6 Hz), a methine at δH 2.20 (qd, J = 7.5, 1.6 Hz), a methoxyl at δH 3.75 (s) and a doublet methyl at δH 1.20 (J = 7.5 Hz) (Table 1). The 13C NMR spectrum displayed seven carbon signals including a keto carbonyl, two olefinic and four aliphatic carbons (Table 1). The planar structure of 5 was confirmed by the HMBC correlations (Figure 2). In the NOE difference experiment, the signal of H-2 was enhanced when H-6 was irradiated, so H-2 and H-3 were in the opposite directions, which was further confirmed by the small coupling constant (J = 1.6 Hz). Compound 5 was subjected to chiral HPLC and separated into (+)-5 and (−)-5. According to the calculated and experimental ECD data (Figure 3), their absolute configurations were determined to be 2S,3S and 2R,3R for 5a and 5b, respectively. 5b had been reported as a synthetic intermediate without 13C NMR, ECD and specific rotation data reported []. Therefore, 5a was a new compound, while 5b was a new natural product.
Compound 6 was obtained as colorless blocks with the molecular formula of C8H12O5 on the basis of its negative HRESIMS. Some articles reported its structure with a set of 1H and 13C NMR data [,], but the compound obtained in our project showed two sets of 1H and 13C NMR data (6a and 6b) (Table 3) with the ratio of about 1:1 in DMSO-d6, about 2:1 in acetone-d6 (only 1H NMR spectrum measured, Figure S42), about 1.4:1 in MeOH-d4. Additionally, only H-6, H-8, and C-5 exhibited two sets of NMR signals, and signals of C-3 and C-4 were too weak to be observed in MeOH-d4 (Figure S43 and S44). The above phenomena suggested 6 was a mixture of two pairs of racemates, and the proportion of anomers of C-4 changed with solvents (Figure 4). The structures and the assignments of the 1H and 13C NMR data of 6a and 6b in DMSO-d6 were completed by 2D NMR spectra (Figure 2).
Table 3.
1H (400 MHz) and 13C (100 MHz) NMR data for 6a and 6b in DMSO-d6.
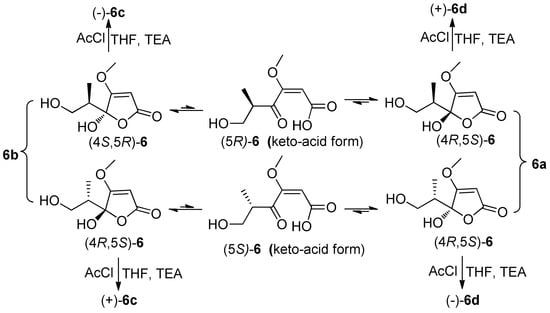
Figure 4.
Tautomeric forms (γ-keto-acids and γ-hydroxyl butenolides) of 6, and the reaction of 6 with AcCl.
In order to explore the case, compound 6 was reacted with AcCl leading to the production of 6c and 6d with optical inactivity (Figure 4). Their molecular formulae of C12H16O7 were obtained on the basis of their HRESIMS. And their planar structures were constructed by the 1H and 13C NMR (Table 4) and HMBC spectra (Figure 4). The single crystal X-ray diffraction using Cu Kα radiation showed 6c to be a centrosymmetric space group P21/C with 4S,5R and 4R,5S configurations (Figure 5), so (±)-6d should be the 4S,5S and 4R,5R configurations. Then they were subjected to chiral HPLC and isolated into (+)/(−)-6c and (+)/(−)-6d, respectively. The calculated and experimental ECD data (Figure 6) proved the absolute configurations of (+)-6c and (−)-6c to be the respective 4R,5S and 4S,5R. Considering their ECD absorptions mainly resulted from C-4 chiral center, the same ECD data of (+)-6c and (+)-6d, and (−)-6c and (−)-6d to the mirror images, implied that (+)-6d and (−)-6d had 4R,5R and 4S,5S configurations, respectively. The δ values of C-1, C-2 and C-4 in 6c were slightly larger than those in 6d with C-3 and C-5 to the contrary (Table 4), and the same behaviors were also observed in 6b and 6a (Table 3), so (±)-6c and (±)-6d should derive from 6b and 6a, separately. Consequently, 6b should be a racemate with the configurations of 4S,5R and 4R,5S, 6a be 4S,5S and 4R,5R.
Table 4.
1H (400 MHz) and 13C (100 MHz) NMR data for (±)-6c and (±)-6d in DMSO-d6.
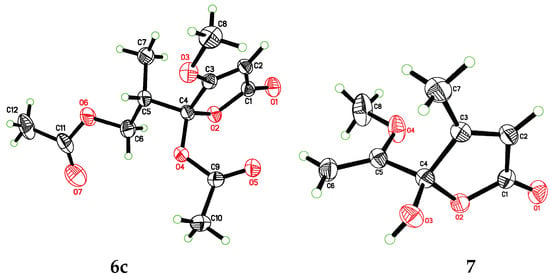
Figure 5.
X-ray ORTEP diagrams of 6c and 7.
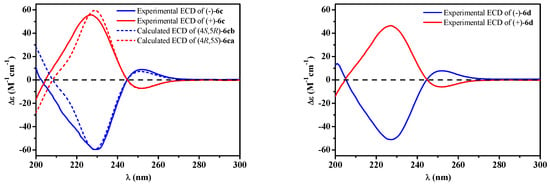
Figure 6.
Experimental [(+)/(–)-6c, (+)/(–)-6d] and calculated [(4S,5R)/(4R,5S)-6c] ECD spectra.
Compound 7 was isolated as colorless blocks. Its NMR data was almost identical with spersclerotioron G, which was reported as an S configuration compound at C-4 chiral center []. However, in our report it was optically inactive and of no Cotton effects in its ECD spectrum, therefore it was a racemic mixture, which was confirmed by a centrosymmetric space group P21/n in the single crystal X-ray diffraction with Cu Kα radiation (Figure 5). Other known compounds were identified as penicillic acid (8) [], dihydropenicillic acid (9) [], orcinol (10) [], and p-hydroxyl benzaldehyde (11) [], by comparison of their spectroscopic data with those in the literature.
Compounds 1–5 were preliminarily evaluated for their cytotoxicity against human promyelocytic leukemia (HL60), human lung adenocarcinoma (A549) and human normal liver (HL-7702) cell lines by the MTT method, with doxorubicin as a positive control (IC50: 0.85, 1.5 and 8.3 µM, respectively). Compound 1 and 3 showed selective cytotoxicity against HL60 (IC50: 6.5 and 12.1 µM, respectively), A549 (IC50: 8.9 and 16.7 µM, respectively), and HL-7702 (IC50: 17.6 and 22.8 µM, respectively) cell lines. The results suggested that compounds 1 and 3 showed stronger cytotoxicity to cancer cells than to non-cancer cell lines. The other compounds were inactive to the tested cell lines (IC50 > 20 µM).
Meanwhile, the antimicrobial assays of compounds 1–9 were screened against Staphylococcus aureus (ATCC 25923), Escherichia coli (ATCC 25922) and Candida albicans (ATCC 10231). Among them, the known compound 8 showed pronounced antimicrobial activity to the tested organisms, while compounds 2 and 3 displayed weak activity against S. aureus and E. coli (Table 5).
Table 5.
Antimicrobial Activity of Compounds 1–9 (MIC μg/mL).
3. Discussion
A. sclerotiorum JH42 not only produced six new compounds (1–3, 4a, 4b and 5a) and a new natural product (5b), but also penicillic acid (8) with a high yield (59 g in 73 g of the crude extract). Since penicillic acid possessed multiple bioactivities, such as antitumor [,,,], antibacterial [,], antimalarial [], phytotoxic [], antiviral [], antifungal properties [], A. sclerotiorum JH42 might be employed as a potential producer to provide penicillic acid in industry for the future development and applications.
Additionally, the γ-hydroxyl butenolides (1–3, 6–7) in this study were all proved to be mixtures of enantiomers. The information implied that γ-hydroxyl butenolides usually exist in a mixture of anomers of C-4, which were inseparable because of their mutual transformation through the γ-keto-acid form. The results have guiding significance for the isolation and structure determination of this kind of compounds.
4. Materials and Methods
4.1. General Experimental Procedures
The optical rotations, ultraviolet (UV), IR and ECD spectra were measured on an Autopol V Plus polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA), a TU-1091 spectrophotometer (Beijing Purkinje General Instrument Co., Beijing, China), Nicolet 6700 spectrophotometer (Thermo Scientific, Waltham, MA, USA) with an attenuated total reflectance (ATR) method, and a Chirascan spectropolarimeter (Applied Photophysics, Leatherhead, United Kingdom), respectively. An Avance 400 (Bruker, Billerica, MS, USA) was used to collect the NMR data. X-ray crystal data were performed on a Bruker Smart 1000 CCD X-ray diffractometer (Bruker Biospin Group, Karlstuhe, Germany). HRESIMS data were acquired on a Q-TOF Ultima GLOBAL GAA076 LC or a 1200RRLC-6520 Accurate-Mass Q-TOF LC/MS mass spectrometer (Agilent, Santa Clara, CA, USA). LC-6AD Liquid Chromatography (Shimadzu, Kyoto, Japan) equipped with an ODS column (HyperClone, 5 μm ODS C18 120 Å, 250 × 10 mm, Phenomenex, 4 mL/min), and a chiral column [ChiralPAK IC, 5 μm cellulose tri(3,5-dichlorophenyl carbamate), 250 × 10 mm, Daicel Chiral Technologies Co. LTD. (Shaihai, China)] was used in the HPLC isolation process. The optical density was measured on a Multiskan FC microplate readers (Thermo Fisher Scientific, Shanghai, China). Silica gel (200−300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), reversed-phase C18 silica gel (Pharmacia Fine Chemical Co., Ltd., Uppsala, Sweden) and sephadex LH-20 (Ge Healthcare Bio-Sciences AB, Uppsala, Sweden), were used in column chromatography.
4.2. Fungal Material
A. sclerotiorum JH42 (Genbank accession No. HQ717801) was isolated from the saline soil collected along the coast of Bohai bay in Zhanhua in August 2008. The working strain was identified according to ITS sequence analysis and assigned the accession number JH42. It was preserved in China General Microbiological Culture Collection Center (Depositary Number: CGMCC NO. 13562).
4.3. Fermentation and Extraction
A. sclerotiorum JH42 was cultured on Petri dishes of potato dextrose agar (PDA) at 28 °C for 7 days. A small spoon of spores was transferred into 500-mL conical flasks containing 180 mL culture medium (decoction of 200 g potato, glucose 20 g, maltose 20 g, mannitol 10 g, yeast extract 3 g, KH2PO4 0.5 g, MgSO4·7H2O 0.3 g, dissolved in 1 L seawater), and cultured at 28 °C for 9 days on a rotary shaker at 170 rpm. The culture broth (34.5 L) was filtered into filtrate and mycelia. The former was extracted with ethyl acetate, while the latter was extracted with methanol. The methanol solution was concentrated under reduced pressure to yield an aqueous solution, which was then extracted with ethyl acetate. The ethyl acetate extracts were merged and evaporated under reduced pressure to give an extract (73 g).
4.4. Purification
The extract was dissolved in acetone and left for crystallization at room temperature by slow evaporation of the solvent to obtain 8 (18.0 g). Then the residue was performed on a silica gel column chromatography with a step gradient of petroleum ether/ethyl acetate (from 1:0 to 0:1, v/v) to afford ten fractions (Fr.s 1–10). Fr. 7 (3.6 g) was separated into seven subfractions (Fr.s 7.1–7.7) by an ODS column eluting with MeOH/H2O gradient (from 20:80 to 100:0, v/v). Fr. 7.2 (0.2 g) was purified by semipreparative HPLC on an ODS column eluting with 15% MeOH to yield 1 (25.2 mg, tR 7.9 min) and 2 (14.2 mg, tR 5.6 min). Fr. 4 (1.1 g) was chromatographed on a Sephadex LH-20 column (MeOH) and then purified by HPLC (60% MeOH) to yield 3 (38.0 mg, tR 22.1 min). Fr. 7.6 (0.1 g) was purified by HPLC (40% MeOH) on an ODS column to yield 4 (12.0 mg, tR 26.8 min), which was further separated by HPLC on a chiral column (n-hexane/isopropanol, 60:40, v/v, 2.0 mL/min) to give 4a (1.8 mg, tR 17.3 min) and 4b (1.7 mg, tR 41.5 min). Fr. 7.3 (0.1 g) was separated by HPLC (15% MeOH) to afford 5 (14.6 mg, tR 12.2 min), which was further separated by HPLC on a chiral column (n-hexane/isopropanol, 60:40, v/v, 2.0 mL/min) to give 5a (2.5 mg, tR 28.6 min) and 5b (3.3 mg, tR 21.3 min). Fr. 6 (1.3 g) was chromatographed on a silica gel column using chloroform/methanol (30:1, v/v) to obtain 6 (0.63 g). Fr. 5 (44.0 g) was crystalized in acetone to give 8 (41.0 g) again, and then the residue was isolated by HPLC (25% MeOH) to yield 7 (16.0 mg, tR 13.8 min) and 9 (27.0 mg, tR 20.3 min). Fr. 8 (0.35 g) was purified by HPLC (15% MeOH) to yield 10 (13.8 mg, tR 27.2 min) and 11 (9.8 mg, tR 21.2 min).
Aspersclerolide A (1): colorless oil (MeOH); UV (MeOH) λmax (log ε): 210 (3.94) nm; IR (ATR) νmax 3374, 1755, 1731, 1652, 1435, 1382, 1360, 1302, 1202, 1149, 1018, 912, 857, 765 cm−1; 1H and 13C NMR data: see Table 1; HRESIMS m/z 155.0339 [M − H]− (calcd for C7H7O4, 155.0344).
Aspersclerolide B (2): colorless oil (MeOH); UV (MeOH) λmax (log ε): 206 (3.88) nm; IR (ATR) νmax 3373, 2943, 1739, 1652, 1438, 1380, 1185, 1114, 1037, 922, 853, 697 cm−1; 1H and 13C NMR data: see Table 1; HRESIMS m/z 187.0610 [M − H]− (calcd for C8H11O5, 187.0601).
Aspersclerolide C (3): colorless solid (MeOH); UV (MeOH) λmax (log ε): 282 (3.35), 221 (4.14), 205 (4.46) nm; IR (ATR) νmax 3369, 3251, 1742, 1706, 1630, 1593, 1511, 1456, 1342, 1293, 1267, 1222, 1142, 1095, 985, 911, 806, 780 cm−1; 1H and 13C NMR data: see Table 2; HRESIMS m/z 293.1021 [M − H]− (calcd for C15H17O6, 293.1020).
(±)-Aspersclerolide D (4): colorless solid (MeOH); UV (MeOH) λmax (log ε): 279 (3.51), 221 (4.13), 210 (4.11) nm; IR (ATR) νmax 3326, 1737, 1679, 1639, 1626, 1594, 1501, 1451, 1373, 1342, 1266, 1173, 1061, 993, 926, 802, 776 cm−1; 1H and 13C NMR data: see Table 2; HRESIMS m/z 277.1071 [M + H]+ (calcd for C15H17O5, 277.1071).
(−)-Aspersclerolide D (4a): colorless solid (MeOH); −39.8 (c 0.090, MeOH); ECD (MeOH) λmax (Δε) 280 (+0.95), 237 (−6.16), 219 (+2.54) nm.
(+)-Aspersclerolide D (4b): colorless solid (MeOH); +36.9 (c 0.084, MeOH); ECD (MeOH) λmax (Δε) 278 (−0.003), 237 (+5.24), 219 (−1.75) nm.
(±)-4-hydroxy-3-methoxy-5-methyl-2-cyclopentenone (5): colorless solid (MeOH); UV (MeOH) λmax (log ε): 246 (3.75) nm; IR (ATR) νmax 3405, 2943, 1712, 1627, 1456, 1316, 1252, 1130, 1074, 1006, 937, 888, 844, 794 cm−1; 1H and 13C NMR data: see Table 1; HRESIMS m/z 143.0704 [M − H]− (calcd for C7H11O3, 143.0703).
(+)-(4S,5S)-4-hydroxy-3-methoxy-5-methyl-2-cyclopentenone (5a): colorless solid (MeOH); +11.11 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 317 (+21.36), 247 (−56.64), 209 (+12.29) nm.
(−)-(4R,5R)-4-hydroxy-3-methoxy-5-methyl-2-cyclopentenone (5b): colorless solid (MeOH); −12.65 (c 0.17, MeOH); ECD (MeOH) λmax (Δε) 317 (−27.20), 248 (+69.81), 210 (−14.32) nm.
6-hydroxyl dihydropenicillic acid (6): colorless blocks (MeOH); UV (MeOH) λmax (log ε): 223 (3.98) nm; IR (ATR) νmax 3309, 3112, 1742, 1709, 1622, 1449, 1414, 1356, 1294, 1268, 1223, 1167, 1094, 1038, 1021, 986, 953, 935, 899, 817, 783, 675 cm−1; 1H and 13C NMR data: see Table 3; HRESIMS m/z 187.0615[M − H]− (calcd for C8H11O5, 187.0606).
(±)-6c: colorless blocks (MeOH); 1H and 13C NMR data: see Table 4; HRESIMS m/z 295.0809 [M + Na]+ (calcd for C12H16O7Na, 295.0788).
(+)-6c: colorless solid (MeOH); +43.90 (c 0.04, MeOH); ECD (MeOH) λmax (Δε) 252 (−7.22), 227 (+55.82) nm.
(−)-6c: colorless solid (MeOH); −46.15 (c 0.03, MeOH); ECD (MeOH) λmax (Δε) nm; 252 (+8.92), 229 (−59.62) nm.
(±)-6d: colorless solid (MeOH); 1H and 13C NMR data: see Table 4; HRESIMS m/z 295.0780 [M + Na]+ (calcd for C12H16O7Na, 295.0788).
(+)-6d: colorless solid (MeOH); +78.57 (c 0.08, MeOH); ECD (MeOH) λmax (Δε) 252 (−6.01), 227 (+46.51) nm.
(−)-6d: colorless solid (MeOH); −80.00 (c 0.06, MeOH); ECD (MeOH) λmax (Δε) 252 (+7.82), 227 (−51.14) nm.
X-ray Single-Crystal Structure Determinations of 6c and 7. Colorless crystals of 6c and 7 were obtained from MeOH. Their structures were solved by direct methods using the SHELXTL software package and refined by least squares minimization. The crystallographic data for 6c (deposition number: CCDC 1912596) and 7 (deposition number: CCDC 1912598) have been deposited in the Cambridge Crystallographic Data Centre. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data for 6c. C12H16O7, Mr = 272.25, Monoclinic, space group P21/c, unit cell dimensions a = 8.6937(3) Å, b = 22.0810(5) Å, c = 7.9551(2) Å, V = 1392.71(7) Å3, Z = 4, Dcalcd = 1.298 g/cm3, F(000) = 576. α = γ = 90.00, β = 114.218(4); A total of 2427 unique reflections were collected, with 2123 reflections greater than I ≥ 2σ (I) (Rint = 0.0165). The structure was solved by direct methods and refined by full-matrix least-squares on F2, with anisotropic displacement parameters for non-hydrogen atoms at final R indices [I > 2σ (I)], R1 = 0.0445, wR2 = 0.1199; R indices (all data), R1 = 0.0503, wR2 = 0.1249.
Crystal data for 7. C8H10O4, Mr = 170.16, Monoclinic, space group P21/n, unit cell dimensions a = 7.6359(3) Å, b = 13.2597(4) Å, c = 8.5653(3) Å, V = 852.49(5) Å3, Z = 4, Dcalcd = 1.326 g/cm3, F(000) = 360. α = γ = 90.00, β = 100.581(2); A total of 1482 unique reflections were collected, with 1394 reflections greater than I ≥ 2σ (I) (Rint = 0.0185). The structure was solved by direct methods and refined by full-matrix least-squares on F2, with anisotropic displacement parameters for non-hydrogen atoms at final R indices [I > 2σ (I)], R1 = 0.0414, wR2 = 0.1115; R indices (all data), R1 = 0.0436, wR2 = 0.1130.
4.5. Preparation and Isolation of (±)-6c and (±)-6d
AcCl (41μL) was added to a solution of 6 (53 mg) and triethylamine (TEA) (79 μL) in THF (5 mL). The mixed solution was stirred at room temperature for 2 h. After removal of the solvent under reduced pressure, 5 mL of distilled water was added into the residue and subsequently extracted with ethyl acetate (5 × 3 mL). The ethyl acetate layer was evaporated under reduced pressure to give a crude product, followed by purification on HPLC with an ODS column (40% MeOH) to give 6c (11.6 mg, tR 15.6 min) and 6d (10.8 mg, tR 16.6 min). 6c and 6d were further separated by HPLC on a chiral column (n-hexane/isopropanol, 60:40, v/v, 2.0 mL/min) to give (+)-6c (5.2 mg, tR 68.2 min), (−)-6c (4.8 mg, tR 48.2 min), (+)-6d (4.4 mg, tR 74.2 min) and (−)-6d (4.1 mg, tR 47.5 min), respectively.
4.6. Biological Assay
The cytotoxic activity against HL60, A549 and HL-7702 cell lines was performed by the MTT method as previously described [].
The antimicrobial assays against S. aureus, E. coli and C. albicans were carried out by the broth microdilution method []. The tested organisms were incubated overnight with shaking (200 rpm) in thermostatic oscillation incubator (37 °C) in Mueller Hinton broth (MHB) and liquid Sabourand medium for the bacteria and the fungus, respectively. The microbial inoculum density was adjusted to 1 × 106 cfu/mL with 0.9% saline solution by comparison with a MacFarland standard. The tested substances and positive drugs were dissolved in methanol to an initial concentration of 40 mg/mL. 4 μL of initial compound solution and 196 μL of MHB (liquid Sabourand medium for fungus) were added into the first well and mixed evenly. Then 100 μL of solution from the first hole, along with 2 μL of methanol and 98 μL of MHB (liquid Sabourand medium for fungus), were transferred to the second hole, and then shaken up as mixture uniform. The repetitive operation was performed to the eleventh one, from which 100 μL of solution well was discarded. Then, 100 μL of microbial suspension was added to the solutions in 96-well to achieve a final volume of 200 µL and final sample concentrations from 400 to 0.39 µg/mL. The blank well was also incubated with only medium under the same conditions. All experiments were carried out in triplicate and with chloramphenicol and ketoconazole as the positive controls. Optical density measurement for bacteria and fungus was recorded at 620 nm after incubation at 37 °C for 12 and 24 h, respectively. The minimal inhibitory concentration (MIC) was defined as the concentration at which the growth was inhibited 80% of the tested microorganisms [].
5. Conclusions
In summary, three new (1–3) and four known (6–9) γ-hydroxyl butenolides, a pair of new enantiomeric spiro-butenolides (4a and 4b), a pair of enantiomeric cyclopentenones (5a new and 5b new natural), along with orcinol (10) and p-hydroxyl benzaldehyde (11), were isolated from A. sclerotiorum JH42. The acquisition of two pairs of enantiomers [(+)/(−)-6c) and (+)/(−)-6d] by the reaction of 6 with AcCl confirmed that 6 was a mixture of two pairs of enantiomers. In addition, the X-ray diffraction data of 7 revealed it was also a racemic mixture. Compound 1 exhibited moderate cytotoxicity against HL60 and A549 cell lines with IC50 values of 6.5 and 8.9 µM, respectively. New compounds 2 and 3 showed weak antibacterial activity against S. aureus and E. coli, while 8 displayed pronounced antimicrobial activity against all the tested organisms.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/14/2642/s1, Figure S1: 1H NMR spectrum (400 MHz) of 1 in DMSO-d6, Figure S2: 13C NMR spectrum (100 MHz) of 1 in DMSO-d6, Figure S3: HMBC spectrum of 1 in DMSO-d6, Figure S4: IR spectrum of 1, Figure S5: UV spectrum of 1 in MeOH, Figure S6: HRESIMS of 1, Figure S7: 1H NMR spectrum (400 MHz) of 2 in MeOH-d4, Figure S8: 13C NMR spectrum (100 MHz) of 2 in MeOH-d4, Figure S9: DEPT 135 spectrum of 2 in MeOH-d4, Figure S10: HSQC spectrum of 2 in MeOH-d4, Figure S11: HMBC spectrum of 2 in MeOH-d4, Figure S12: IR spectrum of 2, Figure S13: UV spectrum of 2 in MeOH, Figure S14: HRESIMS of 2, Figure S15: 1H NMR spectrum (400 MHz) of 3 in acetone-d6, Figure S16: 13C NMR spectrum (100 MHz) of 3 in acetone-d6, Figure S17: HMBC spectrum of 3 in acetone-d6, Figure S18: UV spectrum of 3 in MeOH, Figure S19: IR spectrum of 3, Figure S20: HRESIMS of 3, Figure S21: 1H NMR spectrum (400 MHz) of 4 in acetone-d6, Figure S22: 13C NMR spectrum (100 MHz) of 4 in acetone-d6, Figure S23: DEPT spectrum of 4 in acetone-d6, Figure S24: HMQC spectrumof 4 in acetone-d6, Figure S25: 1H-1H COSY spectrum of 4 in acetone-d6, Figure S26: HMBC spectrum of 4 in acetone-d6, Figure S27: IR spectrum of 4, Figure S28: UV spectrum of 4 in MeOH, Figure S29: HRESIMS of 4, Figure S30: 1H NMR spectrum (400 MHz) of 5 in MeOH-d4, Figure S31: 13C NMR spectrum (100 MHz) of 5 in MeOH-d4, Figure S32: HMBC spectrum of 5 in MeOH-d4, Figure S33: NOE difference spectrum of 5 in MeOH-d4, Figure S34: IR spectrum of 5, Figure S35: UV spectrum of 5 in MeOH, Figure S36: HRESIMS of 5, Figure S37: 1H NMR spectrum (400 MHz) of 6 in DMSO-d6, Figure S38: 13C NMR spectrum (100 MHz) of 6 in DMSO-d6, Figure S39: 1H-1H COSY spectrum of 6 in DMSO-d6, Figure S40: HSQC spectrum of 6 in DMSO-d6, Figure S41: HMBC spectrum of 6 in DMSO-d6, Figure S42: 1H NMR spectrum (400 MHz) of 6 in acetone-d6, Figure S43: 1H NMR spectrum (400 MHz) of 6 in MeOH-d4, Figure S44: 13C NMR spectrum (100 MHz) of 6 in MeOH-d4, Figure S45: HSQC spectrum of 6 in MeOH-d4, Figure S46: HRESIMS of 6, Figure S47: 1H NMR spectrum (400 MHz) of 6c in DMSO-d6, Figure S48: 13C NMR spectrum (100 MHz) of 6c in DMSO-d6, Figure S49: HMBC spectrum of 6c in DMSO-d6, Figure S50: HRESIMS of 6c, Figure S51: 1H NMR spectrum (400 MHz) of 6d in DMSO-d6, Figure S52: 13C NMR spectrum (100 MHz) of 6d in DMSO-d6, Figure S53: HMBC spectrum of 6d in DMSO-d6, Figure S54: HRESIMS of 6d, Attachment S1: Supporting information for the calculated ECD spectra of compounds 4, 5, and 6c.
Author Contributions
Conceptualization, W.-Z.L. and L.-Y.M.; fermentation, compound purification and bioassay, L.-Y.M., H.-B.Z., H.-H.K., M.-J.Z. and H.R.; structural elucidation, writing and funding acquisition, W.-Z.L.; structural elucidation, review and editing, D.-S.L.
Funding
This research was funded by National Natural Science Foundation of China (No. 31270082) and Natural Science Foundation of Shandong Province, China (No. Y2008B17).
Acknowledgments
A. sclerotiorum JH42 was identified by Tianjiao Zhu (Key Laboratory of Marine Drugs, Chinese Ministry of Education; School of Medicine and Pharmacy, Ocean University of China.). We thank Qianqun Gu (Ocean University of China) for giving us the great help and beneficial suggestions in the structure determinations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xu, J.W.; Che, Q.; Zhu, T.J.; Gu, Q.Q.; Li, D.H. The cytotoxics secondary metabolites from South China Sea derived fungus Aspergillus sclertoiorum XJW-56. Chin. J. Mar. Drugs 2014, 33, 13–18. [Google Scholar]
- Motohashi, K.; Inaba, S.; Takagi, M.; Shin-ya, K. JBIR-15, a new aspochracin derivative, isolated from a sponge-derived fungus Aspergillus sclerotiorum Huber Sp080903f04. Biosci. Biotechnol. Biochem. 2009, 73, 1898–1900. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phainuphong, P.; Rukachaisirikul, V.; Saithong, S.; Phongpaichit, S.; Bowornwiriyapan, K.; Muanprasat, C.; Srimaroeng, C.; Duangjai, A.; Sakayaroj, J. Lovastatin analogues from the soil-derived fungus Aspergillus sclerotiorum PSU-RSPG178. J. Nat. Prod. 2016, 79, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Phainuphong, P.; Rukachaisirikul, V.; Tadpetch, K.; Sukpondma, Y.; Saithong, S.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. γ-Butenolide and furanone derivatives from the soil-derived fungus Aspergillus sclerotiorum PSU-RSPG178. Phytochemistry 2017, 137, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhu, H.; Hong, K.; Wang, Y.; Liu, P.; Wang, X.; Peng, X.; Zhu, W. Novel cyclic hexapeptides from marine-derived fungus Aspergillus sclerotiorum PT06-1. Org. Lett. 2009, 11, 5262–5265. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, Z.; Wang, Y.; Hong, K.; Liu, P.; Zhu, W. Cyclic tripeptides from the halotolerant fungus Aspergillus sclerotiorum PT06-1. J. Nat. Prod. 2010, 73, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Whyte, A.C.; Joshi, B.K.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. New cyclic peptide and bisindolyl benzenoid metabolites from the sclerotia of Aspergillus sclerotiorum. J. Nat. Prod. 2000, 63, 1006–1009. [Google Scholar] [CrossRef]
- Whyte, A.C.; Gloer, J.B.; Wicklow, D.T.; Dowdw, P.F. Sclerotiamide: a new member of the paraherquamide class with potent antiinsectan activity from the sclerotia of Aspergillus sclerotiorum. J. Nat. Prod. 1996, 59, 1093–1095. [Google Scholar] [CrossRef]
- Micetich, R.G.; Macdona, J.C. Metabolites of Aspergillus sclerotiorum Huber. J. Chem. Soc. 1964, 1507–1510. [Google Scholar] [CrossRef]
- Varga, J.; Kevei, E.; Rinyu, E.; Téren, J.; Kozakiewicz, Z. Ochratoxin production by Aspergillus species. Appl. Environ. Microbiol. 1996, 62, 4461–4464. [Google Scholar] [CrossRef]
- Bao, J.; Wang, J.; Zhang, X.-Y.; Nong, X.-H.; Qi, S.-H. New furanone derivatives and alkaloids from the co-culture of marine-derived fungi Aspergillus sclerotiorum and Penicillium citrinum. Chem. Biodivers. 2017, 14, e1600327. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Liu, G.; Liu, Q. Spatial and environmental effects on plant communities in the Yellow River Delta, Eastern China. J. For. Res. 2009, 20, 117–122. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, J.; Liu, C.; Zong, S.; Lu, Z. Effect of organic materials on the chemical properties of saline soil in the Yellow River Delta of China. Front. Earth Sci. 2015, 9, 259–267. [Google Scholar] [CrossRef]
- Guan, Y.; Liu, G.; Wang, J. Saline-alkali land in the Yellow River Delta: amelioration zonation based on GIS. J. Geogr. Sci. 2001, 11, 313–320. [Google Scholar]
- Fu, P.; Liu, P.; Qu, H.; Wang, Y.; Chen, D.; Wang, H.; Li, J.; Zhu, W. α-Pyrones and diketopiperazine derivatives from the marine-derived actinomycete Nocardiopsis dassonvillei HR10-5. J. Nat. Prod. 2011, 74, 2219–2223. [Google Scholar] [CrossRef]
- Liu, W.Z.; Ma, L.Y.; Liu, D.S.; Huang, Y.L.; Wang, C.H.; Shi, S.S.; Pan, X.H.; Song, X.D.; Zhu, R.X. Peniciketals A−C, new spiroketals from saline soil derived Penicillium raistrichii. Org. Lett. 2014, 16, 90–93. [Google Scholar] [CrossRef]
- Kang, H.H.; Zhang, H.B.; Zhong, M.J.; Ma, L.Y.; Liu, D.S.; Liu, W.Z.; Ren, H. Potential antiviral xanthones from a coastal saline soil fungus Aspergillus iizukae. Mar. Drugs 2018, 16, 449. [Google Scholar] [CrossRef]
- He, J.; Wijeratne, E.M.; Bashyal, B.P.; Zhan, J.; Seliga, C.J.; Liu, M.X.; Pierson, E.E.; Pierson III, L.S.; VanEtten, H.D.; Gunatilaka, A.A. Cytotoxic and other metabolites of Aspergillus inhabiting the rhizosphere of Sonoran desert plants. J. Nat. Prod. 2004, 67, 1985–1991. [Google Scholar] [CrossRef]
- Birkinshaw, J.H.; Oxford, A.E.; Raistrick, H. Studies in the biochemistry of micro-organisms. Biochem. J. 1936, 30, 394–411. [Google Scholar] [CrossRef]
- Kimura, Y.; Nakahara, S.; Fujioka, S. Aspyrone, a nematocidal compound isolated from the fungus, Aspergillus melleus. Biosci. Biotechnol. Biochem. 1996, 60, 1375–1376. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, L.; Wang, H.; Wang, H.; Wu, H.-H.; Lu, X.; Pei, Y.-H.; Wu, X.; Pan, B.; Hua, H.-M.; et al. A new compound along with seven known compounds from an endophytic fungus Aspergillus sp HS-05. Rec. Nat. Prod. 2013, 7, 320–324. [Google Scholar]
- Matoba, K.; Yamazaki, T. Reduction of some vinylogous esters with lithium aluminum hydride. IV Yakugaku Zasshi. 1972, 92, 213–220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bao, J.; Zhang, X.-Y.; Yao, Q.-F.; Xu, X.-Y.; Nong, X.-H.; Qi, S.H. Secondary metabolites from the co-culture of gorgonian-associated fungi Aspergillus sclertoiorum and Penicillium citrinum. Nat. Prod. Res. Dev. 2014, 26, 1–4. [Google Scholar]
- Niu, D.L.; Wang, L.S.; Zhang, Y.J.; Yan, C.R. Chemical constituents of Acroscyphus sphaerophoroides. Plant Sci. J. 2011, 29, 234–237. [Google Scholar] [CrossRef]
- Zhang, D.W.; Dai, S.J.; Liu, W.; Li, G.H. Chemical constituents from the vines of Pueraria lobata. Chin. J. Nat. Med. 2010, 8, 196–198. [Google Scholar] [CrossRef]
- Montenegro, T.G.C.; Rodrigues, F.A.R.; Jimenez, P.C.; Angelim, A.L.; Melo, V.M.M.; Filho, E.R.; Oliveira, M.C.F.; Costa-Lotufo, L.V. Cytotoxic activity of fungal strains isolated from the ascidian Eudistoma vannamei. Chem. Biodivers. 2012, 9, 2203–2209. [Google Scholar] [CrossRef] [PubMed]
- Vansteelandt, M.; Blanchet, E.; Egorov, M.; Petit, F.; Toupet, L.; Bondon, A.; Monteau, F.; Le Bizec, B.; Thomas, O.P.; Pouchus, Y.F.; et al. Ligerin, an antiproliferative chlorinated sesquiterpenoid from a marine-derived Penicillium strain. J. Nat. Prod. 2013, 76, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.-J.; Xu, L.-L.; Li, Y.-Y.; Han, T.; Zhang, Q.-Y.; Ming, Q.-L.; Rahman, K.; Qin, L.P. Cytotoxic metabolites from the cultures of endophytic fungi from Panax ginseng. Appl. Microbiol. Biotechnol. 2013, 97, 7617–7625. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.-M.; Meng, L.-H.; Wang, B.-G. Polyketides from the marine mangrove-derived fungus Aspergillus ochraceus MA-15 and their activity against aquatic pathogenic bacteria. Phytochem. Lett. 2015, 12, 232–236. [Google Scholar] [CrossRef]
- Martínez-Luis, S.; González, M.C.; Ulloa, M.; Mata, R. Phytotoxins from the fungus Malbranchea aurantiaca. Phytochemistry 2005, 66, 1012–1016. [Google Scholar] [CrossRef]
- Suzuki, S.; Kimura, T.; Saito, F.; Ando, K. Antitumor and antiviral properties of penicillic acid. Agric. Biol. Chem. 1971, 35, 287–290. [Google Scholar] [CrossRef]
- Kang, S.W.; Kim, S.W. New antifungal activity of penicillic acid against Phytophthora species. Biotechnol. Lett. 2004, 26, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, W.; Huang, Y.; Xian, G. Two acid sorbicillin analogues from saline lands-derived fungus Trichoderma sp. J. Antibiot. 2011, 64, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-X.; Li, J.; Zhang, J.-M.; Jin, X.-J.; Yu, B.; Fang, J.-G.; Wu, Q.-X. Isolation, identification, and activity evaluation of chemical constituents from the soil fungus Fusarium avenaceum SF-1502 and endophytic fungus Fusarium proliferatum AF-04. J. Agric. Food Chem. 2019, 67, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Liu, H.; Pan, Y.; Li, J.; Liu, L.; She, Z. Anti-inflammatory activity from the mangrove endophytic fungus Ascomycota sp. CYSK-4. Mar. Drugs 2018, 16, 54. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all the compounds are available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).