Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone

 and
and
Abstract
1. Introduction
2. Results and Discussion
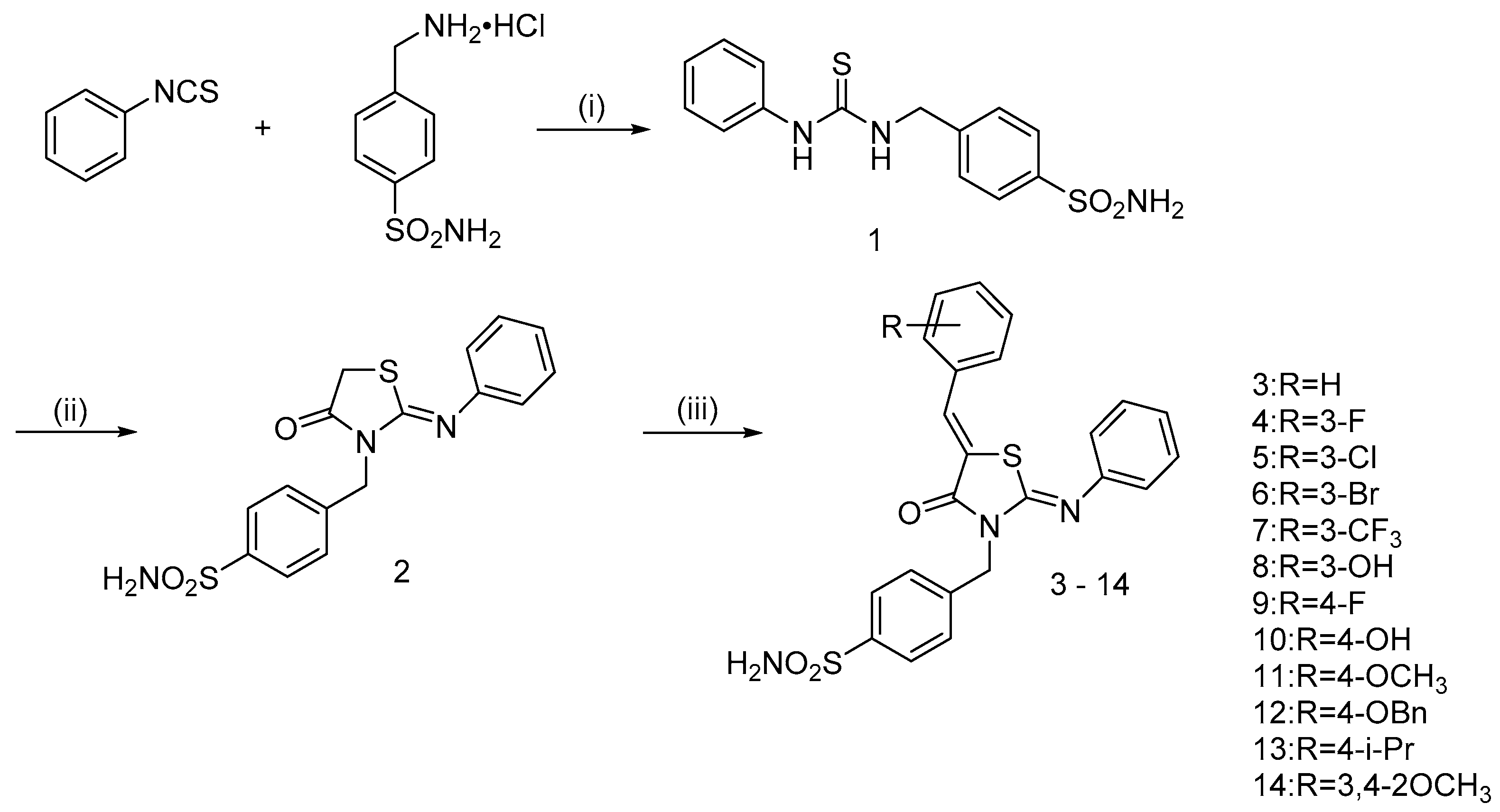
2.1. Chemistry
2.2. hCA Enzyme Inhibition Studies
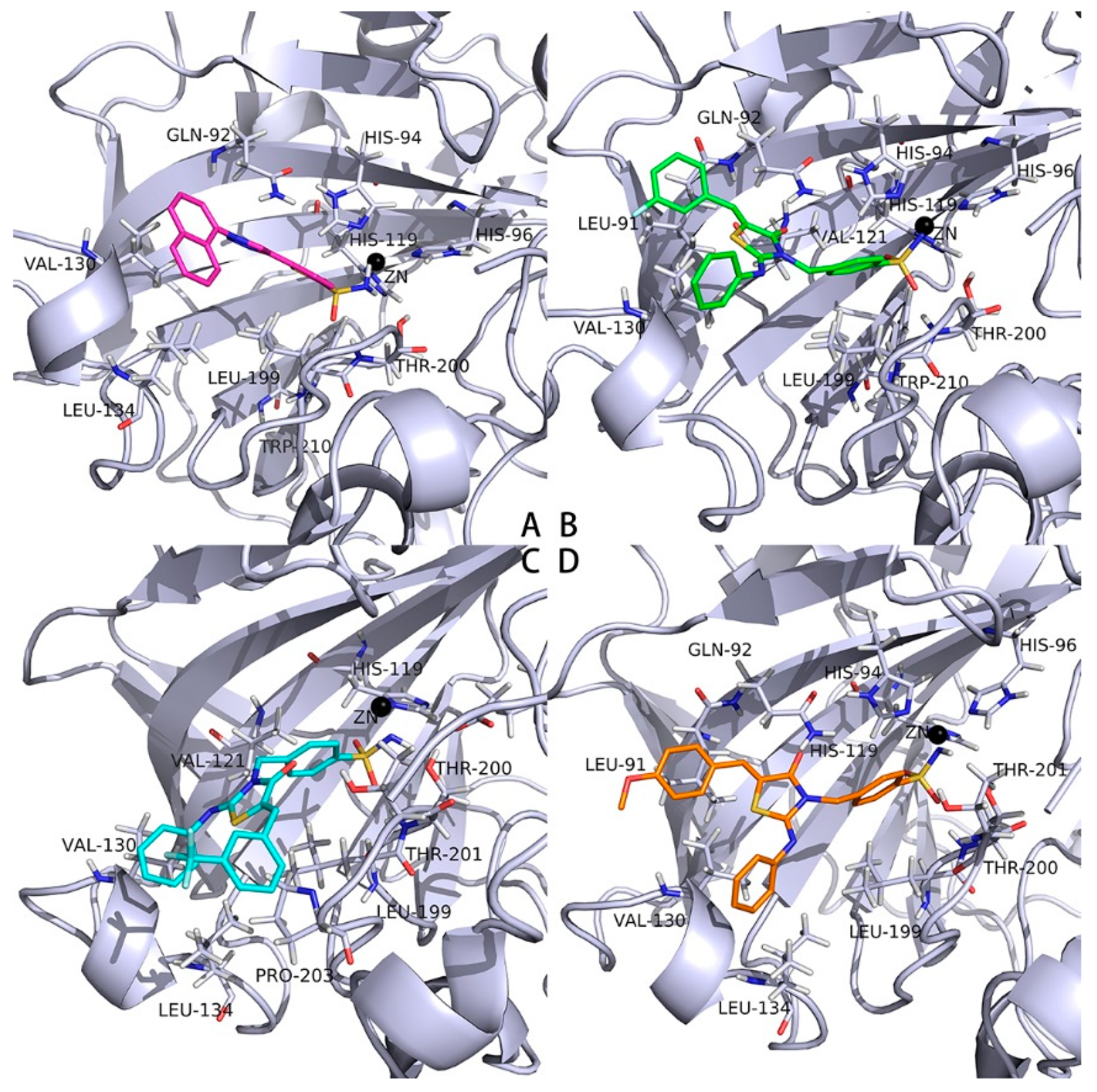
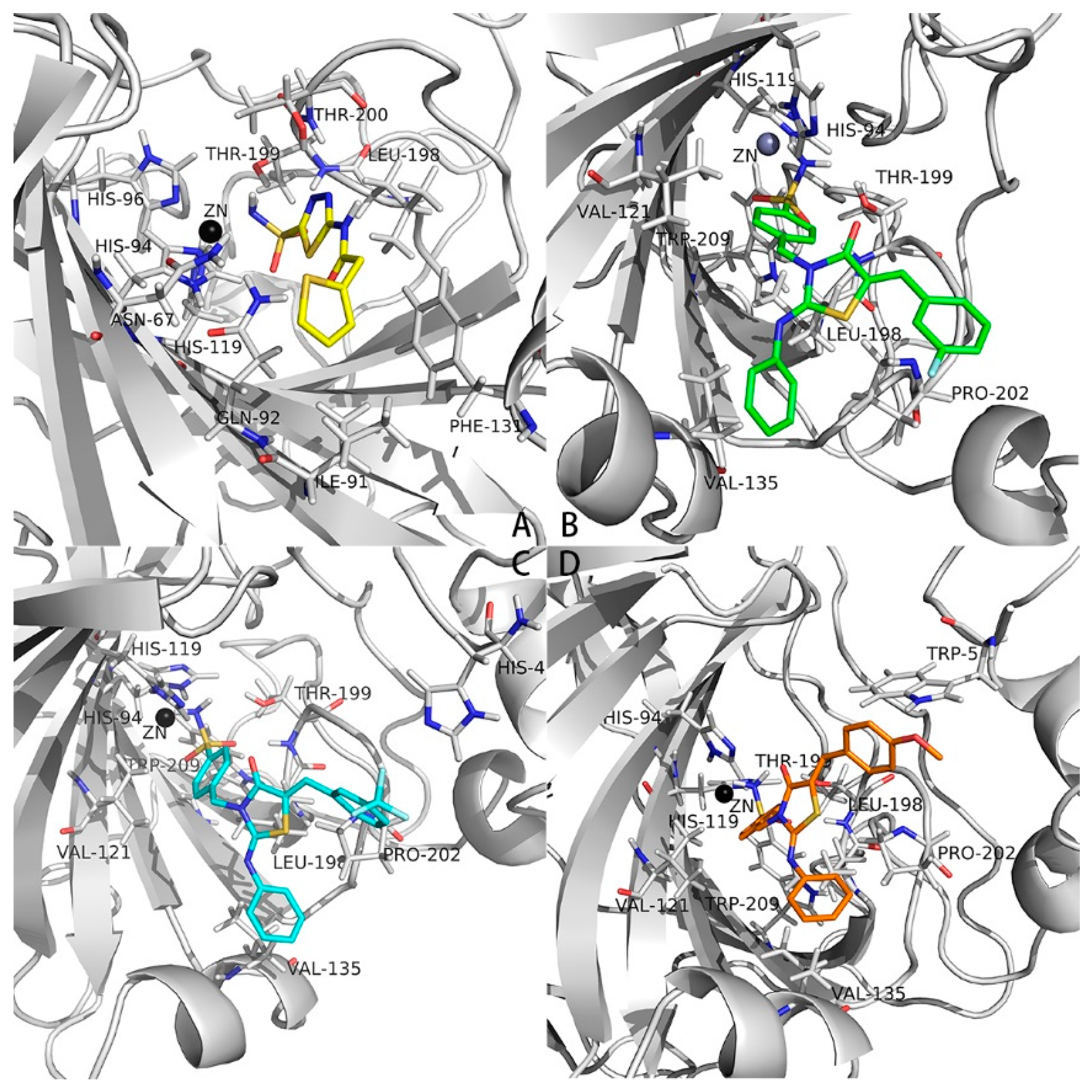
2.3. Docking Studies into the Active Site of hCAs
3. Materials and Methods
3.1. Chemistry
3.1.1. 4-((3-phenylthioureido)methyl)benzenesulfonamide (1)
3.1.2. (Z)-4-((4-oxo-2-(phenylimino)thiazolidin-3-yl)methyl)benzenesulfonamide (2)
3.1.3. General Procedure of Synthesis of 4-(((Z)-5-((Z)-benzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)methyl)benzenesulfonamide hybrids (3–14)
3.2. hCA Enzyme Inhibition Assays
3.3. Single-Crystal Structure
3.4. Preparation of Compound 2–14 for Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stadie, W.C.; O’Brien, H. The catalytic of the hydration of carbon dioxide and dehydration of carbonic acid by an enzyme isolated from red blood cells. J. Biol. Chem. 1933, 103, 521–529. [Google Scholar]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, Subcellular Biochemistry; Frost, S.C., McKenna, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Švastová, E.; Hulíková, A.; Rafajová, M.; Zat’Ovičová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A.; Harris, A.L.; Harris, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Boil. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1377–1378. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 1–13. [Google Scholar] [CrossRef]
- A Phase I, Multi-center, Open-label, Study to Investigate the Safety, Tolerability and Pharmacokinetic of SLC-0111 in Subjects with Advanced Solid Tumours. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02215850 (accessed on 15 September 2017).
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Laurenzana, A.; Carta, F.; Supuran, C.T.; Calorini, L. The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells toconventional chemotherapy. J. Enzym. Inhib. Med. Chem. 2019, 8, 117–123. [Google Scholar]
- Romagnoli, R.; Baraldi, P.G.; Prencipe, F.; Oliva, P.; Baraldi, S.; Salvador, M.K.; Lopez-Carz, L.C.; Brancale, A.; Ferla, S.; Hamel, E.; et al. Synthesis and biological evaluation of 2-methyl-4, 5-disubstituted oxazoles as a novel class of highly potent antitubulin agents. Sci. Rep. 2017, 7, 1–19. Available online: https://www.nature.com/articles/srep46356. (accessed on 13 April 2017). [CrossRef] [PubMed]
- Li, Y.-S.; Hu, D.-K.; Zhao, D.-S.; Liu, X.-Y.; Jin, H.-W.; Song, G.-P.; Cui, Z.-N.; Zhang, L.-H. Design, synthesis and biological evaluation of 2,4-disubstituted oxazole derivatives as potential PDE4 inhibitors. Bioorg. Med. Chem. 2017, 25, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Sun, X.; Liu, Y.; Long, W.; Chen, B.; Shen, S.; Ma, H. Synthesis, crystal structure, biological activity and theoretical calculations of novel isoxazole derivatives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 152, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Tomi, I.H.; Tomma, J.H.; Al-Daraji, A.H.; Al-Dujaili, A.H. Synthesis, characterization and comparative study the microbial activity of some heterocyclic compounds containing oxazole and benzothiazole moieties. J. Saudi Chem. Soc. 2015, 19, 392–398. [Google Scholar] [CrossRef]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sato, M.; Kakinuma, H.; Miyata, N.; Taniguchi, K.; Bando, K.; Koda, A.; Kameo, K. Pyrazole and Isoxazole Derivatives as New, Potent, and Selective 20-Hydroxy-5,8,11,14-eicosatetraenoic Acid Synthase Inhibitors. J. Med. Chem. 2003, 46, 5416–5427. [Google Scholar] [CrossRef]
- Patel, N.B.; Shaikh, F.M. Synthesis and antimicrobial activity of new 4-thiazolidinone derivatives containing 2-amino-6-methoxybenzothiazole. Saudi Pharm. J. 2010, 18, 129–136. [Google Scholar] [CrossRef]
- Shiradkar, M.R.; Ghodake, M.; Bothara, K.G.; Bhandari, S.V.; Nikalje, A.; Akula, K.C.; Desai, N.C.; Burange, P.J. Synthesis and anticonvulsant activity of clubbed thiazolidinoneebarbituric acid and thiazolidinoneetriazole derivatives. Arkivoc 2007, 14, 58–74. [Google Scholar]
- Ottana, R.; Maccari, R.; Giglio, M.; Del Corso, A.; Cappiello, M.; Mura, U.; Cosconati, S.; Marinelli, L.; Novellino, E.; Sartini, S.; et al. Identification of 5-arylidene-4-thiazolidinone derivatives endowed with dual activity as aldose reductase inhibitors and antioxidant agents for the treatment of diabetic complications. Eur. J. Med. Chem. 2011, 46, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent developments and biological activities of thiazolidinone derivatives: A review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar]
- Barreca, M.L.; Balzarini, J.; Chimirri, A.; De Clercq, E.; De Luca, L.; Höltje, H.D.; Höltje, M.; Monforte, A.M.; Monforte, P.; Pannecouque, C.; et al. Design, Synthesis, Structure−Activity Relationships, and Molecular Modeling Studies of 2,3-Diaryl-1,3-thiazolidin-4-ones as Potent Anti-HIV Agents. J. Med. Chem. 2002, 45, 5410–5413. [Google Scholar] [CrossRef] [PubMed]
- Takasu, K.; Pudhom, K.; Kaiser, M.; Brun, R.; Ihara, M. Synthesis and Antimalarial Efficacy of Aza-Fused Rhodacyanines in Vitro and in the P. berghei Mouse Model. J. Med. Chem. 2006, 49, 4795–4798. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Tan, O.; Ozadali, K.; Piskin, K.; Balkan, A. Molecular modeling, synthesis and screening of some new 4-thiazolidinone derivatives with promising selective COX-2 inhibitory activity. Eur. J. Med. Chem. 2012, 57, 59–64. [Google Scholar] [CrossRef]
- Ansari, M.F.; Idrees, D.; Hassan, M.I.; Ahmad, K.; Avecilla, F.; Azam, A. Design, synthesis and biological evaluation of novel pyridine-thiazolidinone derivatives as anticancer agents: Targeting human carbonic anhydrase IX. Eur. J. Med. Chem. 2018, 144, 544–556. [Google Scholar]
- Güzel-Akdemir, Ö.; Angeli, A.; Demir, K.; Supuran, C.T.; Akdemir, A. Novel thiazolidinone-containing compounds, without the well-known sulphonamide zinc-binding group acting as human carbonic anhydrase IX inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 1299–1308. [Google Scholar]
- Li, F.-R.; Fan, Z.-F.; Qi, S.-J.; Wang, Y.-S.; Wang, J.; Liu, Y.; Cheng, M.-S. Design, Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase IX Inhibitory Evaluations of Novel N-Substituted-β-d-Glucosamine Derivatives that Incorporate Benzenesulfonamides. Molecules 2017, 22, 785. [Google Scholar]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Boil. Chem. 1967, 242, 4221–4229. [Google Scholar]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- Biswas, S.; McKenna, R.; Supuran, C.T. Effect of incorporating a thiophene tail in the scaffold of acetazolamide on the inhibition of human carbonic anhydraseisoforms I, II, IX and XII. Bioorg. Med. Chem. Lett. 2013, 23, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An Improved AutoDock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar]
- Discovery Studio User Manual; Accelrys Inc.: San Diego, CA, USA, 2008.
Sample Availability: Samples of the compounds (2)–(14) are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | hCA II (IC50, nM) | hCA IX (IC50, nM) | Docking Scores (Kcal/mol) |
|---|---|---|---|---|
| 2 | / | 266.2 | 242.1 | −5.16 |
| 3 | H | 166.3 | 182.9 | −6.14 |
| 4 | 3-F | 146.5 | 130.5 | −6.68 |
| 5 | 3-Cl | 270.3 | 292.3 | −5.00 |
| 6 | 3-Br | 148.0 | 154.2 | −6.18 |
| 7 | 3-CF3 | 335.0 | 544.4 | −3.24 |
| 8 | 3-OH | 151.3 | 140.9 | −6.90 |
| 9 | 4-F | 216.7 | 246.1 | −5.02 |
| 10 | 4-OH | 160.8 | 144.9 | −6.30 |
| 11 | 4-OCH3 | 115.4 | 117.2 | −6.79 |
| 12 | 4-OBn | 1725 | >10,000 | 0.70 |
| 13 | 4-i-Pr | 692.1 | 1246 | −2.15 |
| 14 | 3,4-2OCH3 | 121.3 | 146.5 | −6.51 |
| Mafenide hydrochloride | 8912 | >30,000 | NA a | |
| AcetazolaMide (AZM) | 30.47 | 88.1 | −5.46 | |
| SLC-0111 | 137.8 | 180.7 | −6.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-P.; Yin, Z.-F.; Li, J.-Y.; Wang, Z.-P.; Wu, Q.-J.; Wang, J.; Liu, Y.; Cheng, M.-S. Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone. Molecules 2019, 24, 2418. https://doi.org/10.3390/molecules24132418
Zhang Z-P, Yin Z-F, Li J-Y, Wang Z-P, Wu Q-J, Wang J, Liu Y, Cheng M-S. Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone. Molecules. 2019; 24(13):2418. https://doi.org/10.3390/molecules24132418
Chicago/Turabian StyleZhang, Zuo-Peng, Ze-Fa Yin, Jia-Yue Li, Zhi-Peng Wang, Qian-Jie Wu, Jian Wang, Yang Liu, and Mao-Sheng Cheng. 2019. "Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone" Molecules 24, no. 13: 2418. https://doi.org/10.3390/molecules24132418
APA StyleZhang, Z.-P., Yin, Z.-F., Li, J.-Y., Wang, Z.-P., Wu, Q.-J., Wang, J., Liu, Y., & Cheng, M.-S. (2019). Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone. Molecules, 24(13), 2418. https://doi.org/10.3390/molecules24132418