NFBTA: A Potent Cytotoxic Agent against Glioblastoma


 , and
, and 
Abstract
1. Introduction
2. Results
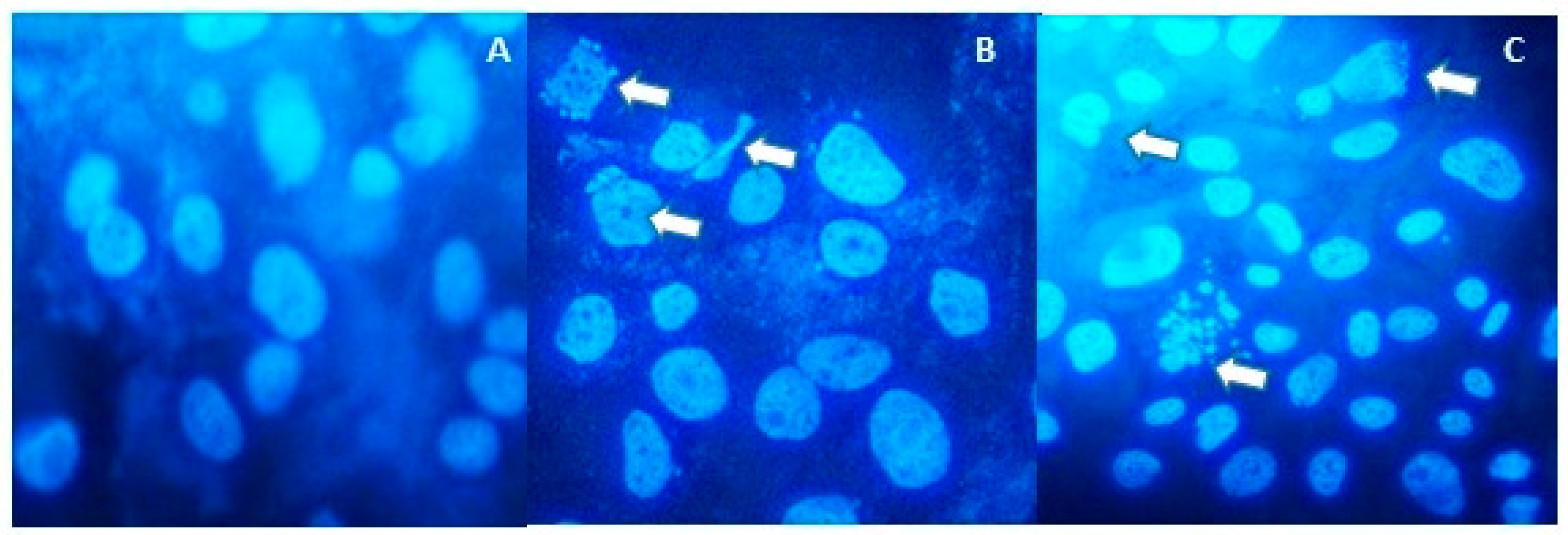
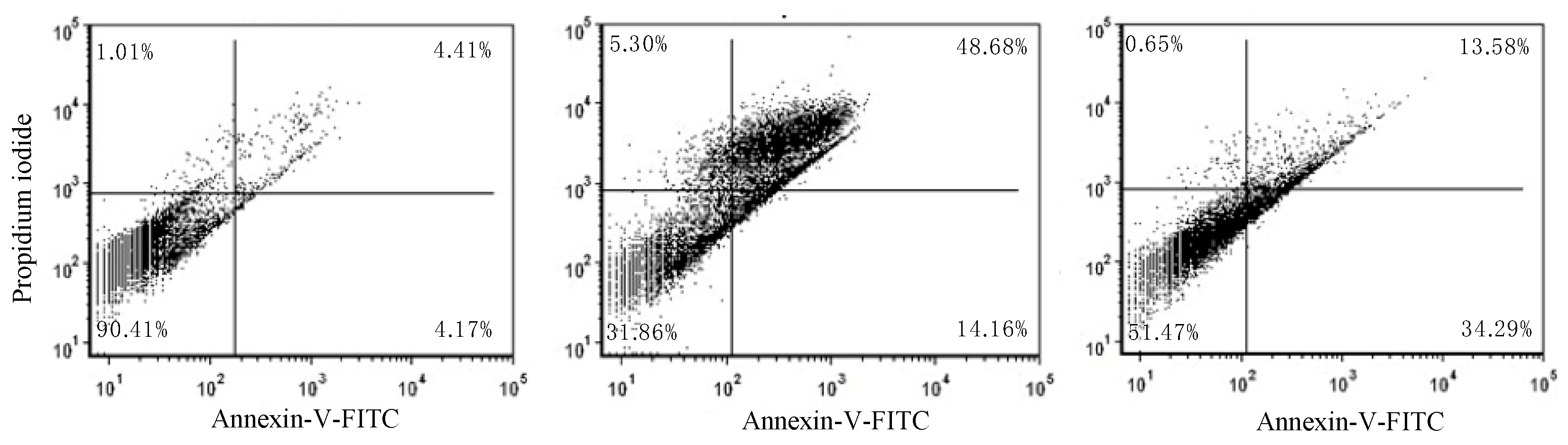
2.1. Anti-Proliferative and Pro-Apoptotic Effect of NFBTA
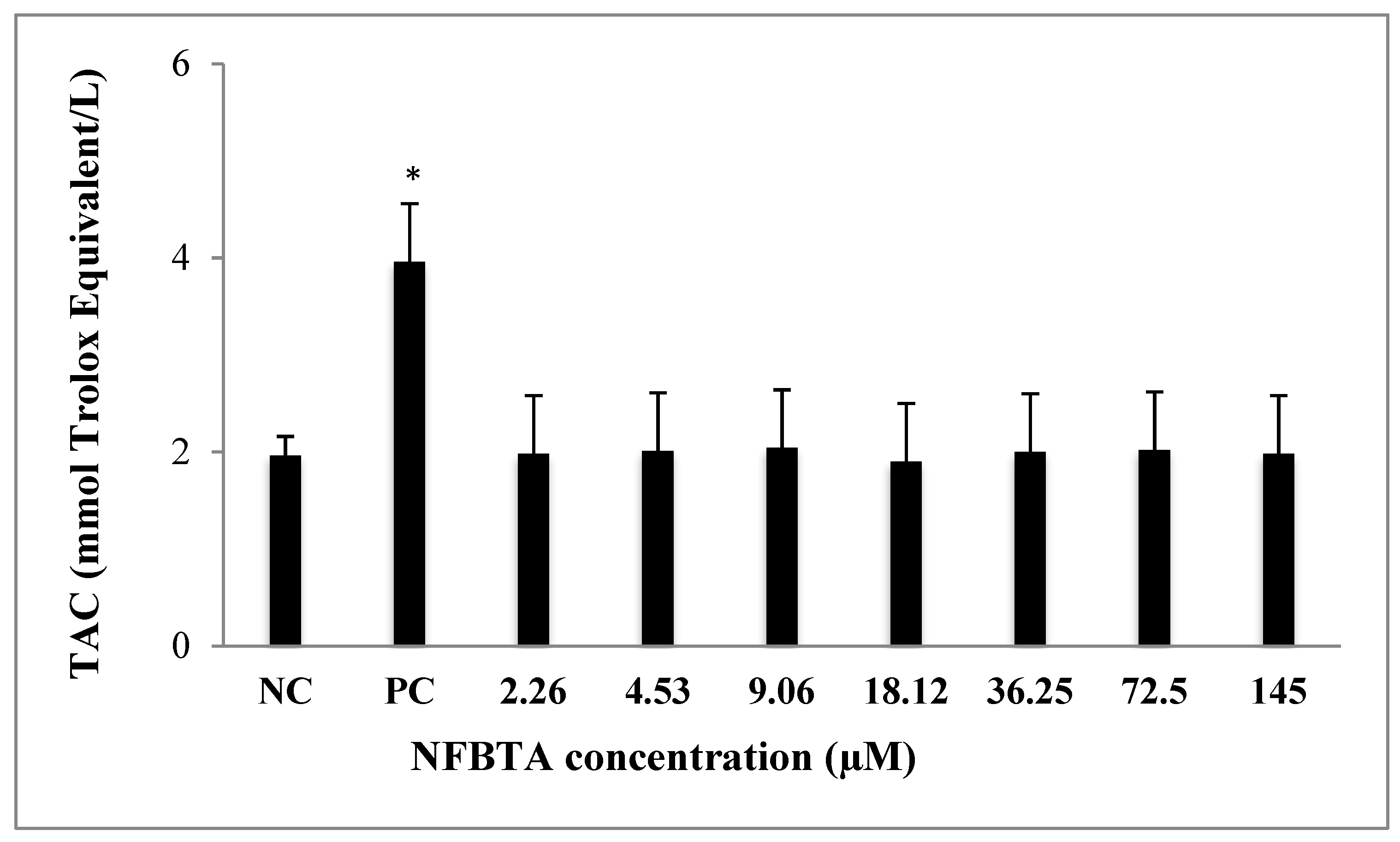
2.2. Oxidative Effects of NFBTA on U87MG Cells
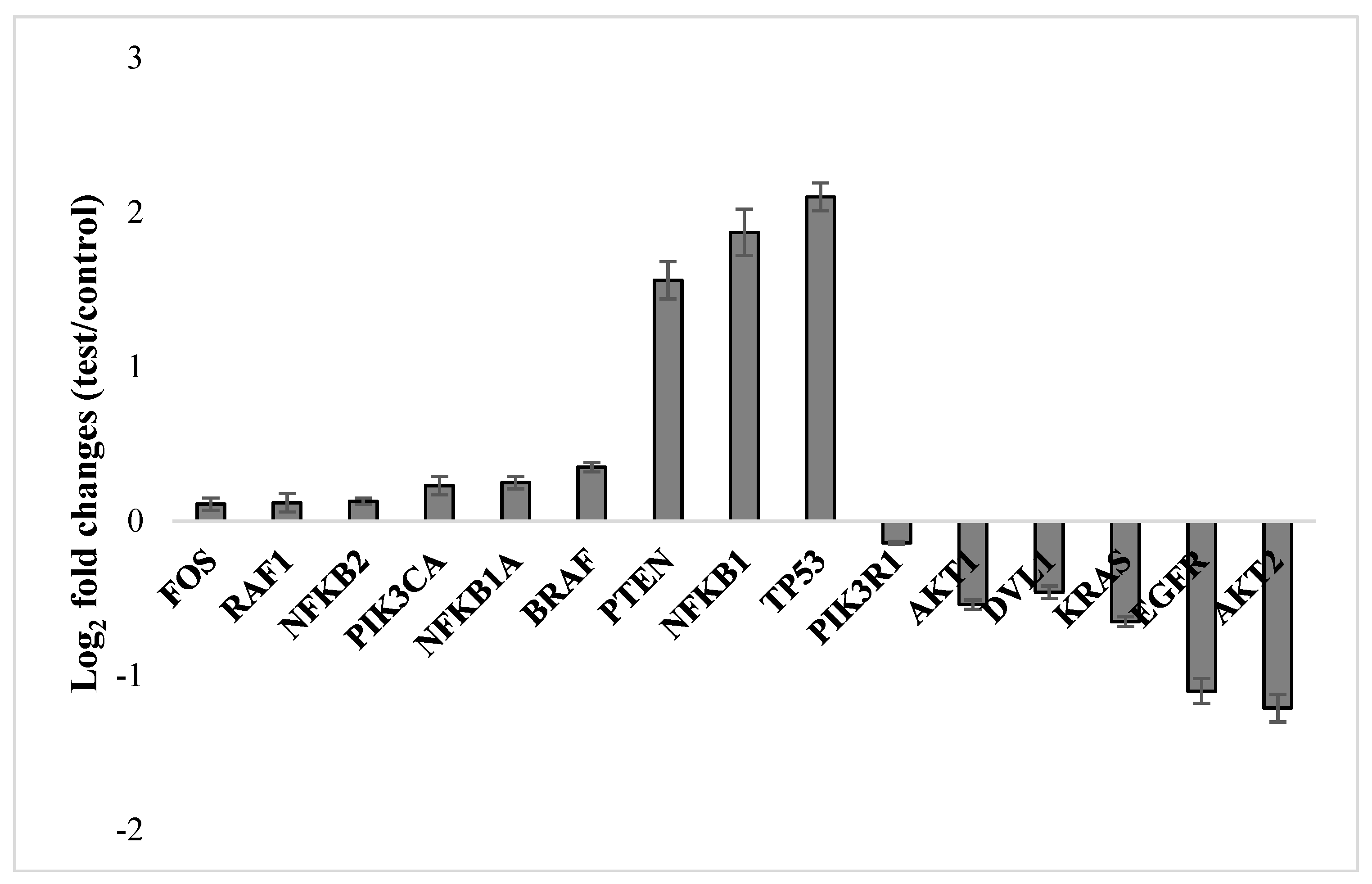
2.3. Molecular Responses in U87MG Cells to NFBTA
2.4. Biosafety Assessments
3. Discussion
4. Materials and Methods
4.1. Preparation of NFBTA
4.2. Cell Cultures and Treatments
4.3. MTT Assay
4.4. LDH Assay
4.5. Total Antioxidant Capacity (TAC) and Total Oxidant Status (TOS) Assays
4.6. Hoechst 33,258 Staining for Apoptosis Determination
4.7. Annexin V-FITC/PI Apoptosis Assay
4.8. qRT-PCR Assay
4.9. Biosafety Evaluation
4.10. SCE Testing
4.11. Nucleic Acid Oxidation
4.12. Statistical Analysis
4.13. General Modeling Workflow
4.14. Computational Target Fishing
4.15. Molecular Docking
4.16. Molecular Dynamics Simulations
4.17. MM-PBSA Calculations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.; Scott, M.P. The developmental biology of brain tumors. Annu. Rev. Neurosci. 2001, 24, 385–428. [Google Scholar] [CrossRef] [PubMed]
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018, 18, 30031–30037. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273–e281. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Population-Based Studies on Incidence, Survival Rates, and Genetic Alterations in Astrocytic and Oligodendroglial Gliomas. J. Neuropathol. Exp. Neurol. 2005, 64, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: current status and future perspectives. BioMed Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef]
- Vital, A.L.; Tabernero, M.D.; Castrillo, A.; Rebelo, O.; Tão, H.; Gomes, F.; Nieto, A.B.; Resende Oliveira, C.; Lopes, M.C.; Orfao, A. Gene expression profiles of human glioblastomas are associated with both tumor cytogenetics and histopathology. Neuro Oncol. 2010, 12, 991–1003. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, G. Differential gene expression analysis in glioblastoma cells and normal human brain cells based on GEO database. Oncol. Lett. 2017, 14, 6040–6044. [Google Scholar] [CrossRef]
- Duan, C.; Zhang, B.; Deng, C.; Cao, Y.; Zhou, F.; Wu, L.; Duan, G. Piperlongumine induces gastric cancer cell apoptosis and G2/M cell cycle arrest both in vitro and in vivo. Tumor Biol. 2016, 37, 10793–10804. [Google Scholar] [CrossRef]
- Wang, F.; Mao, Y.; You, Q.; Hua, D.; Cai, D. Piperlongumine induces apoptosis and autophagy in human lung cancer cells through inhibition of PI3K/Akt/mTOR pathway. Int. J. ImmunopathoL Pharmacol. 2015, 28, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Pan, F.; Li, L.; Liu, Q.R.; Chen, Y.; Xiong, X.X.; Chen, X.Q. Piperlongumine selectively kills glioblastoma multiforme cells via reactive oxygen species accumulation dependent JNK and p38 activation. Biochem. Biophys. Res. Commun. 2013, 437, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.X.; Liu, J.M.; Qiu, X.Y.; Pan, F.; Yu, S.B.; Chen, X.Q. Piperlongumine induces apoptotic and autophagic death of the primary myeloid leukemia cells from patients via activation of ROS-p38/JNK pathways. Acta Pharmacol. Sin. 2015, 36, 362. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.O.; Park, J.A.; Kim, H.A.; Chang, Y.H.; Hong, Y.J.; Park, I.C.; Lee, J.K. Piperlongumine downregulates the expression of HER family in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 486, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Agnihotri, N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019, 109, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Tompkins, V.S.; Son, D.J.; Kamberos, N.L.; Stunz, L.L.; Halwani, A.; Janz, S. Piperlongumine inhibits LMP1/MYC-dependent mouse B-lymphoma cells. Biochem. Biophys. Res. Commun. 2013, 436, 660–665. [Google Scholar] [CrossRef]
- Song, X.; Gao, T.; Lei, Q.; Zhang, L.; Yao, Y.; Xiong, J. Piperlongumine Induces Apoptosis in Human Melanoma Cells Via Reactive Oxygen Species Mediated Mitochondria Disruption. Nutr. Cancer 2018, 70, 502–511. [Google Scholar] [CrossRef]
- Ginzburg, S.; Golovine, K.V.; Makhov, P.B.; Uzzo, R.G.; Kutikov, A.; Kolenko, V.M. Piperlongumine inhibits NF-κB activity and attenuates aggressive growth characteristics of prostate cancer cells. Prostate 2014, 74, 177–186. [Google Scholar] [CrossRef]
- Liu, Q.R.; Liu, J.M.; Chen, Y.; Xie, X.Q.; Xiong, X.X.; Qiu, X.Y.; Pan, F.; Liu, D.; Yu, S.B.; Chen, X.Q. Piperlongumine inhibits migration of glioblastoma cells via activation of ROS-dependent p38 and JNK signaling pathways. Oxid. Med. Cell. Longev. 2014, 2014, 653732. [Google Scholar] [CrossRef]
- Kim, T.H.; Song, J.; Kim, S.H.; Parikh, A.K.; Mo, X.; Palanichamy, K.; Kaur, B.; Yu, J.; Yoon, S.O.; Nakano, I.; et al. Piperlongumine treatment inactivates peroxiredoxin 4,exacerbates endoplasmic reticulum stress, and preferentially kills high-grade glioma cells. Neuro Oncol. 2014, 16, 1354–1364. [Google Scholar] [CrossRef]
- D’Sousa, C.O.; Araujo-Neto, J.H.; Baliza, I.R.S.; Dias, R.B.; Valverde, L.F.; Vidal, M.T.A.; Sales, C.B.S.; Rocha, C.A.G.; Moreira, D.R.M.; Soares, M.B.P.; et al. Novel piplartine-containing ruthenium complexes: synthesis, cell growth inhibition, apoptosis induction and ROS production on HCT116 cells. Oncotarget 2017, 8, 104367–104392. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.U.; Shankaraiah, G.; Kumar, R.S.; Pitke, V.V.; Rao, G.T.; Poornima, B.; Babu, K.S.; Sreedhar, A.S. Synthesis, anticancer, and antibacterial activities of piplartine derivatives on cell cycle regulation and growth inhibition. J. Asian Nat. Prod. Res. 2013, 15, 658–669. [Google Scholar] [CrossRef] [PubMed]
- da Nóbrega, F.R.; Ozdemir, O.; Nascimento, S.C.S.; Barboza, J.N.; Turkez, H.; de Sousa, D.P. Piplartine Analogues and Cytotoxic Evaluation against Glioblastoma. Molecules 2018, 23, 1382. [Google Scholar] [CrossRef]
- Thomas, G. Medicinal Chemistry An Introduction, 2nd ed.; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Kortagere, S.; Ekins, S.; Welsh, W.J. Halogenated ligands and their interactions with amino acids: implications for structure-activity and structure-toxicity relationships. J. Mol. Graph. Model. 2008, 27, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Diao, W.; Tong, X.; Yang, C.; Zhang, F.; Bao, C.; Chen, H.; Liu, L.; Li, M.; Ye, F.; Fan, Q.; et al. Behaviors of Glioblastoma Cells in in Vitro Microenvironments. Sci. Rep. 2019, 9, 85. [Google Scholar] [CrossRef]
- Pei, J.; Moon, K.S.; Pan, S.; Lee, K.H.; Ryu, H.H.; Jung, T.Y.; Kim, I.Y.; Jang, W.Y.; Jung, C.H.; Jung, S. Proteomic Analysis between U87MG and U343MG-A Cell Lines: Searching for Candidate Proteins for Glioma Invasion. Brain Tumor Res. Treat. 2014, 2, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Homer, N.; O’Connor, B.D.; Chen, Z.; Eskin, A.; Lee, H.; Merriman, B.; Nelson, S.F. U87MG decoded: the genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet. 2010, 6, e1000832. [Google Scholar] [CrossRef]
- Régina, A.; Demeule, M.; Ché, C.; Lavallée, I.; Poirier, J.; Gabathuler, R. Antitumor activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br. J. Pharmacol. 2008, 155, 185–197. [Google Scholar] [CrossRef]
- Desai, A.; Vyas, T.; Amiji, M. Cytotoxicity and apoptosis enhancement in brain tumor cells upon coadministration of paclitaxel and ceramide in nanoemulsion formulations. J. Pharm. Sci. 2008, 97, 2745–2756. [Google Scholar] [CrossRef]
- Woo, I.S.; Eun, S.Y.; Kim, H.J. Farnesyl diphosphate synthase attenuates paclitaxel-induced apoptotic cell death in human glioblastoma U87MG cells. Neurosci. Lett. 2010, 474, 115–120. [Google Scholar] [CrossRef]
- Meegan, M.J.; Nathwani, S.; Twamley, B.; Zisterer, D.M.; O’Boyle, N.M. Piperlongumine (piplartine) and analogues: Antiproliferative microtubule-destabilising agents. Eur. J. Med. Chem. 2017, 125, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Capello, T.M.; Martins, E.G.; De Farias, C.F.; Figueiredo, C.R.; Matsuo, A.L.; Passero, L.F.; Oliveira-Silva, D.; Sartorelli, P.; Lago, J.H. Chemical composition and in vitro cytotoxic and antileishmanial activities of extract and essential oil from leaves of Piper cernuum. Nat. Prod. Commun. 2015, 10, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Fofaria, N.M.; Qhattal, H.S.; Liu, X.; Srivastava, S.K. Nanoemulsion formulations for anti-cancer agent piplartine--characterization, toxicological, pharmacokinetics and efficacy studies. Int. J. Pharm. 2016, 498, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Karthick, T.; Joshi, B.D.; Mishra, R.; Tandon, P.; Ayala, A.P.; Ellena, J. Spectroscopic (far or terahertz, mid-infrared and Raman) investigation, thermal analysis and biological activity of piplartine. Spectrochim. Acta A 2017, 184, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Chen, X.; Wang, H. Piperlongumine induces apoptosis and synergizes with cisplatin or paclitaxel in human ovarian cancer cells. Oxid. Med. Cell. Longev. 2014, 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Michel, P.F.; Machado, C.M.L.; Aquino, N.C. Antitumour efficacy of piper tuberculatum and piplartine based on the hollow fiber assay. Planta Med. 2015, 81, 15–19. [Google Scholar] [CrossRef]
- Teng, C.S. Protooncogenes as mediators of apoptosis. Int. Rev. Cytol. 2000, 197, 137–202. [Google Scholar] [PubMed]
- Mikula, M.; Gotzmann, J.; Fischer, A.N.; Wolschek, M.F.; Thallinger, C.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene 2003, 22, 6725–6738. [Google Scholar] [CrossRef]
- Lyustikman, Y.; Momota, H.; Pao, W.; Holland, E.C. Constitutive activation of Raf-1 induces glioma formation in mice. Neoplasia 2008, 10, 501–510. [Google Scholar] [CrossRef]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Ngo, H.T.T.; Pham, T.Q.; Fung, K.M.; Hassell, L. BRAF Mutation is Associated with an Improved Survival in Glioma-a Systematic Review and Meta-analysis. Mol. Neurobiol. 2017, 55, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Chen, Y.H.; Emnett, R.J.; Dahiya, S.; Gutmann, D.H. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev. 2012, 26, 2561–2566. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, Z.; Du, Z.; Wu, B.; Jin, T.; Xu, K.; Xu, L.; Li, M.; Xu, H. The Identification of Key Genes and Pathways in Glioma by Bioinformatics Analysis. J. Immunol. Res. 2017, 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, T.; Perkins, N.D.; Wilson, C.L. NFKB1: a suppressor of inflammation, ageing and cancer. FEBS J. 2016, 283, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Narasimamurthy, R.; Xia, Y.; Myskiw, C.; Soda, Y.; Verma, I.M. Targeting NF-κB in glioblastoma: A therapeutic approach. Sci. Adv. 2016, 2, e1501292. [Google Scholar] [CrossRef]
- Seznec, J.; Weit, S.; Naumann, U. Gene expression profile in a glioma cell line resistant to cell death induced by the chimeric tumor suppressor-1 (CTS-1), a dominant-positive variant of p53--the role of NFkappaB. Carcinogenesis 2010, 31, 411–418. [Google Scholar] [CrossRef]
- Soubannier, V.; Stifani, S. NF-κB Signalling in Glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef]
- Chang, C.J.; Mulholland, D.J.; Valamehr, B.; Mosessian, S.; Sellers, W.R.; Wu, H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol. Cell Biol. 2008, 28, 3281–3289. [Google Scholar] [CrossRef]
- Yang, Z.F.; Yi, J.L.; Li, X.R.; Xie, D.X.; Liao, X.F.; Ma, X. PTEN induces apoptosis and up-regulates p53 expression in HepG2 cells. Zhonghua Gan Zang Bing Za Zhi. 2004, 12, 745–748. [Google Scholar] [PubMed]
- Nakanishi, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. The tumor suppressor PTEN interacts with p53 in hereditary cancer. Int. J. Oncol. 2014, 44, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.O.; Felty, Q.; Parkash, J.; Poppiti, R.; Roy, D. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS ONE 2013, 8, e54206. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1198. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Emdad, L.; Bacolod, M.D.; Kegelman, T.P.; Shen, X.N.; Alzubi, M.A.; Das, S.K.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 interacts with Akt isoform 2 to control glioma growth, survival, and pathogenesis. Cancer Res. 2014, 74, 7321–7332. [Google Scholar] [CrossRef] [PubMed]
- Majewska, E.; Szeliga, M. AKT/GSK3β Signaling in Glioblastoma. Neurochem. Res. 2017, 42, 918–924. [Google Scholar] [CrossRef]
- Xu, H.; Zong, H.; Ma, C.; Ming, X.; Shang, M.; Li, K.; He, X.; Du, H.; Cao, L. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017, 14, 512–516. [Google Scholar] [CrossRef]
- Cacciatore, I.; Fornasari, E.; Marinelli, L.; Eusepi, P.; Ciulla, M.; Ozdemir, O.; Tatar, A.; Turkez, H.; Di Stefano, A. Memantine-derived drugs as pontential antitumor agents for the treatment of glibastoma. Eur. J. Pharm. Sci. 2017, 109, 402–411. [Google Scholar] [CrossRef]
- Tao, T.; Lu, X.; Yao, L.; Wang, J.; Shi, Y.; Luo, H.; Liu, N.; You, Y. Expression of FOS protein in glioma and its effect on the growth of human glioma cells. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2013, 30, 293–296. [Google Scholar]
- Li, J.; Guo, G.; Li, J.; Hao, J.; Zhang, J.; Guo, Y.; Yu, H. The expression and significance of dishevelled in human glioma. J. Surg. Res. 2014, 192, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.L.; Parat, M.O.; Binder, Z.A.; Gallia, G.L.; Riggins, G.J. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget 2011, 2, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zheng, J. Understand spiciness: mechanism of TRPV1 channel activation by capsaicin. Protein Cell 2017, 8, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.V.; Al-Refae, K.; Wölk, G.; Bonatz, G.; Altmüller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer (Dove. Med Press) 2016, 8, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Lin, G.B.; Lin, S.H.; Lu, C.H.; Hsieh, C.H.; Ma, B.L.; Chao, C.Y. Static magnetic field enhances the anticancer efficacy of capsaicin on HepG2 cells via capsaicin receptor TRPV1. PLoS ONE 2018, 13, e0191078. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Sieglitz, F.L.; Bernardes, G.J. Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents. Chem. Soc. Rev. 2016, 45, 6130–6137. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Övey, İ.S. Involvement of apoptosis and calcium accumulation through TRPV1 channels in neurobiology of epilepsy. Neuroscience 2015, 293, 55–66. [Google Scholar]
- Nersesyan, Y.; Asuthkar, S.; Velpula, K.K.; Sun, X.; Demirkhanyan, L.; Zakharian, E. Role of TRPV1 Channels in Glioma Cell Viability and Survival. Biophys. J. 2015, 108, 124. [Google Scholar] [CrossRef][Green Version]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 2, 977–990. [Google Scholar] [CrossRef]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.G.H.; Méndez, L.Y.V.; Kouznetsov, V.V.; Stashenko, E.E. Efficient Synthesis of New N-Benzyl- or N-(2-Furylmethyl) cinnamamides Promoted by the ‘Green’ Catalyst Boric Acid, and Their Spectral Analysis. Synthesis 2008, 3, 377–382. [Google Scholar] [CrossRef]
- Rajan, P.; Vedernikova, I.; Cos, P.; Berghe, D.V.; Augustyns, K.; Haemers, A. Synthesis and Evaluation of Caffeic Acid Amides as Antioxidants. Bioorg. Med. Chem. Lett. 2001, 11, 215–217. [Google Scholar] [CrossRef]
- Laserra, S.; Basit, A.; Sozio, P.; Marinelli, L.; Fornasari, E.; Cacciatore, I.; Ciulla, M.; Turkez, H.; Geyikoglu, F.; Di Stefano, A. Solid lipid nanoparticles loaded with lipoyl-memantine codrug: preparation and characterization. Int. J. Pharm. 2015, 485, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.; Fornasari, E.; Di Stefano, A.; Turkez, H.; Arslan, M.E.; Eusepi, P.; Ciulla, M.; Cacciatore, I. (R)-α-Lipoyl-Gly-l-Pro-l-Glu dimethyl ester as dual acting agent for the treatment of Alzheimer’s disease. Neuropeptides 2017, 66, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Péon, A.; Naulaerts, S.; Ballester, P.J. Predicting the Reliability of Drug-target Interaction Predictions with Maximum Coverage of Target Space. Sci. Rep. 2017, 7, 3820. [Google Scholar] [CrossRef]
- Nickel., J.; Gohlke, B.O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, 26–31. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Cruz-Monteagudo, M.; Borges, F.; Paz-y-Miño, C.; Cordeiro, M.N.D.S.; Rebelo, I.; Perez-Castillo, Y. Efficient and biologically relevant consensus strategy for Parkinson’s disease gene prioritization. BMC Med. Genom. 2016, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Tejera, E.; Cruz-Monteagudo, M.; Burgos, G.; Sánchez, M.E.; Sánchez-Rodríguez, A.; Pérez-Castillo, Y. Consensus strategy in genes prioritization and combined bioinformatics analysis for preeclampsia pathogenesis. BMC Med. Genom. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Bienert., S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA [Internet]. Santa Fe, NM: OpenEye Scientific Software. Available online: http://www.eyesopen.com (accessed on 1 April 2019).
- OpenEye Scientific Software. QUACPAC [Internet]. Santa Fe, NM: OpenEye Scientific Software. Available online: http://www.eyesopen.com (accessed on 1 April 2019).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018. University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Botello-Smith, W.M.; Luo, R. Applications of MMPBSA to Membrane Proteins I: Efficient Numerical Solutions of Periodic Poisson-Boltzmann Equation. J. Chem. Inf. Model. 2015, 55, 2187–2199. [Google Scholar] [CrossRef]
- Greene, D.; Botello-Smith, W.M.; Follmer, A.; Xiao, L.; Lambros, E.; Luo, R. Modeling Membrane Protein-Ligand Binding Interactions: The Human Purinergic Platelet Receptor. J. Phys. Chem. B 2016, 120, 12293–12304. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | U87MG Cells | HPAEpiC Cells | SI Value |
|---|---|---|---|
| NFBTA | 6.666 ± 0.78 | 97.385 ± 3.83 | 14.621 |
| Paclitaxel | 2.527 ± 0.37 | 21.545 ± 1.46 | 8.525 |
| Concentrations (µM) | TAC (mmol Trolox Equiv./L) | TOS (μmol H2O2 Equiv./L) |
|---|---|---|
| NC | 4.3 ± 0.4 | 7.1 ± 0.8 |
| PC | 11.8 ± 0.7* | 28.3 ± 1.6* |
| 2.26 | 4.5 ± 0.4 | 6.9 ± 0.4 |
| 4.53 | 4.5 ± 0.6 | 6.9 ± 0.5 |
| 9.06 | 5.2 ± 0.6* | 7.0 ± 0.6 |
| 18.12 | 5.5 ± 0.6* | 6.9 ± 0.6 |
| 36.25 | 5.7 ± 0.5* | 7.1 ± 0.5 |
| 72.5 | 7.1 ± 0.7* | 7.1 ± 0.6 |
| 145 | 8.3 ± 0.7* | 7.2 ± 0.6 |
| Concentrations (µM) | SCEs/Cell | pmol 8-OH-dG/μg DNA |
|---|---|---|
| NC | 5.9 ± 0.7 | 1.0 ± 0.2 |
| PC | 13.8 ± 1.8* | 4.3 ± 0.3* |
| 2.26 | 5.2 ± 0.6 | 1.0 ± 0.2 |
| 4.53 | 4.9 ± 0.5 | 1.1 ± 0.1 |
| 9.06 | 5.4 ± 0.6 | 1.1 ± 0.2 |
| 18.12 | 5.8 ± 0.4 | 0.9 ± 0.2 |
| 36.25 | 6.1 ± 0.7 | 1.1 ± 0.2 |
| 72.5 | 5.9 ± 0.6 | 1.2 ± 0.2 |
| 145 | 6.3 ± 0.8 | 1.1 ± 0.2 |
| Target | ||||
|---|---|---|---|---|
| MMP-2 | MMP-9 | HSP90 | ||
| CHEMPLP | Fitness | 99.18 | 101.71 | 58.22 |
| Z-score | 2.71 | 1.33 | 2.54 | |
| GoldScore | Fitness | 71.59 | 44.64 | 35.29 |
| Z-score | 2.45 | 0.97 | 1.90 | |
| ChemScore | Fitness | 43.01 | 30.36 | 21.71 |
| Z-score | 2.73 | 1.72 | 2.62 | |
| ASP | Fitness | 52.24 | 52.23 | 28.15 |
| Z-score | 2.36 | 0.94 | 2.18 | |
| Consensus Z-score | 10.25 | 4.97 | 9.24 | |
| RMSD (Å) (a) | 0.89 | 1.87 | 0.56 | |
| Target | |||||
|---|---|---|---|---|---|
| MMP-2 | MMP-9 | HSP90 | TRPV1 | ||
| CHEMPLP | Fitness | 69.81 | 83.32 | 56.67 | 54.05 |
| Z-score | 2.09 | 1.38 | 2.51 | 1.59 | |
| GoldScore | Fitness | 31.34 | 38.04 | 27.28 | 25.44 |
| Z-score | 0.57 | 0.02 | 0.82 | 0.57 | |
| ChemScore | Fitness | 17.30 | 25.99 | 11.81 | 12.78 |
| Z-score | 1.51 | 2.93 | 0.82 | 0.19 | |
| ASP | Fitness | 33.92 | 35.29 | 19.82 | 28.71 |
| Z-score | 1.53 | 0.69 | −0.55 | 3.42 | |
| Aggregated Z-score | 5.69 | 5.02 | 3.60 | 5.77 | |
| MMP-2 | MMP-9 | HSP90 | TRPV1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LIG A | LIG B | LIG C | LIG D | All Ligs | ||||||||||||
| Component | Average | Std. | Average | Std. | Average | Std. | Average | Std. | Average | Std. | Average | Std. | Average | Std. | Average | Std. |
| VDWAALS | −31.20 | 2.71 | −30.75 | 3.45 | −35.36 | 2.49 | −37.48 | 3.29 | −38.48 | 2.57 | −26.69 | 1.95 | −48.12 | 2.99 | −150.77 | 5.55 |
| EEL | −54.57 | 6.81 | −54.33 | 6.22 | −6.84 | 7.37 | 0.02 | 0.19 | 0.94 | 0.27 | −0.12 | 0.33 | 1.07 | 0.22 | 1.90 | 0.59 |
| EPB | 73.54 | 9.17 | 66.93 | 5.29 | 34.23 | 7.73 | ||||||||||
| ENPOLAR | −23.86 | 1.24 | −24.10 | 1.67 | −27.04 | 1.64 | −4.29 | 0.16 | −4.48 | 0.09 | −3.48 | 0.20 | −4.51 | 0.12 | −16.76 | 0.33 |
| EDISPER | 41.76 | 1.39 | 42.63 | 2.91 | 46.83 | 1.66 | ||||||||||
| ΔG gas | −85.77 | 7.47 | −85.08 | 6.08 | −42.19 | 7.48 | −37.46 | 3.26 | −37.54 | 2.58 | −26.81 | 1.92 | −47.05 | 2.98 | −148.87 | 5.51 |
| ΔG solv | 91.43 | 9.25 | 85.46 | 5.78 | 54.01 | 7.59 | −4.29 | 0.16 | −4.48 | 0.09 | −3.48 | 0.20 | −4.51 | 0.12 | −16.76 | 0.33 |
| ΔG TOTAL | 5.67 | 7.78 | 0.39 | 3.95 | 11.82 | 6.31 | −41.76 | 3.27 | −42.02 | 2.57 | −30.29 | 2.03 | −51.56 | 3.00 | −165.63 | 5.55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turkez, H.; Nóbrega, F.R.d.; Ozdemir, O.; Bezerra Filho, C.d.S.M.; Almeida, R.N.d.; Tejera, E.; Perez-Castillo, Y.; Sousa, D.P.d. NFBTA: A Potent Cytotoxic Agent against Glioblastoma. Molecules 2019, 24, 2411. https://doi.org/10.3390/molecules24132411
Turkez H, Nóbrega FRd, Ozdemir O, Bezerra Filho CdSM, Almeida RNd, Tejera E, Perez-Castillo Y, Sousa DPd. NFBTA: A Potent Cytotoxic Agent against Glioblastoma. Molecules. 2019; 24(13):2411. https://doi.org/10.3390/molecules24132411
Chicago/Turabian StyleTurkez, Hasan, Flávio Rogério da Nóbrega, Ozlem Ozdemir, Carlos da Silva Maia Bezerra Filho, Reinaldo Nóbrega de Almeida, Eduardo Tejera, Yunierkis Perez-Castillo, and Damião Pergentino de Sousa. 2019. "NFBTA: A Potent Cytotoxic Agent against Glioblastoma" Molecules 24, no. 13: 2411. https://doi.org/10.3390/molecules24132411
APA StyleTurkez, H., Nóbrega, F. R. d., Ozdemir, O., Bezerra Filho, C. d. S. M., Almeida, R. N. d., Tejera, E., Perez-Castillo, Y., & Sousa, D. P. d. (2019). NFBTA: A Potent Cytotoxic Agent against Glioblastoma. Molecules, 24(13), 2411. https://doi.org/10.3390/molecules24132411