
Synthesis, Crystal Structure, and Biological Evaluation of Fused Thiazolo[3,2-a]Pyrimidines as New Acetylcholinesterase Inhibitors

 , , , and
, , , and
Abstract
:
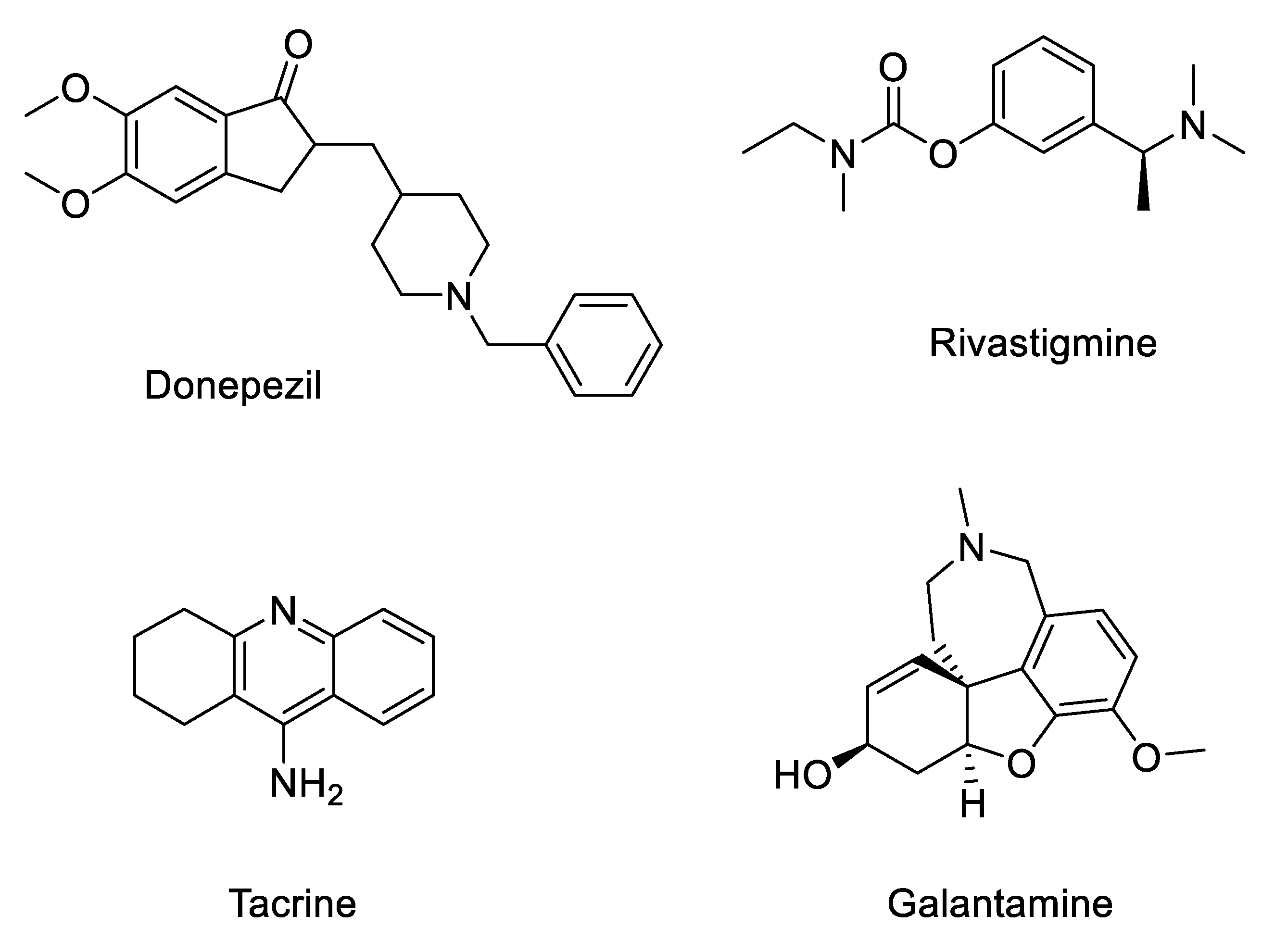
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Structural Characterization
2.2.1. NMR Data
2.2.2. X-Ray Diffraction Studies
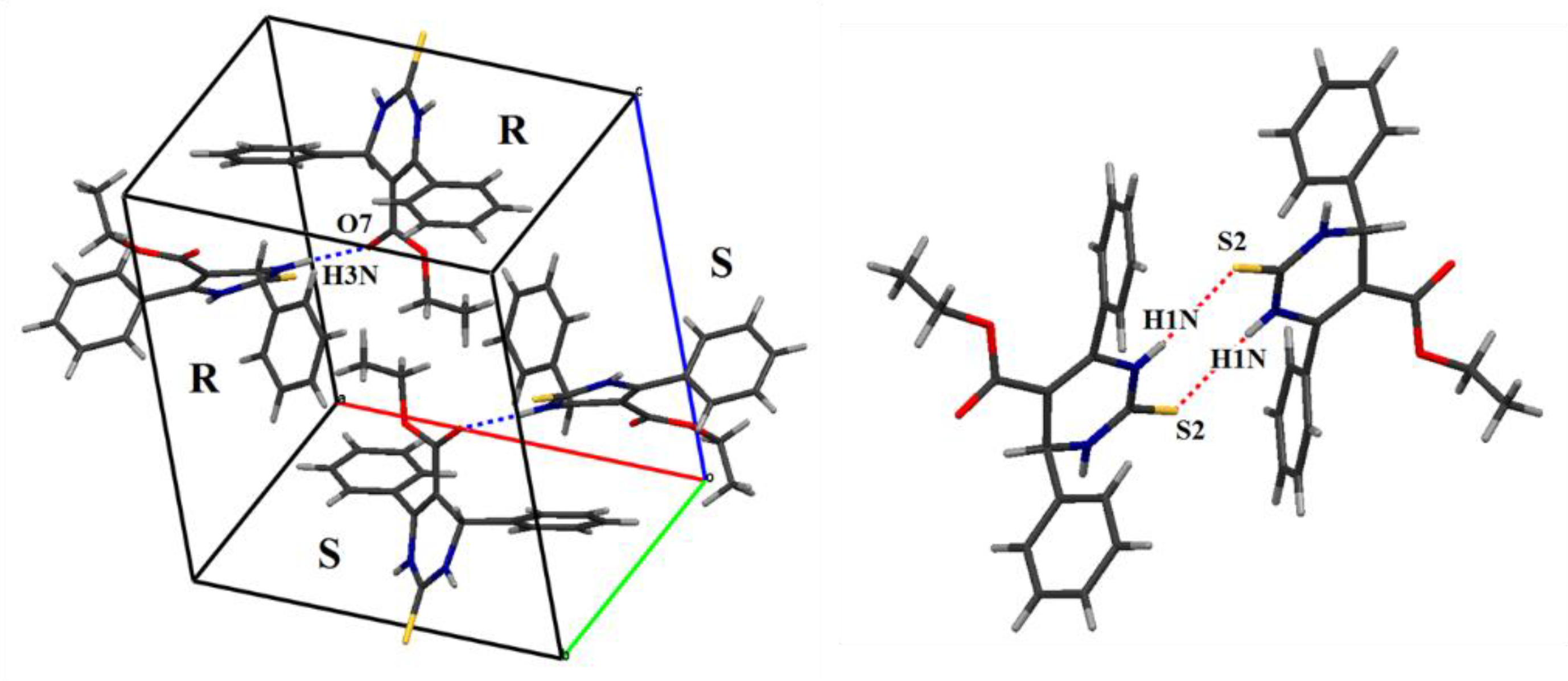
2.3. Supramolecular Interactions
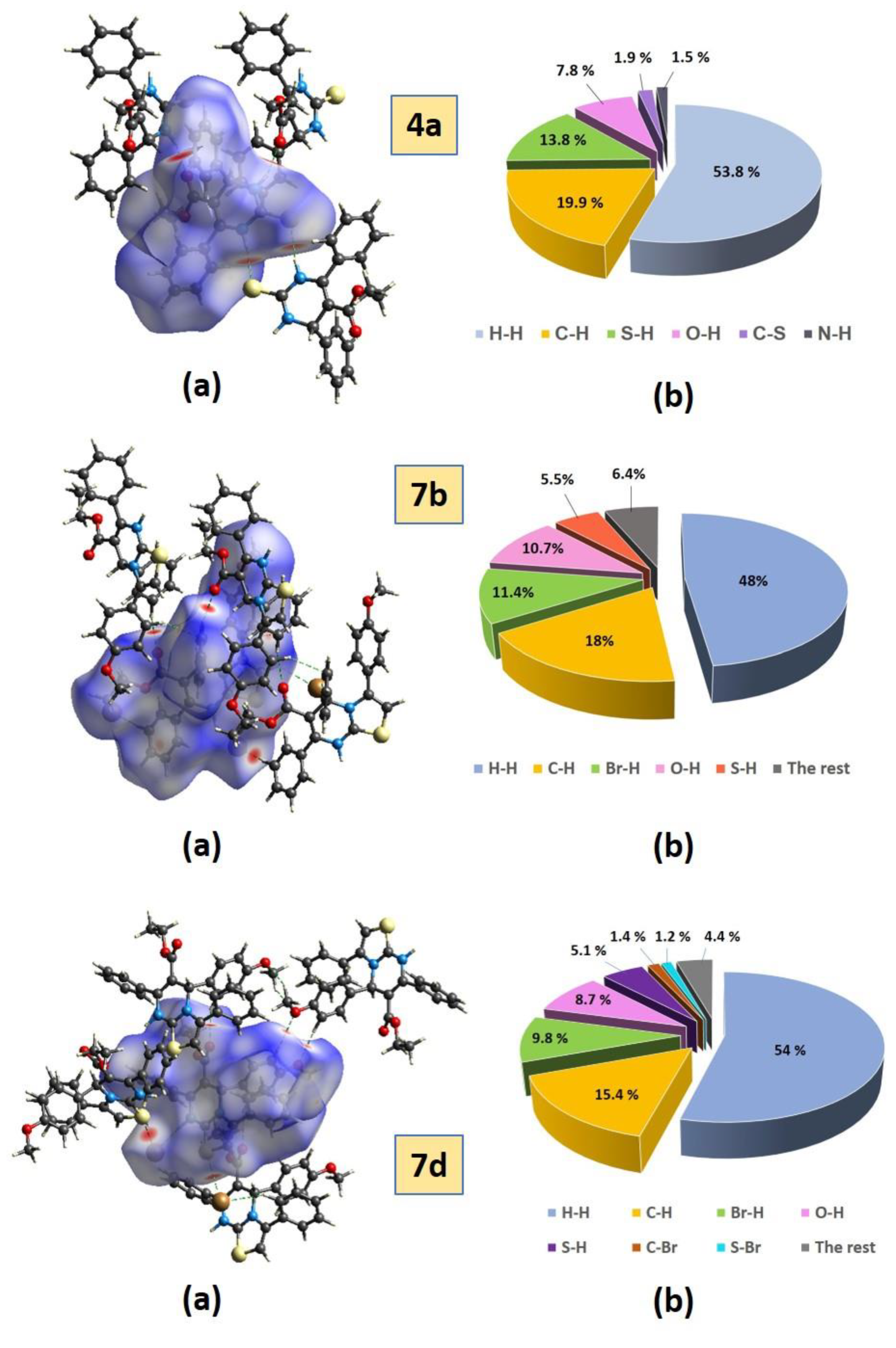
2.3.1. Hirshfeld Surface Analysis
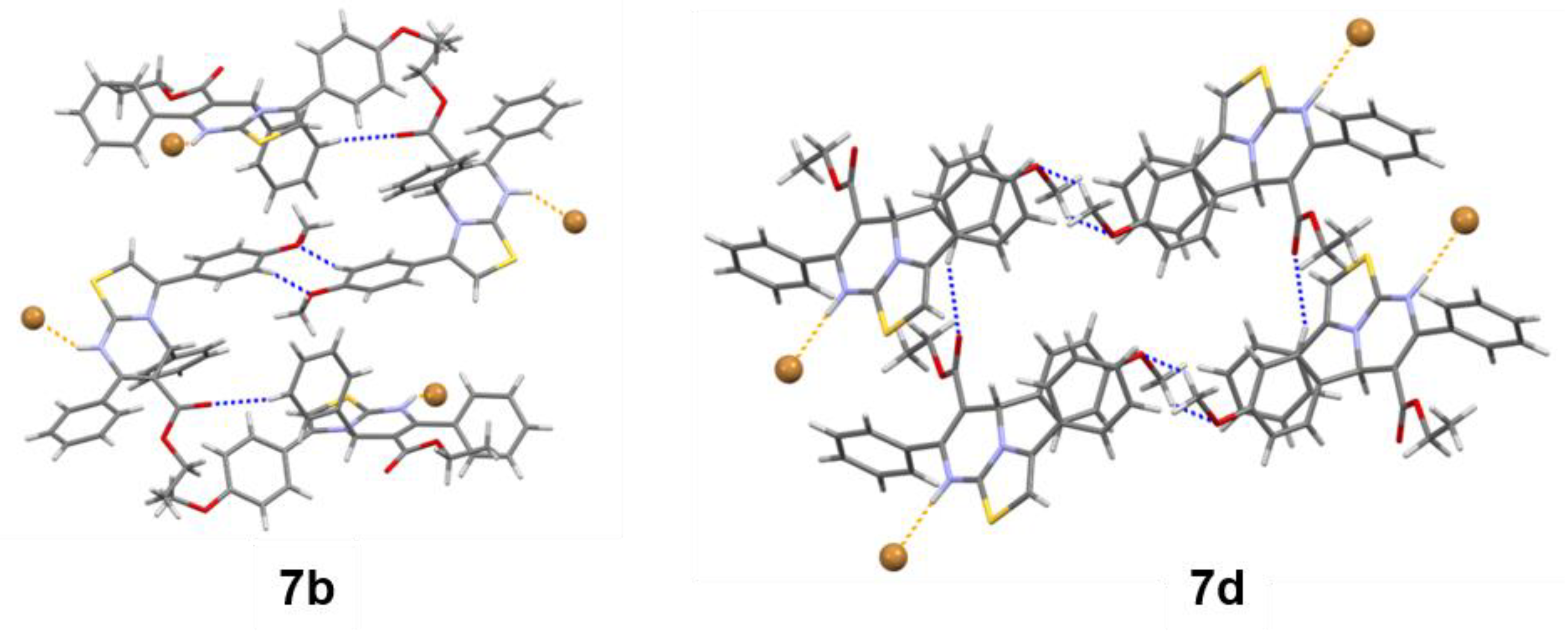
2.3.2. Crystal Packings
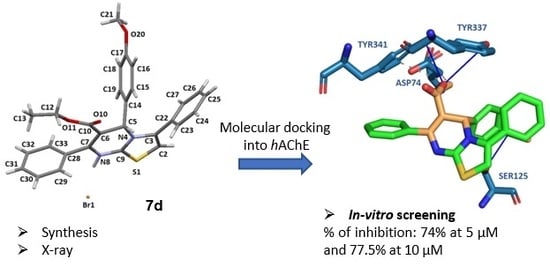
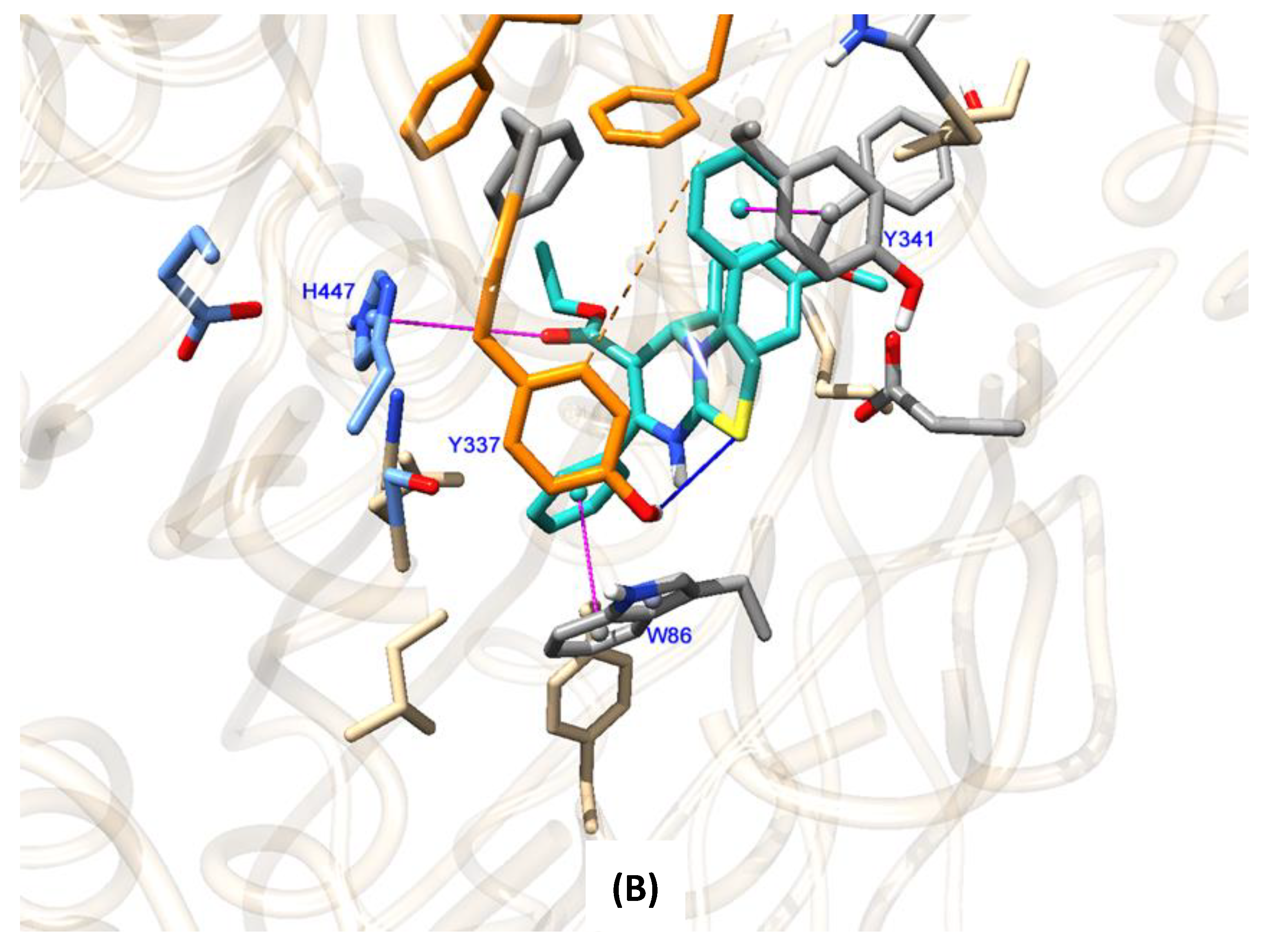
2.4. Molecular Docking Studies to Human Acetylcholinesterase
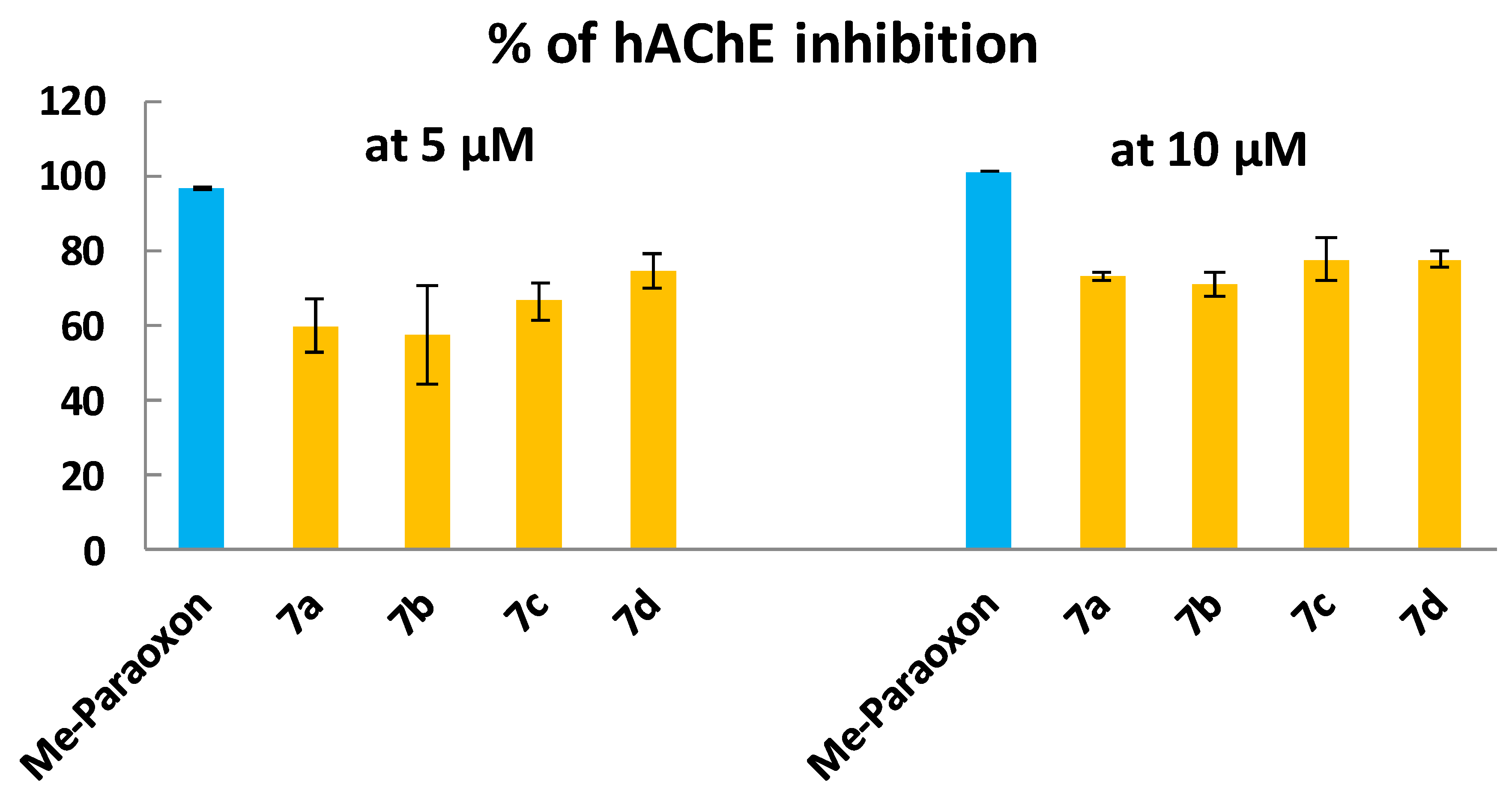
2.5. In Vitro hAChE Inhibition
3. Materials and Methods
3.1. X-Ray Crystallographic Analysis
3.2. General Procedure for the Preparation of DHPMs 4a,b
3.2.1. Ethyl 4,6-diphenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a)
3.2.2. Ethyl 4-(4-methoxyphenyl)-6-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4b)
3.2.3. Synthesis of the Thiazolo[3,2-a]pyrimidine Bromides 7a–d and the Neutralized Compound 9
3.2.4. 6-(Ethoxycarbonyl)-3,5,7-triphenyl-5H-thiazolo[3,2-a]pyrimidin-8-ium bromide (7a)
3.2.5. 6-(Ethoxycarbonyl)-3-(4-methoxyphenyl)-5,7-diphenyl-5H-thiazolo[3,2-a]pyrimidin-8-ium bromide (7b)
3.2.6. 6-(Ethoxycarbonyl)-3-(4-fluorophenyl)-5,7-diphenyl-5H-thiazolo[3,2-a]pyrimidin-8-ium bromide (7c)
3.2.7. 6-(Ethoxycarbonyl)-5-(4-methoxyphenyl)-3,7-diphenyl-5H-thiazolo[3,2-a]pyrimidin-8-ium bromide (7d)
3.2.8. Ethyl 3-(4-methoxyphenyl)-5,7-diphenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate (9)
3.3. Molecular Docking Studies
3.4. Enzyme Inhibition Activity
3.4.1. Enzyme and Assay Reagents
3.4.2. In Vitro Acetylcholinesterase Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Alzheimer Report 2018. Available online: https://www.alz.co.uk/research/world-report (accessed on 10 March 2019).
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Martinez, A. Peripheral and dual binding site acetylcholinesterase inhibitors: Implications in treatment of Alzheimer’s disease. Mini Rev. Med. Chem. 2001, 1, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Gallego-Sandín, S.; Villarroya, M.; García, A.G.; López, M.G. Unequal neuroprotection afforded by the acetylcholinesterase inhibitors galantamine, donepezil, and rivastigmine in SH-SY5Y neuroblastoma cells: role of nicotinic receptors. J. Pharmacol. Exp. Ther. 2005, 315, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Chen, L.; Zhang, L.; Liu, S.; Cheong, C. Design, synthesis, and biological evaluation of 5H-thiazolo[3,2-a]pyrimidine derivatives as a new type of acetylcholinesterase inhibitors. Arkivoc 2008, 2008, 266–277. [Google Scholar]
- Kolb, S.; Mondésert, O.; Goddard, M.L.; Jullien, D.; Villoutreix, B.O.; Ducommun, B.; Garbay, C.; Braud, E. Development of novel thiazolopyrimidines as CDC25B phosphatase inhibitors. ChemMedChem 2009, 4, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Jun, K.Y.; Lee, E.; Kim, S.; Kwon, Y.; Kim, K.; Na, Y. Ethyl 2-(benzylidene)-7-methyl-3-oxo-2,3-dihydro-5H-thiazolo[3,2-a] pyrimidine-6-carboxylate analogues as a new scaffold for protein kinase casein kinase 2 inhibitor. Bioorganic Med. Chem. 2014, 22, 4553–4565. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Chen, C.; Liu, H.; Zheng, L.; Tong, Y.; Qu, D.; Han, S. Biological evaluation of halogenated thiazolo[3,2-a]pyrimidin-3-one carboxylic acid derivatives targeting the YycG histidine kinase. Eur. J. Med. Chem. 2014, 87, 500–507. [Google Scholar] [CrossRef]
- Geist, J.G.; Lauw, S.; Illarionova, V.; Illarionov, B.; Fischer, M.; Gräwert, T.; Rohdich, F.; Eisenreich, W.; Kaiser, J.; Groll, M.; et al. Thiazolopyrimidine inhibitors of 2-methylerythritol 2,4-cyclodiphosphate synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. ChemMedChem 2010, 5, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, C.N.; Bodade, R.G.; Dawane, B.S.; Konda, S.G.; Khandare, N.T. Synthesis and biological activity of pyrazolo[3,4–d]thiazolo[3,2-a]pyrimidin-4-one derivatives: in silico approach. J. Enzyme Inhib. Med. Chem. 2010, 25, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; El-Messery, S.M.; Abbas, A. Synthesis and anticancer activity of new thiazolo[3,2-a]pyrimidines: DNA binding and molecular modeling study. Bioorg. Chem. 2017, 74, 41–52. [Google Scholar] [CrossRef]
- Abdel Moty, S.G.; Hussein, M.A.; Abdel Aziz, S.A.; Abou-Salim, M.A. Design and synthesis of some substituted thiazolo[3,2-a]pyrimidine derivatives of potential biological activities. Saudi Pharm. J. 2016, 24, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jin, Y.; Fu, W.; Xiao, S.; Feng, C.; Fang, B.; Gu, Y.; Li, C.; Zhao, Y.; Liu, Z.; et al. Design, synthesis, and structure-activity relationship analysis of thiazolo[3,2-a]pyrimidine derivatives with anti-inflammatory activity in acute lung injury. ChemMedChem 2017, 12, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Tozkoparan, B.; Yarim, M.; Saraç, S.; Ertan, M.; Kelicen, P.; Altinok, G.; Demirdamar, R. Studies on synthesis, chromatographic resolution, and antiinflammatory activities of some 2-thioxo-1,2,3,4-tetrahydropyrimidines and their condensed derivatives. Arch. Pharm. (Weinheim) 2000, 333, 415–420. [Google Scholar] [CrossRef]
- Sawant, R.L.; Bansode, C.A.; Wadekar, J.B. In vitro anti-inflammatory potential and QSAR analysis of oxazolo/thiazolo pyrimidine derivatives. Med. Chem. Res. 2013, 22, 1884–1892. [Google Scholar] [CrossRef]
- Pan, B.; Huang, R.; Zheng, L.; Chen, C.; Han, S.; Qu, D.; Zhu, M.; Wei, P. Thiazolidione derivatives as novel antibiofilm agents: Design, synthesis, biological evaluation, and structure-activity relationships. Eur. J. Med. Chem. 2011, 46, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Banothu, J.; Khanapur, M.; Basavoju, S.; Bavantula, R.; Narra, M.; Abbagani, S. Synthesis, characterization and biological evaluation of fused thiazolo[3,2-a]pyrimidine derivatives. RSC Adv. 2014, 4, 22866. [Google Scholar] [CrossRef]
- Gali, R.; Banothu, J.; Porika, M.; Velpula, R.; Hnamte, S.; Bavantula, R.; Abbagani, S.; Busi, S. Indolylmethylene benzo[h]thiazolo[2–b]quinazolinones: Synthesis, characterization and evaluation of anticancer and antimicrobial activities. Bioorg. Med. Chem. Lett. 2014, 24, 4239–4242. [Google Scholar] [CrossRef]
- Abu-Hashem, A.A.; Youssef, M.M.; Hussein, H.A.R. Synthesis, antioxidant, antituomer activities of some new thiazolopyrimidines, pyrrolothiazolopyrimidines and triazolopyrrolothiazolopyrimidines derivatives. J. Chinese Chem. Soc. 2011, 58, 41–48. [Google Scholar] [CrossRef]
- Ergan, E.; Akbas, E.; Levent, A.; Sahin, E.; Konus, M.; Seferoglu, N. Synthesis, theoretical calculation, electrochemistry and total antioxidant capacity of 5-benzoyl-6-phenyl-4-(4-methoxyphenyl)-1,2,3,4-tetrahydro-2-thioxopyrimidine and derivatives. J. Mol. Struct. 2017, 1136, 231–243. [Google Scholar] [CrossRef]
- Mohamed, S.F.; Flefel, E.M.; Amr, A.E.G.E.; Abd El-Shafy, D.N. Anti-HSV-1 activity and mechanism of action of some new synthesized substituted pyrimidine, thiopyrimidine and thiazolopyrimidine derivatives. Eur. J. Med. Chem. 2010, 45, 1494–1501. [Google Scholar] [CrossRef]
- Ravendra Babu, K.; Koteswara Rao, V.; Nanda Kumar, Y.; Polireddy, K.; Venkata Subbaiah, K.; Bhaskar, M.; Lokanatha, V.; Naga Raju, C. Identification of substituted [3, 2-a] pyrimidines as selective antiviral agents: Molecular modeling study. Antiviral Res. 2012, 95, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Amr, A.E.-G.E.; Maigali, S.S.; Abdulla, M.M. Synthesis, and analgesic and antiparkinsonian activities of thiopyrimidine, pyrane, pyrazoline, and thiazolopyrimidine derivatives from 2-chloro-6-ethoxy-4-acetylpyridine. Monatshefte für Chemie-Chem. Mon. 2008, 139, 1409–1415. [Google Scholar] [CrossRef]
- Batool, I.; Saeed, A.; Qureshi, I.Z.; Kalsoom, S.; Razzaq, A. Synthesis, molecular docking and biological evaluation of new thiazolopyrimidine carboxylates as potential antidiabetic and antibacterial agents. Res. Chem. Intermed. 2016, 42, 1139–1163. [Google Scholar] [CrossRef]
- Jeanneau-Nicolle, E.; Benoit-Guyod, M.; Namil, A.; Leclerc, G. New thiazolo[3,2-a]pyrimidine derivatives, synthesis and structure-activity relationships. Eur. J. Med. Chem. 1992, 27, 115–120. [Google Scholar] [CrossRef]
- Fatima, S.; Sharma, A.; Saxena, R.; Tripathi, R.; Shukla, S.K.; Pandey, S.K.; Tripathi, R.; Tripathi, R.P. One pot efficient diversity oriented synthesis of polyfunctional styryl thiazolopyrimidines and their bio-evaluation as antimalarial and anti-HIV agents. Eur. J. Med. Chem. 2012, 55, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Ruiu, S.; Marchese, G.; Saba, P.L.; Gessa, G.L.; Pani, L. The 5-HT2 antagonist ritanserin blocks dopamine re-uptake in the rat frontal cortex. Mol. Psychiatry 2000, 5, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.H.; Kapur, S.; Houle, S.; DaSilva, J.; Owczarek, B.; Brown, G.M.; Wilson, A.A.; Kennedy, S.H. Prefrontal Cortex 5-HT2 Receptors in Depression: An [18F]Setoperone PET Imaging Study. Am. J. Psychiatry 1999, 156, 1029–1034. [Google Scholar] [CrossRef]
- Serrar, H.; Galai, M.; Benhiba, F.; Ouakki, M.; Benzekri, Z.; Boukhris, S.; Hassikou, A.; Souizi, A.; Oudda, H.; Ebn Touhami, M. Two derivatives of 7-amino-thiazolo[3,2-a]pyrimidine as inhibitors of mild steel corrosion in 1.0M HCl solution: Part I: Synthesis of inhibitors and electrochemical study. J. Chem. Technol. Metall. 2018, 53, 324–335. [Google Scholar]
- Lashmanova, E.A.; Kirdyashkina, A.I.; Slepukhin, P.A.; Shiryaev, A.K. Oxidation of thiazolo[3,2-a]pyrimidin-3(2H)-ones with DMSO and Lawesson’s reagent. Tetrahedron Lett. 2018, 59, 1099–1103. [Google Scholar] [CrossRef]
- Elmaghraby, A.M.; Mousa, I.A.; Harb, A.A.; Mahgoub, M.Y. Three Component Reaction: An Efficient Synthesis and Reactions of 3,4-Dihydropyrimidin-2(1H)-Ones and Thiones Using New Natural Catalyst. ISRN Org Chem. 2013. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalt. Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- Kitaigorodskii, A.I. Organic Chemical Crystallography; Consultants Bureau: New York, NY, USA, 1961. [Google Scholar]
- Wolff, D.J.; Grimwood, J.J.; McKinnon, M.J.; Turner, D.; Jayatilaka, M.A.S. Crystal Explorer 3.1; University of Western Australia: Perth, Australia, 2012. [Google Scholar]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Bourne, Y.; Taylor, P.; Radić, Z.; Marchot, P. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 2003, 22, 1–12. [Google Scholar] [CrossRef]
- Colletier, J.; Fournier, D.; Greenblatt, H.M.; Stojan, J.; Sussman, J.L.; Zaccai, G.; Silman, I.; Weik, M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006, 25, 2746–2756. [Google Scholar] [CrossRef]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ogura, H.; Kosasa, T.; Kuriya, Y.; Yamanishi, Y. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find. Exp. Clin. Pharmacol. 2000, 22, 609–613. [Google Scholar] [CrossRef]
- Armarego WLF, C.C. Purification of Laboratory Chemicals, 7th ed.; Elsevier Inc.: Oxford, UK, 2013. [Google Scholar]
- SMART and SAINT. Control and Integration Software; Bruker AXS: Madison, WI, USA, 2004. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. IUCr SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2014, C71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Sherif, S.M.; Youssef, M.M.; Mobarak, K.M. A Convenient Synthesis of Thiazolopyrimidines, Thiazolodipyrimidines and Heterocyclo thiazolopyrimidines. Tetrahedron 1993, 49, 9561–9572. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.M.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4a–b and 7a–d are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | 4a | 4b | 7a | 7b | 7c | 7d | 9 |
|---|---|---|---|---|---|---|---|
| 1H-NMR | 5.27 | 5.22 | 6.44 | 6.41 | 6.36 | 6.73 | 6.27 |
| 13C-NMR | 54.08 | 53.52 | 59.87 | 59.63 | 59.93 | 59.41 | 58.36 |
| Comp. |  |  |
|---|---|---|
| C2 | 113.00 | 103.04 |
| C5 | 59.63 | 58.36 |
| C6 | 103.10 | 100.57 |
| C7 | 144.24 | 156.26 |
| C9 | 160.70 | 166.53 |
| C=O | 164.74 | 167.04 |
| Bond | Bond Length [Å] | Angle | Angle [°] |
| N(1)-C(2) | 1.360(3) | C(2)-N(1)-C(6) | 123.1(2) |
| N(1)-C(6) | 1.387(3) | N(3)-C(2)-N(1) | 116.4(2) |
| S(2)-C(2) | 1.680(3) | N(3)-C(2)-S(2) | 121.9(2) |
| N(3)-C(2) | 1.327(3) | N(1)-C(2)-S(2) | 121.70(18) |
| N(3)-C(4) | 1.469(3) | C(2)-N(3)-C(4) | 126.8(2) |
| C(4)-C(5) | 1.508(3) | N(3)-C(4)-C(5) | 108.95(18) |
| C(5)-C(6) | 1.353(3) | N(3)-C(4)-C(20) | 110.4(2) |
| Torsion Angle (R-molecule) | Angle [°] | C(5)-C(4)-C(20) | 113.3(2) |
| O(7)-C(7)-C(5)-C(4) | −11.5(4) | C(6)-C(5)-C(7) | 125.1(2) |
| C(6)-C(5)-C(4) | 120.6(2) | ||
| C(7)-C(5)-C(4) | 114.2(2) | ||
| C(5)-C(6)-C(30) | 126.6(2) | ||
| N(1)-C(6)-C(30) | 113.9(2) | ||
| C(5)-C(6)-N(1) | 119.5(2) | ||
| Angles between Planes | Angles [°] | ||
| N1-C2-N3- C4-C5-C6 | C20-…-C25 | 85.68(13) | |
| N1-C2-N3- C4-C5-C6 | C30-…-C35 | 66.15(17) | |
| Molecule | 7b | 7d |
|---|---|---|
| Bond Length [Å] | ||
| S(1)-C(2) | 1.731(4) | 1.717(4) |
| S(1)-C(9) | 1.712(3) | 1.706(4) |
| C(3)-C(2) | 1.335(5) | 1.339(6) |
| N(4)-C(3) | 1.425(4) | 1.410(5) |
| N(4)-C(5) | 1.503(4) | 1.488(5) |
| C(5)-C(6) | 1.520(5) | 1.514(5) |
| C(6)-C(7) | 1.339(5) | 1.340(5) |
| N(8)-C(7) | 1.417(4) | 1.394(5) |
| N(8)-C(9) | 1.340(4) | 1.335(5) |
| N(4)-C(9) | 1.337(4) | 1.332(4) |
| Angles [°] | ||
| N(4)-C(3)-C(ring) | 122.3(3) | 122.7(4) |
| C(2)-C(3)-C(ring) | 126.8(3) | 126.2(4) |
| C(3)-N(4)-C(5) | 123.5(3) | 123.9(3) |
| C(9)-N(4)-C(3) | 113.2(3) | 112.6(3) |
| C(9)-N(4)-C(5) | 121.7(3) | 123.5(3) |
| N(4)-C(5)-C(6) | 108.7(3) | 109.3(3) |
| N(4)-C(5)-C(ring) | 108.7(3) | 111.2(3) |
| C(6)-C(5)-C(ring) | 114.0(3) | 110.6(3) |
| C(7)-C(6)-C(10) | 126.0(3) | 122.2(3) |
| C(7)-C(6)-C(5) | 123.4(3) | 124.0(3) |
| C(10)-C(6)-C(5) | 110.6(3) | 113.7(3) |
| C(6)-C(7)-N(8) | 119.1(3) | 119.6(3) |
| C(6)-C(7)-C(ring) | 129.7(3) | 127.1(3) |
| N(8)-C(7)-C(ring) | 111.1(3) | 113.3(3) |
| C(9)-N(8)-C(7) | 120.5(3) | 120.8(3) |
| N(8)-C(9)-N(4) | 122.3(3) | 122.7(3) |
| N(8)-C(9)-S(1) | 125.0(3) | 123.9(3) |
| N(4)-C(9)-S(1) | 112.7(3) | 113.4(3) |
| Torsion Angles [°] | ||
| O(10)-C(10)-C(6)-C(5) | 21.9(6) | −32.7(6) |
| Angles between planes [°] | ||
| Center ring a–C5 phenyl ring b | 89.49(12) | 83.33(11) |
| Center ring a–C7 phenyl ring c | 127.57(12) | 74.09(14) |
| Center ring a–C3 phenyl ring d | 55.68(12) | 56.66(12) |
| D-H…A | H…A [Å] | D…A [Å] | D-H…A [°] | Symmetry Operation |
|---|---|---|---|---|
| Compound 4a | ||||
| N(3)-H(3N)...O(7) | 2.02 | 2.872(2) | 159 | ½ − x,1/2 + y,1/2 − z |
| N(1)-H(1N)...S(2) | 2.52 | 3.337(2) | 173 | −x,1 − y,1 − z |
| Compound 7b | ||||
| N8-H8...Br1a | 2.48 | 3.199(3) | 141 | x,y,z |
| C32-H32...Br1b | 2.81 | 3.742(4) | 175 | x, −1 + y, z |
| C18-H18...O10 | 2.49 | 3.397(6) | 166 | 1 − x, 1/2 + y, 1/2 − z |
| C24-H24...O26 | 2.60 | 3.507(6) | 166 | 1 − x, 1 − y, 1 − z |
| Compound 7d | ||||
| N8-H8...Br1a | 2.28 | 3.167(3) | 165 | x, y, z |
| C19-H19...O10 | 2.51 | 3.372(5) | 153 | x, −1 + y, z |
| C21-H21C...O20 | 2.66 | 3.456(6) | 140 | 2 − x, −y, 1 − z |
| Compound | 4a | 7b | 7d |
|---|---|---|---|
| Absorption coefficient (mm−1) | 0.194 | 1.694 | 1.680 |
| F(000) | 712 | 1128 | 1128 |
| Crystal size (mm) | 0.30 × 0.15 × 0.15 | 0.200 × 0.100 × 0.040 | 0.150 × 0.060 × 0.040 |
| θ range (°) | 2.776 to 26.497 | 2.808 to 26.358 | 2.873 to 26.440 |
| Index ranges | –16 ≤ h ≤16, –10 ≤ k ≤ 10, −20 ≤ l ≤ 20 | −18 ≤ h ≤18, −10 ≤ k ≤10, −19 ≤ l ≤ 26 | −16 ≤ h ≤ 16, −8 ≤ k ≤ 8, −35 ≤ l ≤ 35 |
| Reflections collected | 49,517 | 14,223 | 99,174 |
| Independent reflections | 3618 [R(int) = 0.0299] | 5233 [R(int) = 0.0695] | 5395 [R(int) = 0.2244] |
| Completeness to Θ max (%) | 98.4 | 99.6 | 99.9 |
| Data/restraints/parameters | 3618/1/219 | 5233/0/316 | 5395/1/320 |
| Goodness-of-fit on F2 | 1.051 | 0.955 | 1.054 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0711. wR2 = 0.1922 | R1 = 0.0491, wR2 = 0.1000 | R1 = 0.0491, wR2 = 0.0873 |
| R indices (all data) | R1 = 0.0842, wR2 = 0.2063 | R1 = 0.1095, wR2 = 0.1210 | R1 = 0.1299, wR2 = 0.1163 |
| Extinction coefficient | n/a | n/a | n/a |
| Largest diff. peak/hole (e. Å−3) | 0.872/−0.992 | 0.300 and −0.501 | 0.610 and −0.503 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahgoub, M.Y.; Elmaghraby, A.M.; Harb, A.-E.A.; Ferreira da Silva, J.L.; Justino, G.C.; Marques, M.M. Synthesis, Crystal Structure, and Biological Evaluation of Fused Thiazolo[3,2-a]Pyrimidines as New Acetylcholinesterase Inhibitors. Molecules 2019, 24, 2306. https://doi.org/10.3390/molecules24122306
Mahgoub MY, Elmaghraby AM, Harb A-EA, Ferreira da Silva JL, Justino GC, Marques MM. Synthesis, Crystal Structure, and Biological Evaluation of Fused Thiazolo[3,2-a]Pyrimidines as New Acetylcholinesterase Inhibitors. Molecules. 2019; 24(12):2306. https://doi.org/10.3390/molecules24122306
Chicago/Turabian StyleMahgoub, Mohamed Y., Awatef M. Elmaghraby, Abd-Elfttah A. Harb, João L. Ferreira da Silva, Gonçalo C. Justino, and M. Matilde Marques. 2019. "Synthesis, Crystal Structure, and Biological Evaluation of Fused Thiazolo[3,2-a]Pyrimidines as New Acetylcholinesterase Inhibitors" Molecules 24, no. 12: 2306. https://doi.org/10.3390/molecules24122306