Solid State and Solution Study on the Formation of Inorganic Anion Complexes with a Series of Tetrazine-Based Ligands

 ,
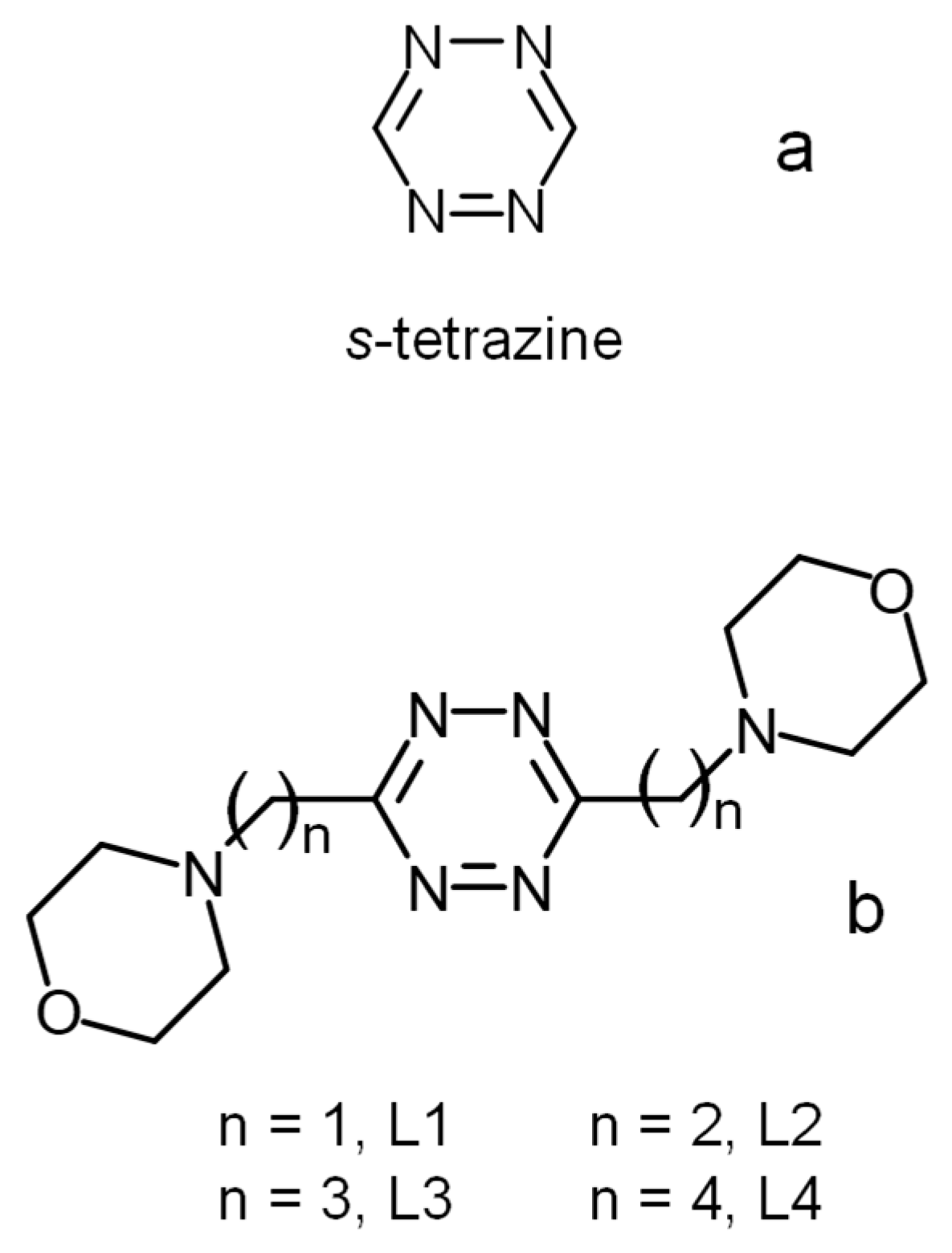
,  ,
,  , , and
, , and
Abstract
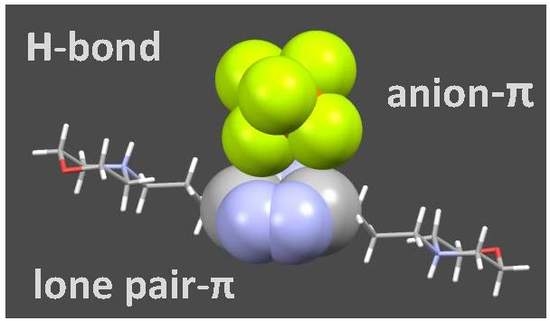
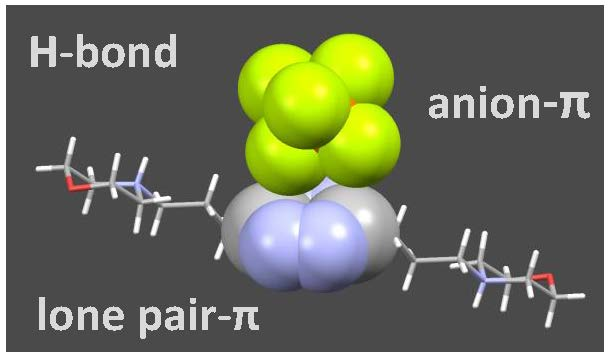
:
1. Introduction
2. Results and Discussion
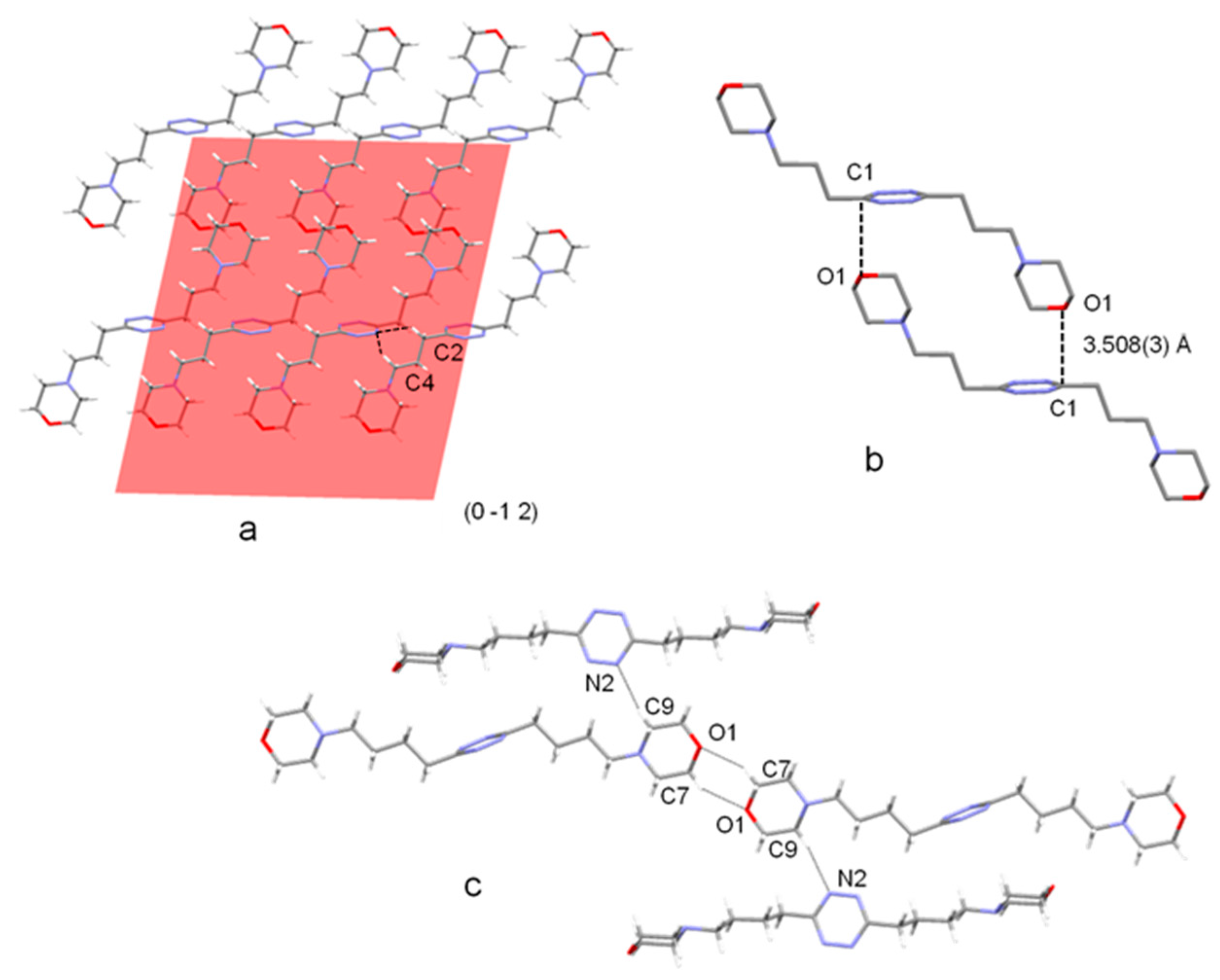
2.1. Crystal Structure of L3 and L4 Free Ligands
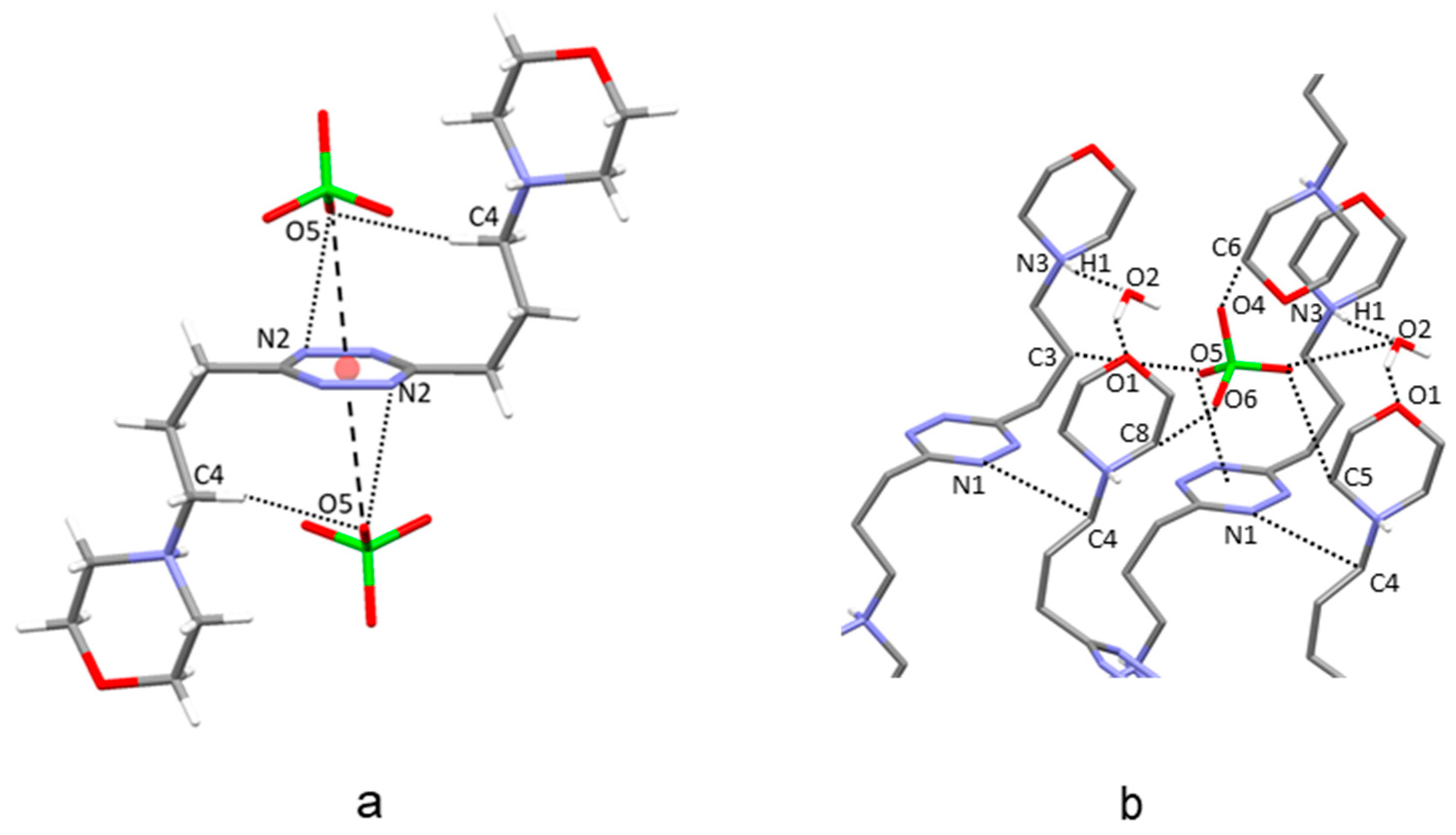
2.2. Crystal Structure of H2L3(ClO4)2∙2H2O
2.3. Crystal Structure of H2L4(ClO4)2∙2H2O
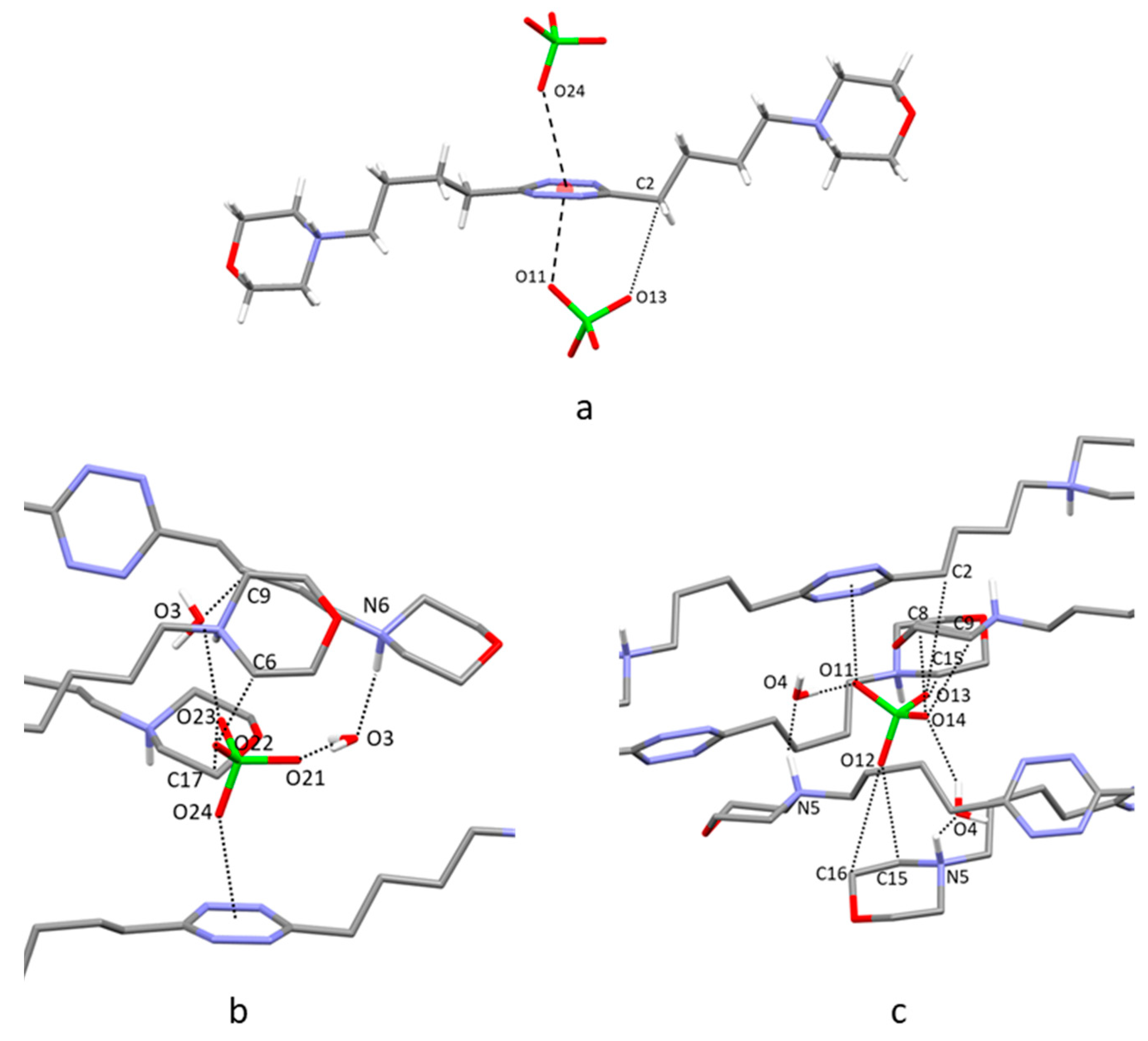
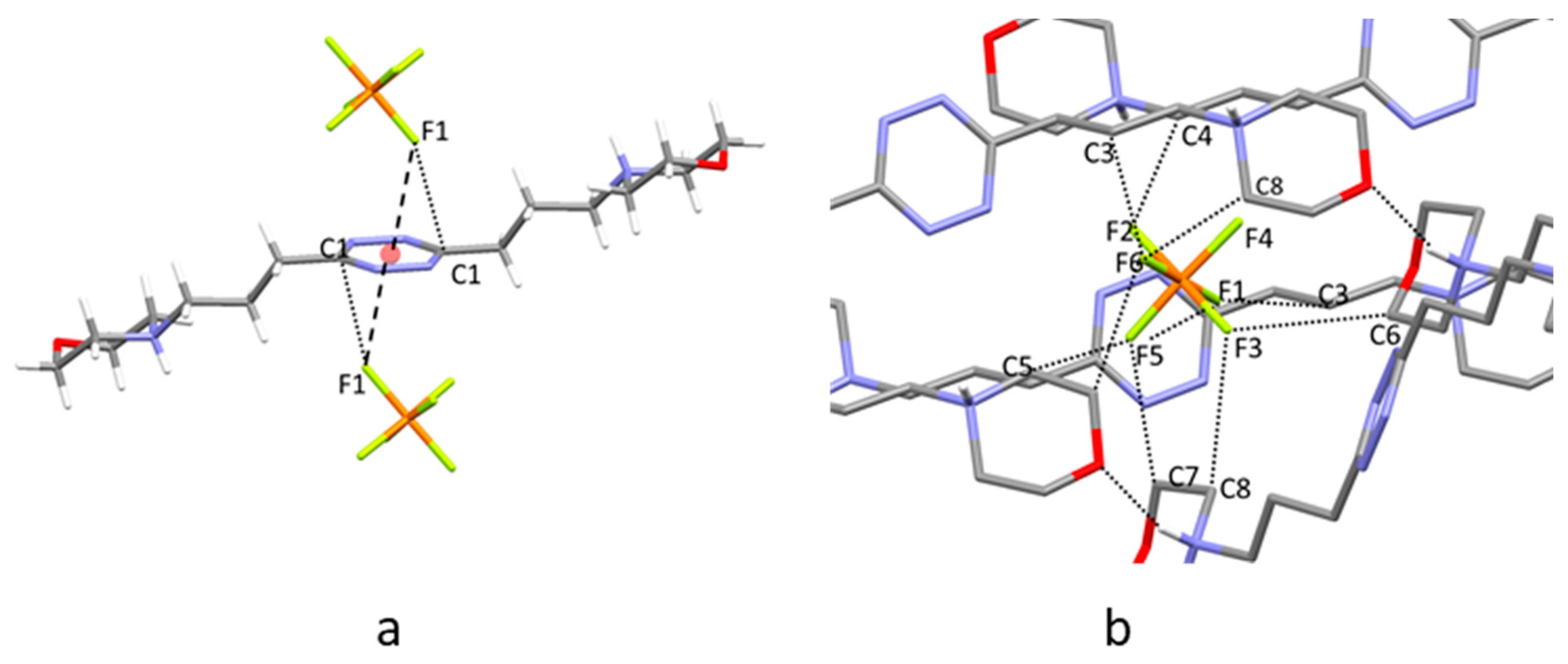
2.4. Crystal Structure of H2L3(PF6)2
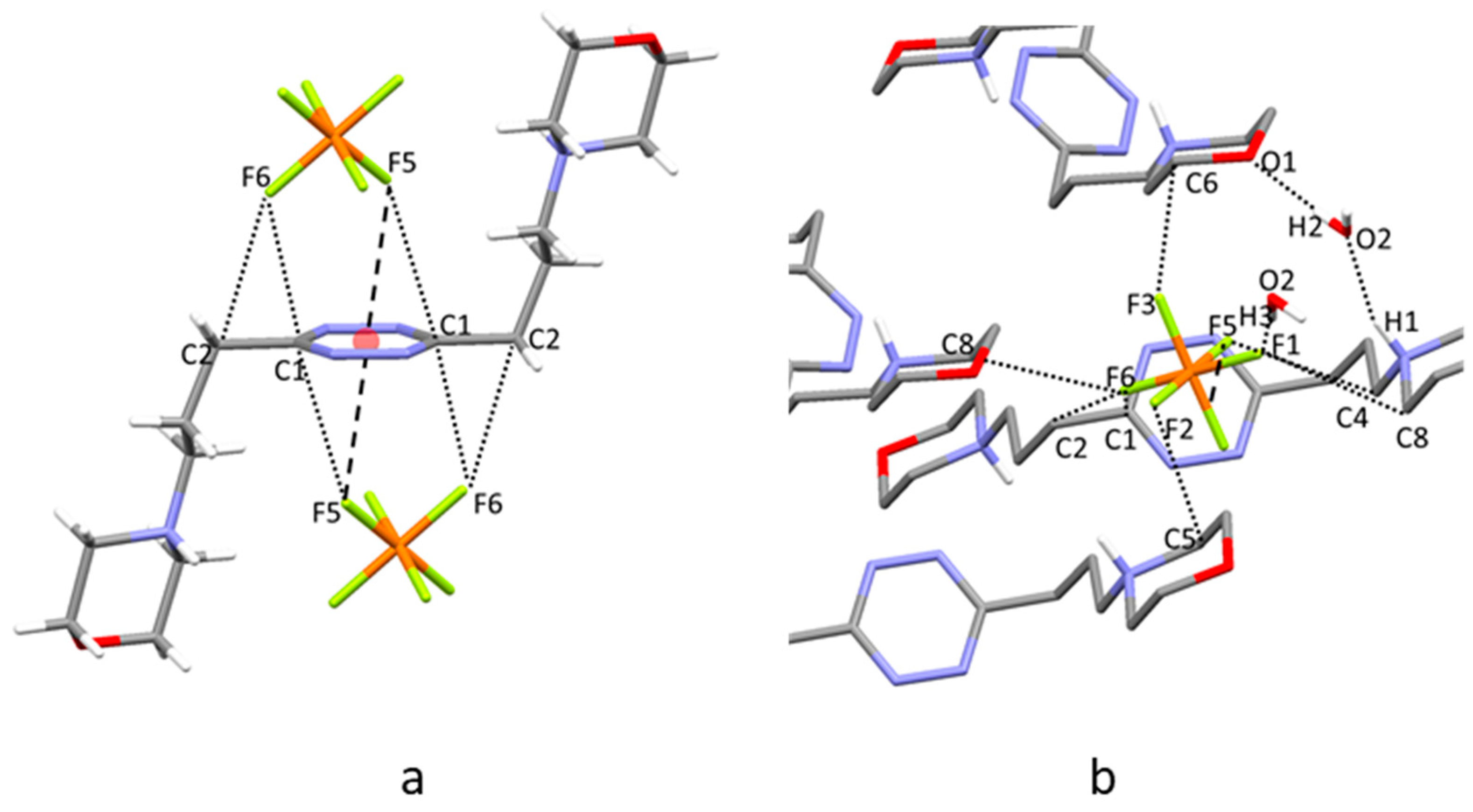
2.5. Crystal Structure of H2L3(PF6)2∙2H2O
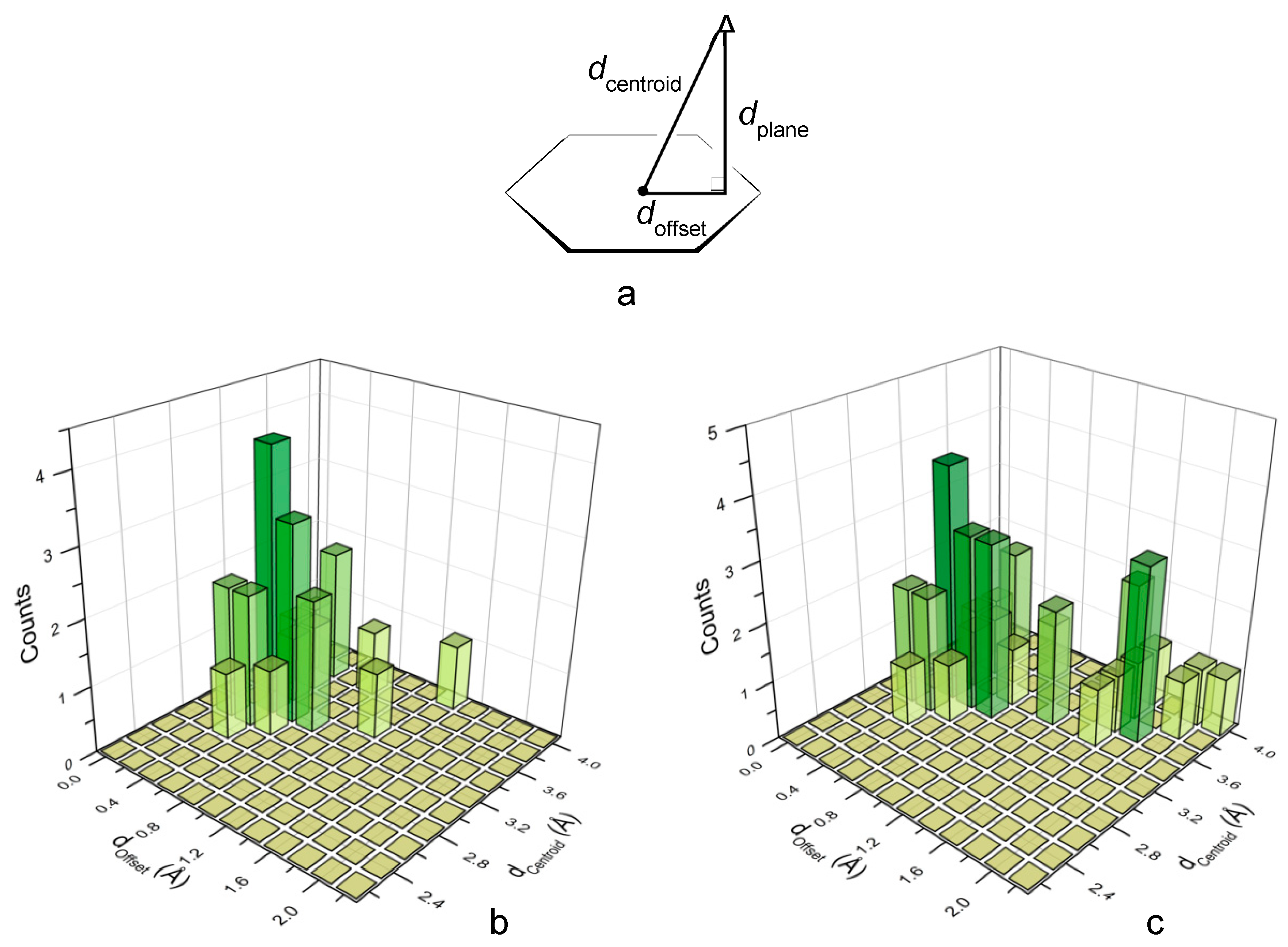
2.6. Analysis of the Crystal Structures of Anion Complexes
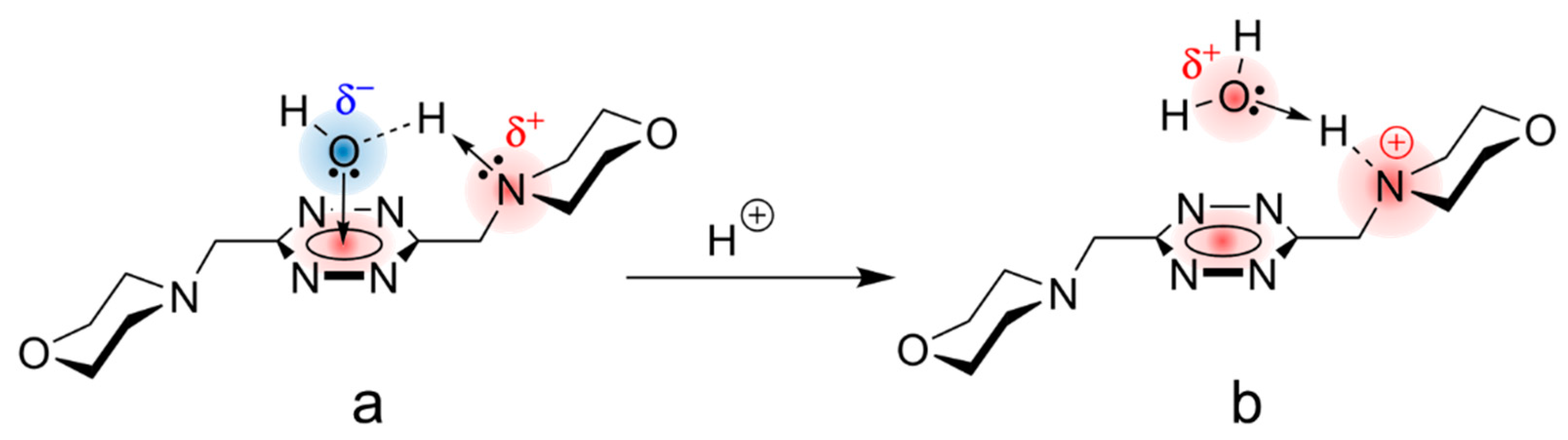
2.7. Ligand Protonation in Solution
2.8. Anion Binding in Solution
3. Materials and Methods
3.1. Materials
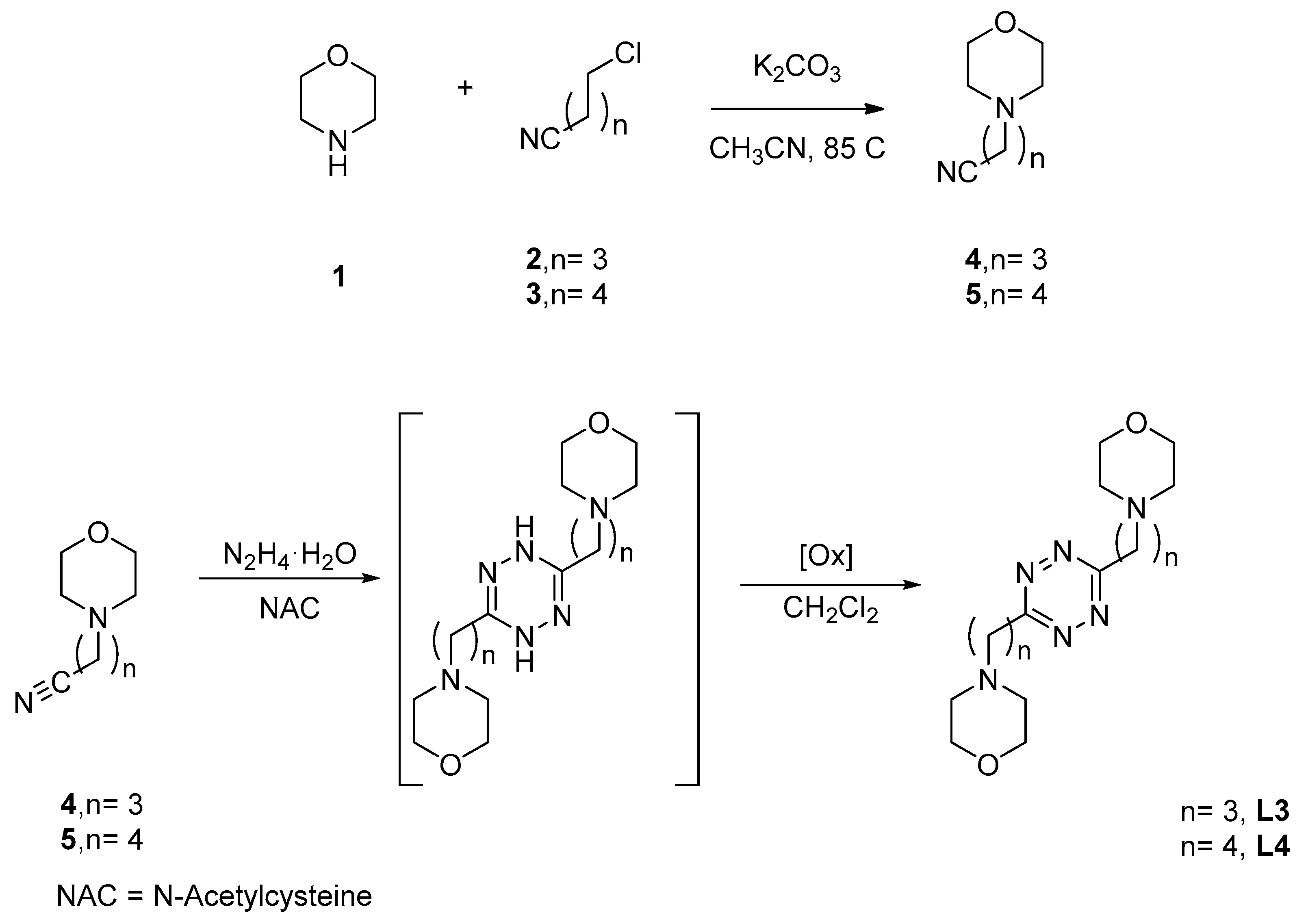
3.2. Preparation of ω-(morpholin-4-yl)nitrile Precursors
3.3. Preparation of 3,6-di(morpholin-4-y)alkyl-s-tetrazines, L3 and L4
3.4. Potentiometric Measurements
3.5. Isothermal Titration Calorimetry
3.6. X-ray Structure Analyses
3.7. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clavier, G.; Audebert, P. s-Tetrazines as Building Blocks for New Functional Molecules and Molecular Materials. Chem. Rev. 2010, 110, 3299–3314. [Google Scholar] [CrossRef] [PubMed]
- Garau, C.; Quiñonero, D.; Frontera, A.; Costa, A.; Ballester, P.; Deyà, P.M.; Costa, A.; Ballester, P. s-Tetrazine as a new binding unit in molecular recognition of anions. Chem. Phys. Lett. 2003, 370, 7–13. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Badria, Z.; Marek, R. Multi-center covalency: Revisiting the nature of anion–π interactions. Phys. Chem. Chem. Phys. 2015, 17, 30670–30679. [Google Scholar] [CrossRef] [PubMed]
- Badri, Z.; Foroutan-Nejad, C.; Kozelkabc, J.; Marek, R. On the non-classical contribution in lone-pair–π interaction: IQA perspective. Phys. Chem. Chem. Phys. 2015, 17, 26183–26190. [Google Scholar] [CrossRef] [PubMed]
- Marek, R.; Novák, M.; Foroutan-Nejad, C. Modulating Electron Sharing in Ion-π-Receptors via Substitution and External Electric Field: A Route toward Bond Strengthening. J. Chem. Theory Comput. 2016, 12, 3788–3795. [Google Scholar] [CrossRef]
- Campos-Fernández, C.S.; Clerac, R.; Koomen, J.M.; Russell, D.H.; Dunbar, K.R. Fine-Tuning the Ring-Size of Metallacyclophanes: A Rational Approach to Molecular Pentagons. J. Am. Chem. Soc. 2001, 123, 773–774. [Google Scholar] [CrossRef] [PubMed]
- Campos-Fernández, C.S.; Schottel, B.L.; Chifotides, H.T.; Bera, J.K.; Bacsa, J.; Koomen, J.M.; Russell, D.H.; Dunbar, K.R. Anion Template Effect on the Self-Assembly and Interconversion of Metallacyclophanes. J. Am. Chem. Soc. 2005, 127, 12909–12923. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; López de la Torre, M.D.; Pichierri, F.; Bianchi, A.; Melguizo, M. Anion Complexes with Tetrazine-Based Ligands: Formation of Strong Anion−π Interactions in Solution and in the Solid State. Inorg. Chem. 2016, 55, 8013–8024. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; Giorgi, C.; López de la Torre, M.D.; Pichierri, F.; Bianchi, A.; Melguizo, M. Halide and hydroxide anion binding in water. Dalton Trans. 2018, 47, 3329–3338. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; López de la Torre, M.D.; Mariani, P.; Pichierri, F.; Bianchi, A.; Melguizo, M. Iodide and triiodide anion complexes involving anion–π interactions with a tetrazine-based receptor. Dalton Trans. 2017, 46, 4518–4529. [Google Scholar] [CrossRef]
- Savastano, M.; García-Gallarín, C.; López de la Torre, M.D.; Pichierri, F.; Bazzicalupi, C.; Bianchi, A.; Melguizo, M. Interplay between salt bridge, hydrogen bond and anion-π interactions in thiocyanate binding. Inorg. Chim. Acta 2018, 470, 133–138. [Google Scholar] [CrossRef]
- Liu, H.-B.; Zhang, Q.; Wang, M.-X. Synthesis, Structure, and Anion Binding Properties of Electron-Deficient Tetrahomocorona[4]arenes: Shape Selectivity in Anion-π Interactions. Angew. Chem. Int. Ed. 2018, 57, 6536–6540. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Bazzicalupi, C.; Mariani, P.; Bianchi, A. Network Formation via Anion Coordination: Crystal Structures Based on the Interplay of Non-Covalent Interactions. Molecules 2018, 23, 572. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-Y.; Wang, D.-X.; Wang, M.-X. Synthesis, Structure, and Properties of Corona[6]arenes and Their Assembly with Anions in the Crystalline State. J. Org. Chem. 2018, 83, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Arranz-Mascarós, P.; Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Godino-Salido, M.L.; Gutierrez-Valero, M.D.; Lopez-Garzón, R.; Savastano, M. Thermodynamics of Anion−π Interactions in Aqueous Solution. J. Am. Chem. Soc. 2012, 135, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R. Binding and removal of octahedral, tetrahedral, square planar and linear anions in water by means of activated carbon functionalized with a pyrimidine-based anion receptor. RSC Adv. 2014, 4, 58505–58513. [Google Scholar] [CrossRef]
- Arranz, P.; Bianchi, A.; Cuesta, R.; Giorgi, C.; Godino, M.L.; Gutierrez, M.D.; Lopez, R.; Santiago, A. Binding and Removal of Sulfate, Phosphate, Arsenate, Tetrachloromercurate, and Chromate in Aqueous Solution by Means of an Activated Carbon Functionalized with a Pyrimidine-Based Anion Receptor (HL). Crystal Structures of [H3L(HgCl4)]·H2O and [H3L(HgBr4)]·H2O Showing Anion−π Interactions. Inorg. Chem. 2010, 49, 9321–9332. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; De La Torre, M.D.L.; Bianchi, A.; Melguizo, M. Supramolecular forces and their interplay in stabilizing complexes of organic anions: Tuning binding selectivity in water. Org. Chem. Front. 2019, 6, 75–86. [Google Scholar] [CrossRef]
- Berryman, O.B.; Bryantsev, V.S.; Stay, D.P.; Johnson, D.W.; Hay, B.P. Structural Criteria for the Design of Anion Receptors: The Interaction of Halides with Electron-Deficient Arenes. J. Am. Chem. Soc. 2007, 129, 48–58. [Google Scholar] [CrossRef]
- Bencini, A.; Bianchi, A.; Garcia-España, E.; Micheloni, M.; Ramirez, J.A. Proton coordination by polyamine compounds in aqueous solution. Coord. Chem. Rev. 1999, 188, 97–156. [Google Scholar] [CrossRef]
- Odelius, M.; Kirchner, B.; Hutter, J. s-Tetrazine in Aqueous Solution: A Density Functional Study of Hydrogen Bonding and Electronic Excitations. J. Phys. Chem. A 2004, 108, 2044–2052. [Google Scholar] [CrossRef] [Green Version]
- Zaitseva, K.V.; Varfolomeev, M.A.; Solomonov, B.N. Hydrogen bonding of aliphatic and aromatic amines in aqueous solution: Thermochemistry of solvation. Russ. J. Gen. Chem. 2012, 82, 1669–1674. [Google Scholar] [CrossRef]
- Korenaga, T.; Shoji, T.; Onoue, K.; Sakai, T. Demonstration of the existence of intermolecular lone pair⋯π interaction between alcoholic oxygen and the C6F5 group in organic solvent. Chem. Commun. 2009, 31, 4678. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Im, S.Y.; Park, S.J.; Im, H.J.; Lee, S.W. Conversion of Ni Nd and Ni Tb compartment compounds into one-dimensional coordination polymers or tetranuclear dimers. Polyhedron 2016, 117, 231–243. [Google Scholar] [CrossRef]
- Mayans, J.; Escuer, A.; Font-Bardia, M. Linked Nickel Metallacrowns from a Phosphonate/2-Pyridyloximate Blend of Ligands: Structure and Magnetic Properties. Inorg. Chem. 2016, 55, 3161–3168. [Google Scholar] [CrossRef]
- Wei, S.Y.; Wang, J.L.; Zhang, C.S.; Xu, X.-T.; Zhang, X.X.; Wang, J.X.; Xing, Y.-H. d7/d8 Metal Complexes Constructed from 2,6-Bis(2-benzimidazolyl)pyridyl or 2,6-Di-(pyrazol-3-yl)pyridine Derivatives: Synthesis, Structure, Characterization, and Photocatalytic Activity. ChemPlusChem 2015, 80, 549–558. [Google Scholar] [CrossRef]
- Guan, Q.-L.; Liu, Z.; Wei, W.-J.; Xing, Y.-H.; Liu, J.; Zhang, R.; Hou, Y.-N.; Wang, X.; Bai, F.-Y. Synthesis, structure, spectroscopy of four novel supramolecular complexes and cytotoxicity study by application of multiple parallel perfused microbioreactors. New J. Chem. 2014, 38, 3258–3268. [Google Scholar] [CrossRef]
- Yang, L.; Xin, L.; Gu, W.; Liao, S.; Du, P.; Tian, J.; Zhang, Y.; Lv, R.; Wei, X.; Liu, X.; et al. Synthesis, Characterizations, Magnetism and Thermal Degradation of a 2-Fold Interpenetrated 3D Cobalt-Organic Framework. Chin. J. Chem. 2014, 32, 227–232. [Google Scholar] [CrossRef]
- Alexandropoulos, D.I.; Manos, M.J.; Papatriantafyllopoulou, C.; Mukherjee, S.; Tasiopoulos, A.J.; Perlepes, S.P.; Christou, G.; Stamatatos, T.C. “Squaring the clusters”: A MnIII4NiII4 molecular square from nickel(ii)-induced structural transformation of a MnII/III/IV12 cage. Dalton Trans. 2012, 41, 4744–4747. [Google Scholar] [CrossRef]
- Derikvand, Z.; Dorosti, N.; Hassanzadeh, F.; Shokrollahi, A.; Mohammadpour, Z.; Azadbakht, A. Three new supramolecular compounds of copper (II), cobalt (II) and zirconium (IV) with pyridine-2,6-dicarboxylate and 3,4-diaminopyridine: Solid and solution states studies. Polyhedron 2012, 43, 140–152. [Google Scholar] [CrossRef]
- Duong, A.; Maris, T.; Wuest, J.D. Using Pyridinyl-Substituted Diaminotriazines to Bind Pd(II) and Create Metallotectons for Engineering Hydrogen-Bonded Crystals. Inorg. Chem. 2011, 50, 5605–5618. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Xia, M.Z.; Wang, F.Y.; Heng, L.J.; Yang, T.H. Synthesis and Crystal Structure of Nickel(II) Complex Derived from Benzotriazole-4-Sulfonate. Asian J. Chem. 2011, 23, 3755–3756. [Google Scholar]
- Jian, F.F.; Wang, J.; Huang, L.H.; Wang, X.; Xiao, H.L. Two supramolecular microporous frameworks stabilized by hydroxyl anionic water cluster. J. Mol. Struct. 2010, 973, 136–143. [Google Scholar] [CrossRef]
- Strotmeyer, K.P.; Fritsky, I.O.; Pritzkow, H.; Krämer, R. Self-assembly of a molecular figure-of-eight strip. Chem. Commun. 2004, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Bergman, S.D.; Goldberg, I.; Barbieri, A.; Barigelletti, F.; Kol, M. Mononuclear and Dinuclear Complexes of Dibenzoeilatin: Synthesis, Structure, and Electrochemical and Photophysical Properties. Inorg. Chem. 2004, 43, 2355–2367. [Google Scholar] [CrossRef]
- Zheng, Y.Q.; Lin, J.L.; Sun, J. Crystal structure of disodium di-mu-hydroxo-bis(chloro 1,10-phenanthroline-N,N‘-copper (II)) dihydroxide, Na2[Cu2Cl2(Phen)2(OH)2](OH)2. Z. Krist. New Cryst. St. 2001, 216, 330–338. [Google Scholar]
- Boury, B.; Carre, F.; Corriu, R.J.P.; Núñez, R. New sodium organobis(silantriolates). Chem. Commun. 1998, 2309–2310. [Google Scholar] [CrossRef]
- Hu, N.H. A hydroxide inclusion complex of a methylene-bridged tetrapyrimidinium macrocycle. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1994, 50, 2082–2085. [Google Scholar] [CrossRef]
- Hatakeyama, J.; Kobayashi, T.; Watanabe, T. Amine Compounds, Resist Compositions and Patterning Process. U.S. Patent 6,749,988, 15 June 2004. [Google Scholar]
- Mengel, A.; Müller, T.; Bärfacker, L.; Hitchcock, M.; Cleve, A.; Briem, H.; Siemeister, G.; Fernandez-Montalvan, A.E.; Schröder, J.; Mönning, U. Benzyl substituted indazoles as bub1 kinase inhibitors. U.S. Patent Application No. 15/512,474, 28 September 2017. [Google Scholar]
- Santarelli, S.; Bazzicalupi, C.; Bianchi, A.; Biver, T.; Giorgi, C.; Savastano, M. Formation of Double-Strand Dimetallic Helicates with a Terpyridine-Based Macrocycle. Inorg. Chem. 2014, 53, 12215–12224. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Simultaneous Calculation of Equilibrium Constants and Standard Formation Enthalpies from Calorimetric Data for Systems with Multiple Equilibria in Solution. J. Solut. Chem. 2008, 37, 467–476. [Google Scholar] [CrossRef]
- CrysAlisPro, Version 1.171.35.11; Agilent Technologies: Yarnton, UK, 2011.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR 92—A program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jaguar, Version 9.1; Schrödinger Suite Release 2016-1; Schrödinger, LLC: New York, NY, USA, 2016.
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L1 | L2 | L3 | L4 | ||
|---|---|---|---|---|---|
| L + H+ = HL+ | −logK | 4.45 (3) a | 6.19 (1) a | 7.18 (3) c | 7.59 (6) c |
| ΔG° | −25.4 (2) a | −35.32 (6) a | −41.0 (2) | −43.3 (4) | |
| ΔH° | −4.6 (4) a | −26.8 (4) a | −41.6 (2) | −39.6 (1) | |
| TΔS° | 20.8 (4) a | 8.5 (4) a | −0.6 (4) | 3.7 (5) | |
| HL+ + H+ = H2L2+ | logK | 3.45 (3) a | 5.37 (1) a | 6.54 (3) c | 6.84 (6) c |
| ΔG° | −19.7 (2) a | −30.64 (6) a | −37.3 (2) c | −39.0 (4) c | |
| ΔH° | 4.6 (4) a | −18.4 (4) a | −47.9 (2) c | −39.7 (1) c | |
| TΔS° | 24.3 (4) a | 12.2 (4) a | −10.6 (4) c | −0.7 (5) c | |
| L + OH− = [(L)OH]− | logK | 2.60 (7) b | 1.76 (7) b | 2.69 (3) c | 2.40 (6) c |
| ΔG° | −14.8 (4) b | −10.0 (4) b | −15.3 (2) c | −13.7 (3) c | |
| ΔH° | −25.1 (4) b | −18.8 (4) b | −9.6 (2) c | −9.5 (1) c | |
| TΔS° | −10.3 (8) b | −8.8 (8) b | 5.7 (4) c | 4.2 (4) c | |
| L + ClO4− = [(L)ClO4]− | logK | 1.98 (5) a | 2.26 (9) c | 1.1 (1) c | |
| HL+ + ClO4− = [(HL)ClO4] | logK | 2.07 (9) a | 2.26 (5) a | 2.2 (1) c | 1.1 (2) c |
| H2L2+ + ClO4− = [(H2L)ClO4]+ | logK | 2.31 (8) a | 2.51 (4) a | 2.1 (1) c | 1.6 (1) c |
| L + PF6− = [(L)PF6]− | logK | 1.96 (8) a | 3.07 (8) a | 2.30 (9) c | |
| HL+ + PF6− = [(HL)PF6] | logK | 2.67 (7) a | 3.17 (7) a | 2.2 (1) c | |
| H2L2+ + PF6− = [(H2L)PF6]+ | logK | 2.98 (7) a | 3.39 (8) a | 2.4 (1) c |
| (a) | (b) | (c) | (d) | (e) | (f) | |
|---|---|---|---|---|---|---|
| Empirical formula | C16H28N6O2 | C18H32N6O2 | C16H34Cl2N6O12 | C18H38Cl2N6O12 | C16H30F12N6O2P2 | C16H34F12N6O4P2 |
| Formula weight | 336.44 | 364.50 | 573.39 | 601.44 | 628.40 | 664.43 |
| Temperature (K) | 150 | 298 | 150 | 150 | 150 | 150 |
| space group | P -1 | P 21/c | P 21/c | P 21/c | P 21/n | P 21/c |
| a (Å) | 6.6983 (6) | 13.5805 (3) | 13.7608 (5) | 14.4546 (3) | 7.7907 (3) | 13.4690 (4) |
| b (Å) | 8.6859 (8) | 5.1482 (1) | 7.3316 (3) | 17.0531 (4) | 8.8430 (3) | 7.8954 (2) |
| c (Å) | 8.7604 (9) | 14.2994 (3) | 13.3194 (7) | 11.0345 (2) | 18.2505 (8) | 13.6108 (4) |
| α (°) | 109.119 (9) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 95.224 (8) | 104.627 (2) | 111.655 (5) | 97.932 (2) | 96.454 (4) | 112.191 (4) |
| γ (°) | 109.872 (8) | 90 | 90 | 90 | 90 | 90 |
| Volume (Å3) | 440.99 (8) | 967.34 (4) | 1248.94 (10) | 2693.93 (10) | 1249.37 (8) | 1340.20 (7) |
| Z | 1 | 2 | 2 | 4 | 2 | 2 |
| Independent reflections/R(int) | 1619/0.0454 | 1849/0.0271 | 2125/0.0837 | 4991/0.0693 | 6350/0.0333 | 2458/0.0895 |
| μ (mm−1) | 0.704 (Cu-Kα) | 0.679 (Cu-Kα) | 2.983 (Cu-Kα) | 2.793 (Cu-Kα) | 0.292 (Mo-Kα) | 2.604 (Cu-Kα) |
| R indices [I>2σ(I)] a | R1 = 0.0698 | R1 = 0.0416 | R1 = 0.0801 | R1 = 0.0599 | R1 = 0.0410 | R1 = 0.0548 |
| wR2 = 0.1995 | wR2 = 0.1077 | wR2 = 0.2447 | wR2 = 0.1513 | wR2 = 0.1017 | wR2 = 0.0964 | |
| R indices (all data) a | R1 = 0.1012 | R1 = 0.0504 | R1 = 0.1409 | R1 = 0.1006 | R1 = 0.0634 | R1 = 0.1162 |
| wR2 = 0.2168 | wR2 = 0.1152 | wR2 = 0.3190 | wR2 = 0.1917 | wR2 = 0.1161 | wR2 = 0.1217 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savastano, M.; García-Gallarín, C.; Giorgi, C.; Gratteri, P.; López de la Torre, M.D.; Bazzicalupi, C.; Bianchi, A.; Melguizo, M. Solid State and Solution Study on the Formation of Inorganic Anion Complexes with a Series of Tetrazine-Based Ligands. Molecules 2019, 24, 2247. https://doi.org/10.3390/molecules24122247
Savastano M, García-Gallarín C, Giorgi C, Gratteri P, López de la Torre MD, Bazzicalupi C, Bianchi A, Melguizo M. Solid State and Solution Study on the Formation of Inorganic Anion Complexes with a Series of Tetrazine-Based Ligands. Molecules. 2019; 24(12):2247. https://doi.org/10.3390/molecules24122247
Chicago/Turabian StyleSavastano, Matteo, Celeste García-Gallarín, Claudia Giorgi, Paola Gratteri, Maria Dolores López de la Torre, Carla Bazzicalupi, Antonio Bianchi, and Manuel Melguizo. 2019. "Solid State and Solution Study on the Formation of Inorganic Anion Complexes with a Series of Tetrazine-Based Ligands" Molecules 24, no. 12: 2247. https://doi.org/10.3390/molecules24122247