
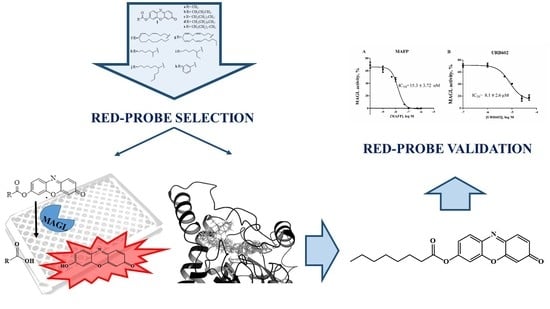
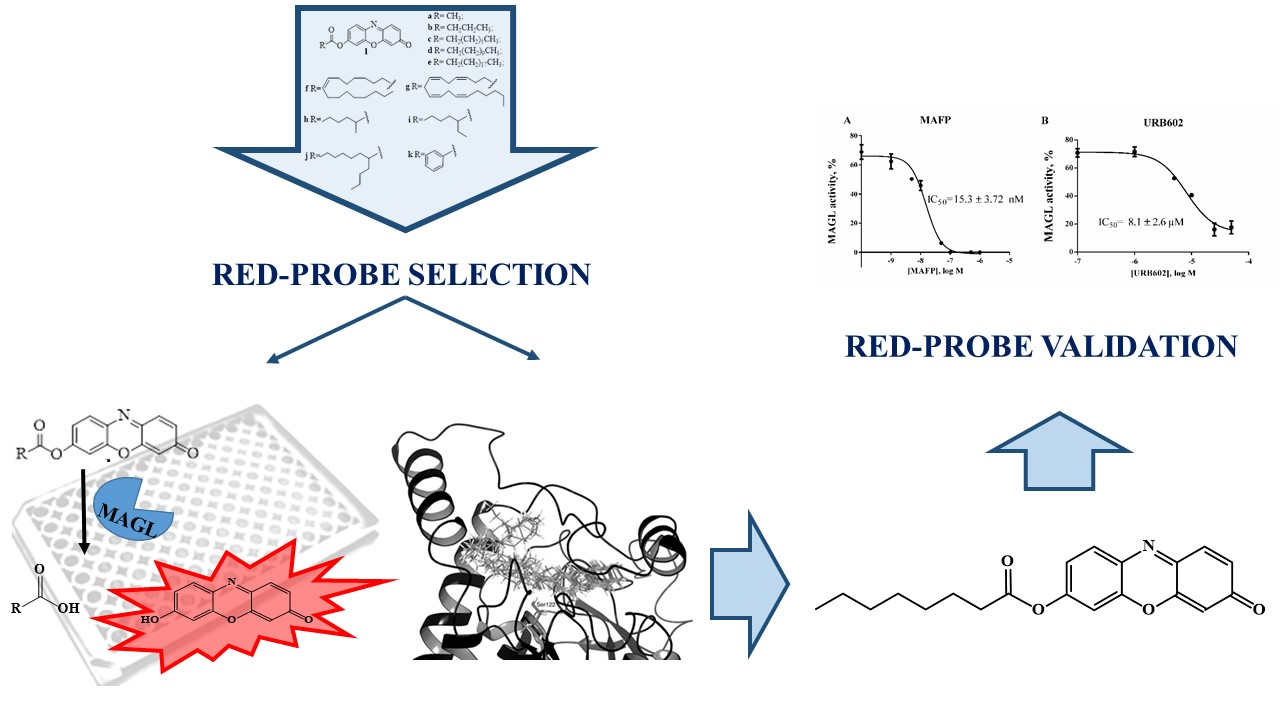
Set-Up and Validation of a High Throughput Screening Method for Human Monoacylglycerol Lipase (MAGL) Based on a New Red Fluorescent Probe

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Results
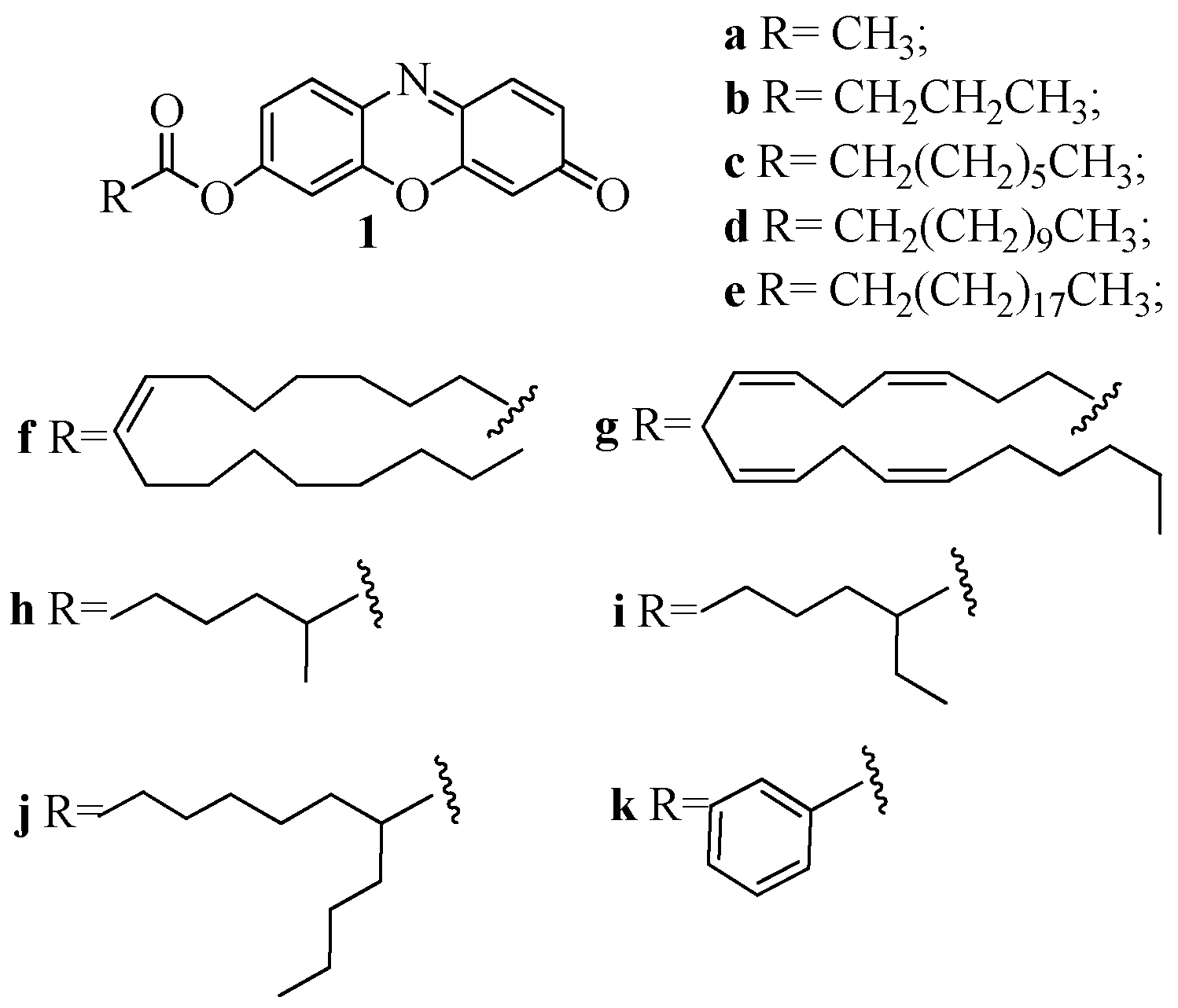
2.1. Synthesis of 1a–k
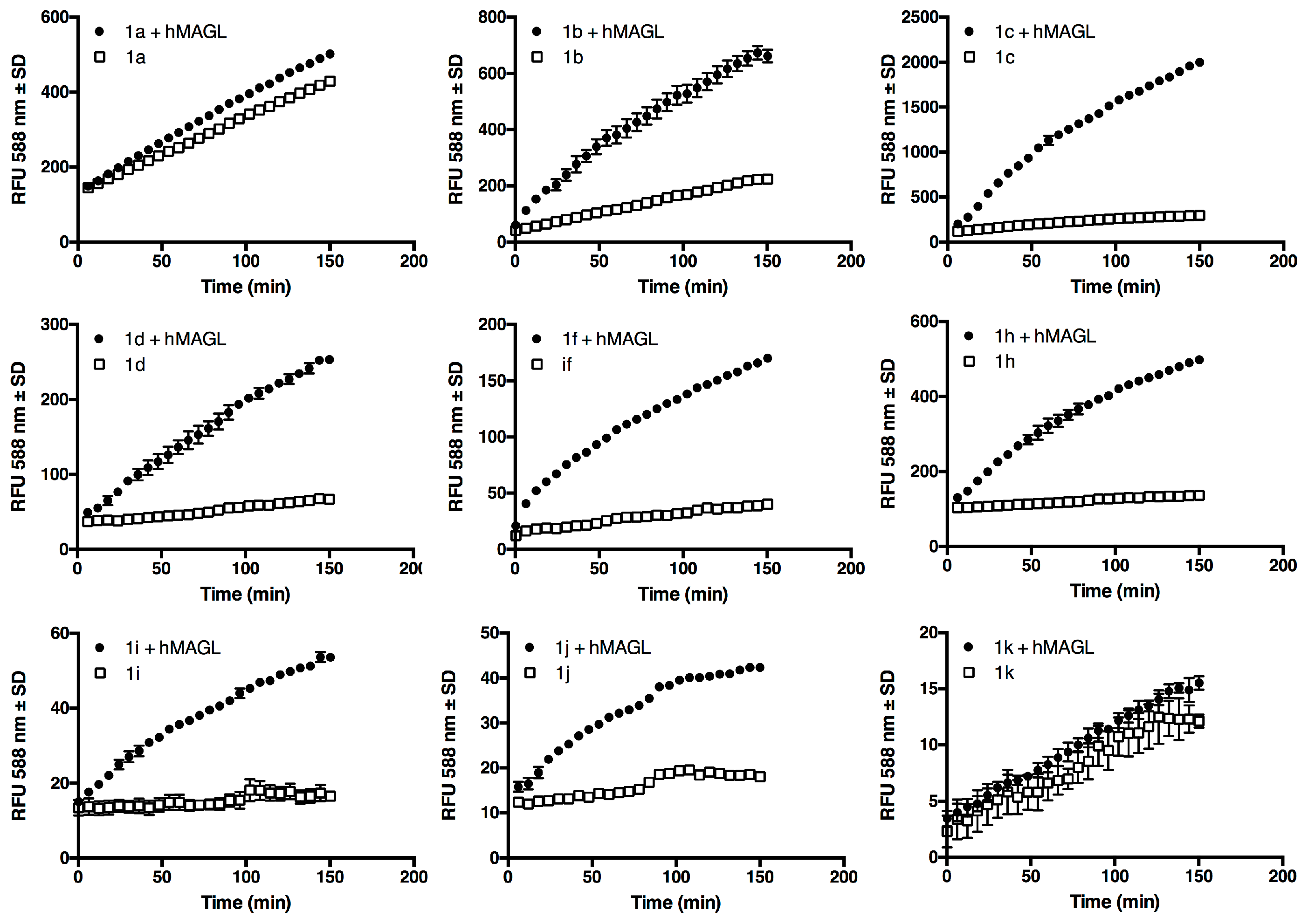
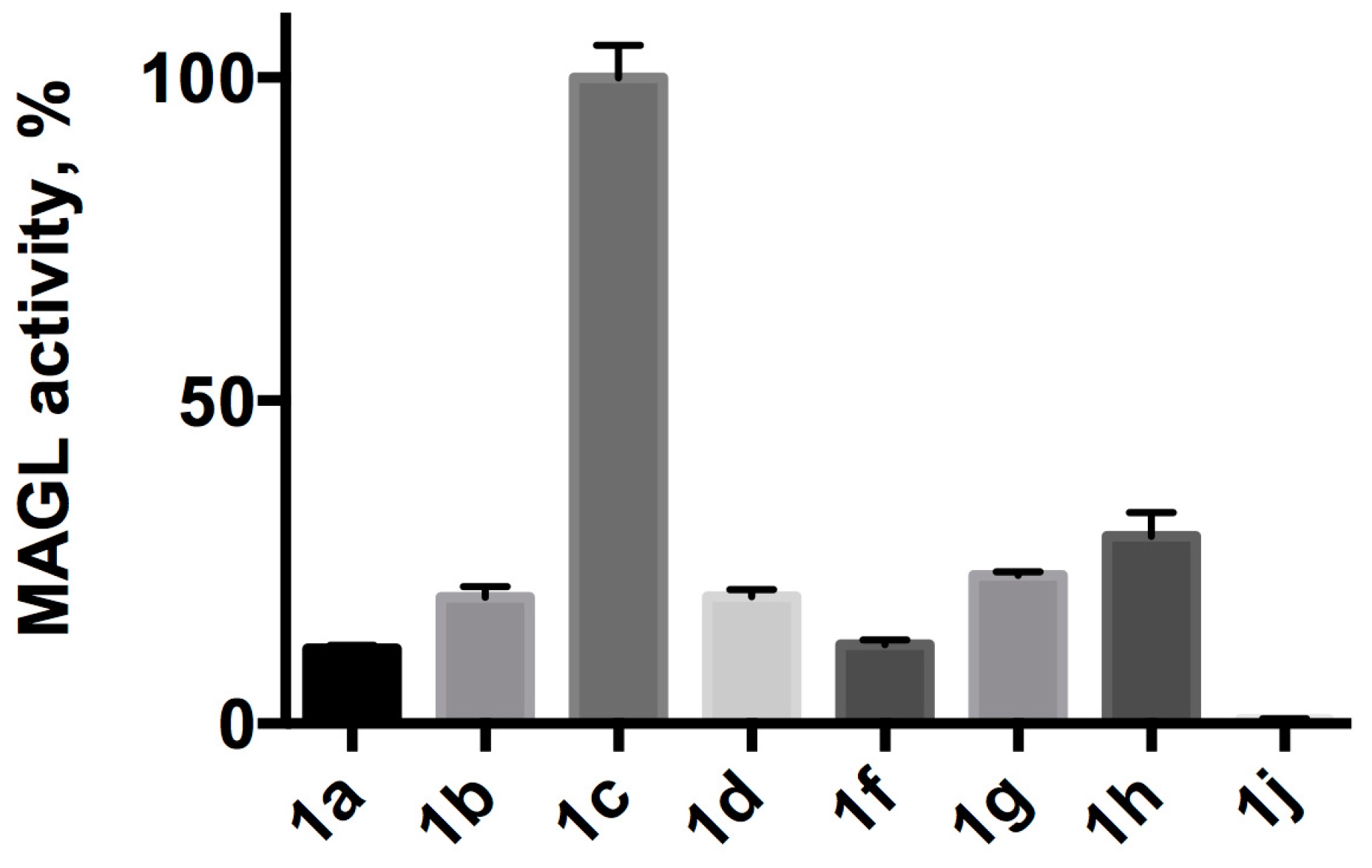
2.2. Substrates Screening
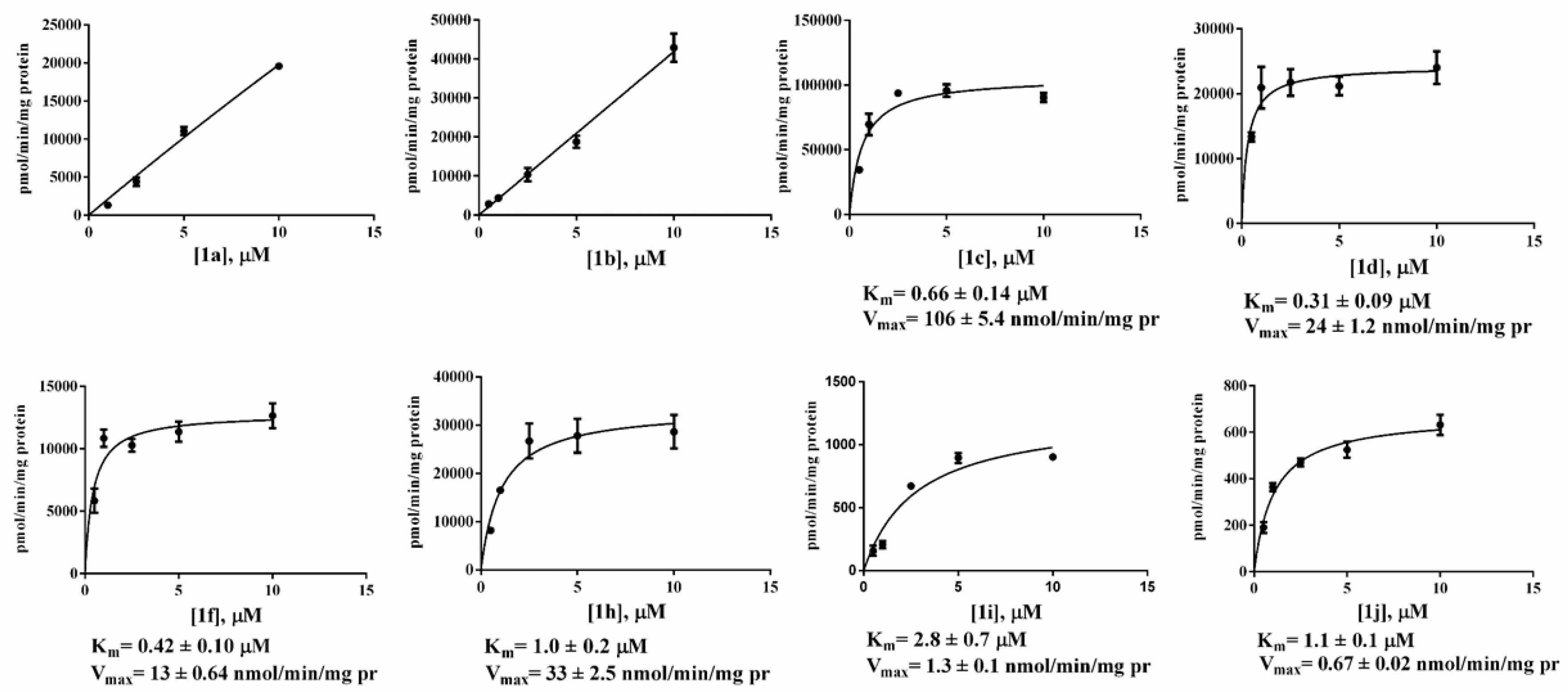
2.3. Kinetic Studies
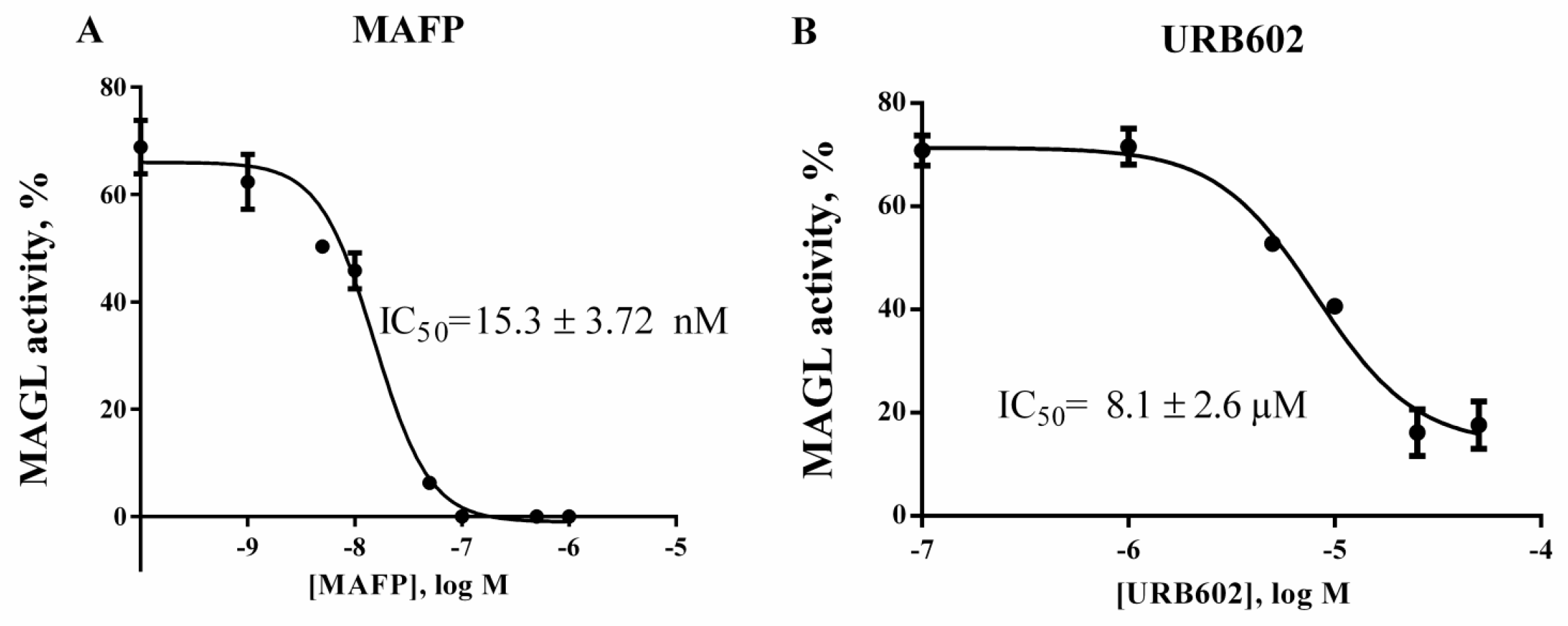
2.4. Validation of 1c for Screening Assay
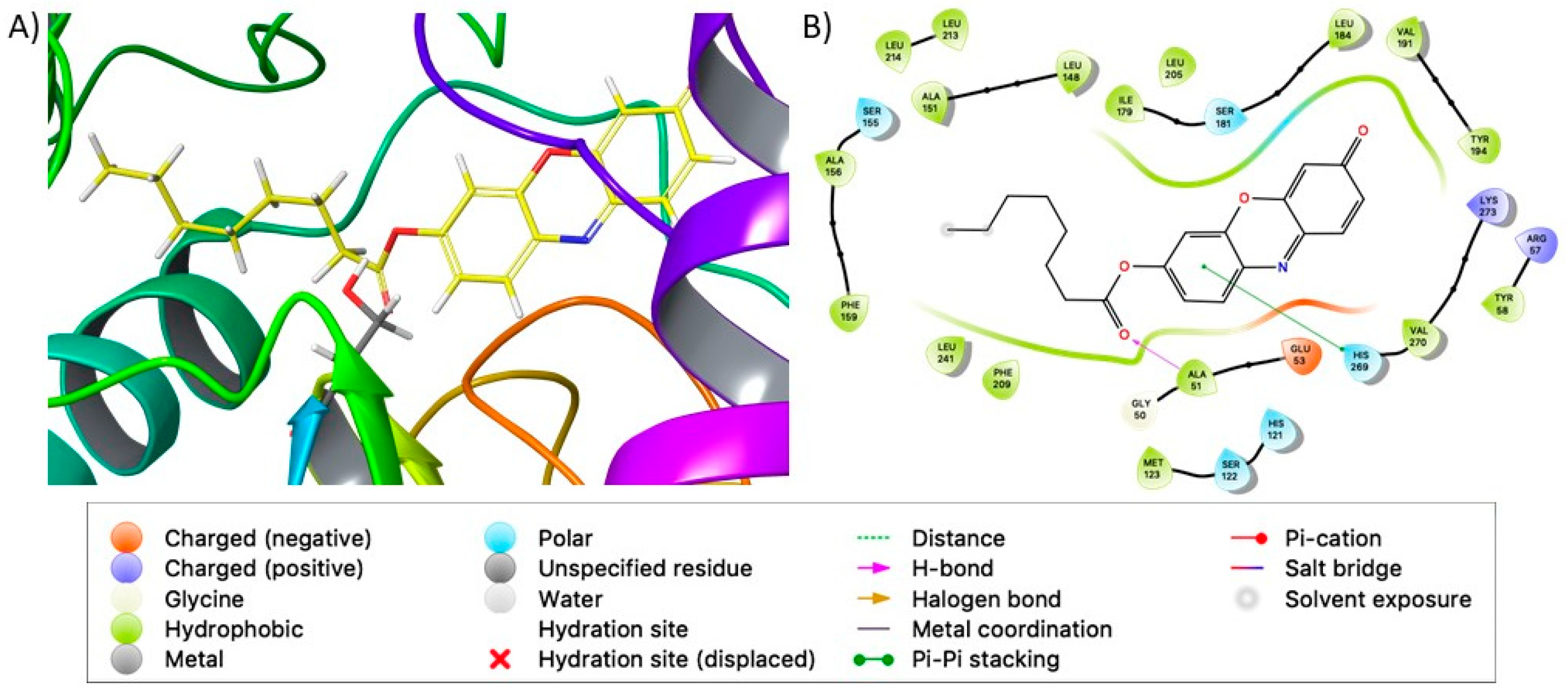
2.5. Docking Studies
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Synthesis
4.2.1. Synthesis of 7-Hydroxyresorufinyl-Derivatives 1a–1g
4.2.2. Synthesis of Resorufin Esters 1h–1k
4.3. Stability of the Substrates
4.4. Substrates Screening
4.5. Kinetic Assays of hMAGL
4.6. Data Analysis
4.7. Validation of 1c for Screening Assay
4.8. In Silico Molecular Docking Simulations
4.9. Dynamic Light Scattering Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, A.H.; Salem, V.; Ghatei, M.A. Rimonabant: From RIO to Ban. J. Obes. 2011, 2011, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tuo, W.; Leleu-Chavain, N.; Spencer, J.; Sansook, S.; Millet, R.; Chavatte, P. Therapeutic Potential of Fatty Acid Amide Hydrolase, Monoacylglycerol Lipase, and N-Acylethanolamine Acid Amidase Inhibitors. J. Med. Chem. 2017, 60, 4–46. [Google Scholar] [CrossRef] [PubMed]
- Savinainen, J.R.; Saario, S.M.; Laitinen, J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. 2012, 204, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labar, G.; Bauvois, C.; Muccioli, G.G.; Wouters, J.; Lambert, D.M. Disulfiram is an Inhibitor of Human Purified Monoacylglycerol Lipase, the Enzyme Regulating 2-Arachidonoylglycerol Signaling. ChemBioChem 2007, 8, 1293–1297. [Google Scholar] [CrossRef]
- Saario, S.M.; Savinainen, J.R.; Laitinen, J.T.; Järvinen, T.; Niemi, R. Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem. Pharmacol. 2004, 67, 1381–1387. [Google Scholar] [CrossRef]
- King, A.R.; Lodola, A.; Carmi, C.; Fu, J.; Mor, M.; Piomelli, D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. Br. J. Pharmacol. 2009, 157, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Muccioli, G.G.; Labar, G.; Lambert, D.M. CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. ChemBioChem 2008, 9, 2704–2710. [Google Scholar] [CrossRef]
- Wang, Y.; Chanda, P.; Jones, P.G.; Kennedy, J.D. A Fluorescence-Based Assay for Monoacylglycerol Lipase Compatible with Inhibitor Screening. Assay Drug Dev. Technol. 2008, 6, 387–393. [Google Scholar] [CrossRef]
- Holtfrerich, A.; Makharadze, T.; Lehr, M. High-performance liquid chromatography assay with fluorescence detection for the evaluation of inhibitors against human recombinant monoacylglycerol lipase. Anal. Biochem. 2010, 399, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Lauria, S.; Casati, S.; Ciuffreda, P. Synthesis and characterization of a new fluorogenic substrate for monoacylglycerol lipase and application to inhibition studies. Anal. Bioanal. Chem. 2015, 407, 8163–8167. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-N.; Huang, Z.-P.; Gu, Q.-L.; Zhi, Z.-E.; Yang, Y.-H.; He, L.; Chen, K.-L.; Wang, J.-X. Synthesis and biological evaluation of novel tanshinone IIA derivatives for treating pain. Chin. J. Nat. Med. 2018, 16, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, Z.; Guo, R.; Qian, J.; Zhang, H.; Zhang, J.; Zhao, X.; Wang, S.; Wang, Y. Ononin, sec-O-β-d-glucosylhamaudol and astragaloside I: antiviral lead compounds identified via high throughput screening and biological validation from traditional Chinese medicine Zhongjing formulary. Pharmacol. Res. 2019, 145, 104248. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, A.; Jadhav, A.; Thomas, C.J.; Wang, Y.; Huang, R.; Southall, N.T.; Shinn, P.; Smith, J.; Austin, C.P.; Auld, D.S.; et al. Fluorescence Spectroscopic Profiling of Compound Libraries. J. Med. Chem. 2008, 51, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Sakurai, Y.; Ma, S.-F.; Watanabe, H.; Yamaotsu, N.; Hirono, S.; Kurono, Y.; Kragh-Hansen, U.; Otagiri, M. Esterase-Like Activity of Serum Albumin: Characterization of Its Structural Chemistry Using p-Nitrophenyl Esters as Substrates. Pharm. Res. 2004, 21, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Bisswanger, H. Enzyme assays. Perspect. Sci. 2014, 1, 41–55. [Google Scholar] [CrossRef] [Green Version]
- King, A.R.; Duranti, A.; Tontini, A.; Rivara, S.; Rosengarth, A.; Clapper, J.R.; Astarita, G.; Geaga, J.A.; Luecke, H.; Mor, M.; et al. URB602 Inhibits Monoacylglycerol Lipase and Selectively Blocks 2-Arachidonoylglycerol Degradation in Intact Brain Slices. Chem. Biol. 2007, 14, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Matuszak, N.; Muccioli, G.G.; Labar, G.; Lambert, D.M. Synthesis and in Vitro Evaluation of N-Substituted Maleimide Derivatives as Selective Monoglyceride Lipase Inhibitors. J. Med. Chem. 2009, 52, 7410–7420. [Google Scholar] [CrossRef]
- Lauria, S.; Perrotta, C.; Casati, S.; Di Renzo, I.; Ottria, R.; Eberini, I.; Palazzolo, L.; Parravicini, C.; Ciuffreda, P. Design, synthesis, molecular modelling and in vitro cytotoxicity analysis of novel carbamate derivatives as inhibitors of Monoacylglycerol lipase. Bioorgan. Med. Chem. 2018, 26, 2561–2572. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, N.; Duan, T.; Kesner, P.; Blaskovits, F.; Liu, J.; Lu, Y.; Tong, L.; Gao, F.; Harris, C.; et al. Monoacylglycerol lipase inhibitors produce pro- or antidepressant responses via hippocampal CA1 GABAergic synapses. Mol. Psychiatry 2017, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, M.; Mandenius, C.-F. Fluorescent cell-based sensing approaches for toxicity testing. Anal. Bioanal. Chem. 2010, 398, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Bornscheuer, U.T. High-throughput assays for lipases and esterases. Biomol. Eng. 2005, 22, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Matsu-Ura, S.; Nishida, M.; Yamauchi, Y.; Ohmori, H. Assessment of acyl groups and reaction conditions in the competition between perhydrolysis and hydrolysis of acyl resorufins for developing an indicator reaction for fluorometric analysis of hydrogen peroxide. Chem. Pharm. Bull. 2002, 50, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Henault, M.; Khougaz, K.; Fortin, L.J.; Ouellet, M.; Melnyk, R.; Partridge, A. Resorufin butyrate as a soluble and monomeric high-throughput substrate for a triglyceride lipase. J. Biomol. Screen. 2012, 17, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; Heckman, L.M.; Lavis, L.D. The chemistry of small-molecule fluorogenic probes. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1–34. [Google Scholar]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Long, J.Z.; Cravatt, B.F. The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease. Chem. Rev. 2011, 111, 6022–6063. [Google Scholar] [CrossRef] [Green Version]
- Simon, G.M.; Cravatt, B.F. Activity-based Proteomics of Enzyme Superfamilies: Serine Hydrolases as a Case Study. J. Biol. Chem. 2010, 285, 11051–11055. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.; Ware, T.B.; Lee, H.-C.; Hsu, K.-L. Lipid-metabolizing serine hydrolases in the mammalian central nervous system: endocannabinoids and beyond. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2019, 1864, 907–921. [Google Scholar] [CrossRef]
- Schalk-Hihi, C.; Schubert, C.; Alexander, R.; Bayoumy, S.; Clemente, J.C.; Deckman, I.; DesJarlais, R.L.; Dzordzorme, K.C.; Flores, C.M.; Grasberger, B.; et al. Crystal structure of a soluble form of human monoglyceride lipase in complex with an inhibitor at 1.35 A° resolution. Protein Sci. 2011, 20, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds Ola and Olad8 are available from the authors. Samples of loading compounds are not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | LogD at pH 7.4 a | Km (μM) | Vmax (nmol/min/mg protein) | Docking Score | MM-GBSA | Fitting Model | r2 |
|---|---|---|---|---|---|---|---|
| 1a | 1.69 | n/a | n/a | −6.7 | −52.4 | n/a | n/a |
| 1b | 2.83 | n/a | n/a | −7.7 | −60.5 | n/a | n/a |
| 1c | 4.61 | 0.66 ± 0.14 | 106 ± 5.4 | −8.2 | −72.0 | M-M | 0.8488 |
| 1d | 6.39 | 0.31 ± 0.09 | 24 ± 1.2 | −10.5 | −81.4 | M-M | 0.6731 |
| 1e | 9.94 | n/a | n/a | −9.8 | −67.8 | n/a | n/a |
| 1f | 8.69 | 0.42 ± 0.1 | 13 ± 0.64 | −10.8 | −73.2 | M-M | 0.7391 |
| 1g | 8.50 | 0.87 ± 0.13 | 25.8 ± 0.88 | −10.7 | −76.0 | M-M | 0.9908 |
| 1h | 4.26 | 1.09 ± 0.02 | 33 ± 2.509 | −8.5 | −60.3 | M-M | 0.8859 |
| 1i | 4.71 | 2.8 ± 0.7 | 1.3 ± 0.1 | −8.8 | −62.8 | M-M | 0.9236 |
| 1j | 6.49 | 1.1 ± 0.1 | 0.67 ± 0.03 | −8.4 | −65.4 | M-M | 0.9450 |
| 1k | 3.74 | n/a | n/a | −8.9 | −63.2 | n/a | n/a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miceli, M.; Casati, S.; Ottria, R.; Di Leo, S.; Eberini, I.; Palazzolo, L.; Parravicini, C.; Ciuffreda, P. Set-Up and Validation of a High Throughput Screening Method for Human Monoacylglycerol Lipase (MAGL) Based on a New Red Fluorescent Probe. Molecules 2019, 24, 2241. https://doi.org/10.3390/molecules24122241
Miceli M, Casati S, Ottria R, Di Leo S, Eberini I, Palazzolo L, Parravicini C, Ciuffreda P. Set-Up and Validation of a High Throughput Screening Method for Human Monoacylglycerol Lipase (MAGL) Based on a New Red Fluorescent Probe. Molecules. 2019; 24(12):2241. https://doi.org/10.3390/molecules24122241
Chicago/Turabian StyleMiceli, Matteo, Silvana Casati, Roberta Ottria, Simone Di Leo, Ivano Eberini, Luca Palazzolo, Chiara Parravicini, and Pierangela Ciuffreda. 2019. "Set-Up and Validation of a High Throughput Screening Method for Human Monoacylglycerol Lipase (MAGL) Based on a New Red Fluorescent Probe" Molecules 24, no. 12: 2241. https://doi.org/10.3390/molecules24122241