A Comprehensive In Silico Method to Study the QSTR of the Aconitine Alkaloids for Designing Novel Drugs


Abstract
:1. Introduction
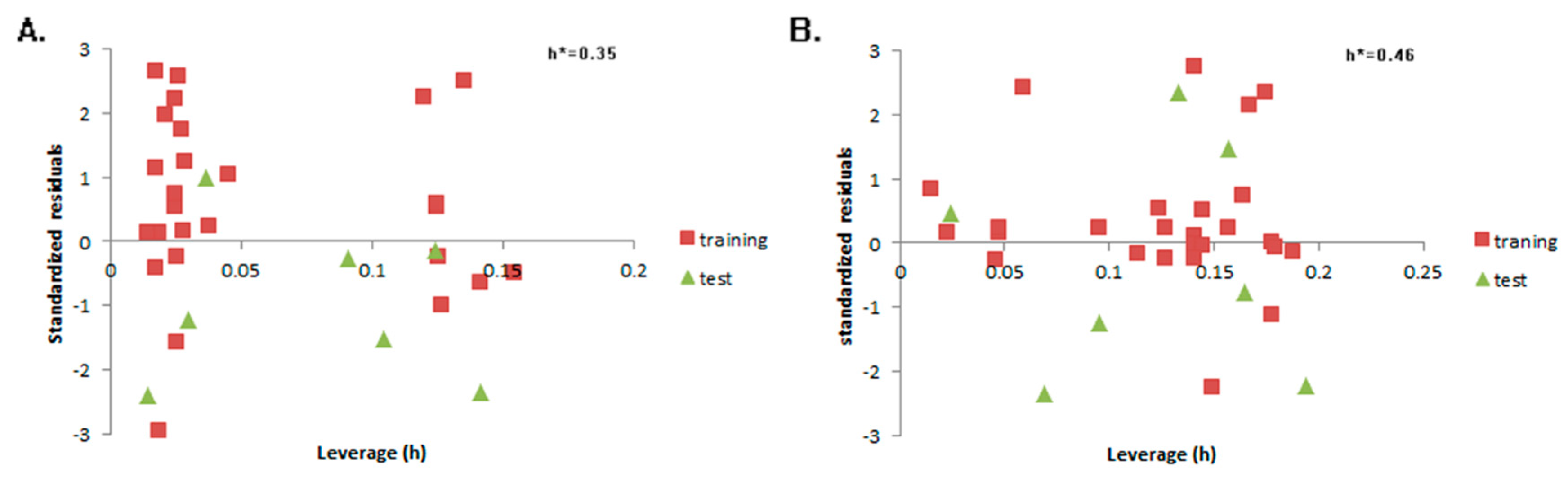
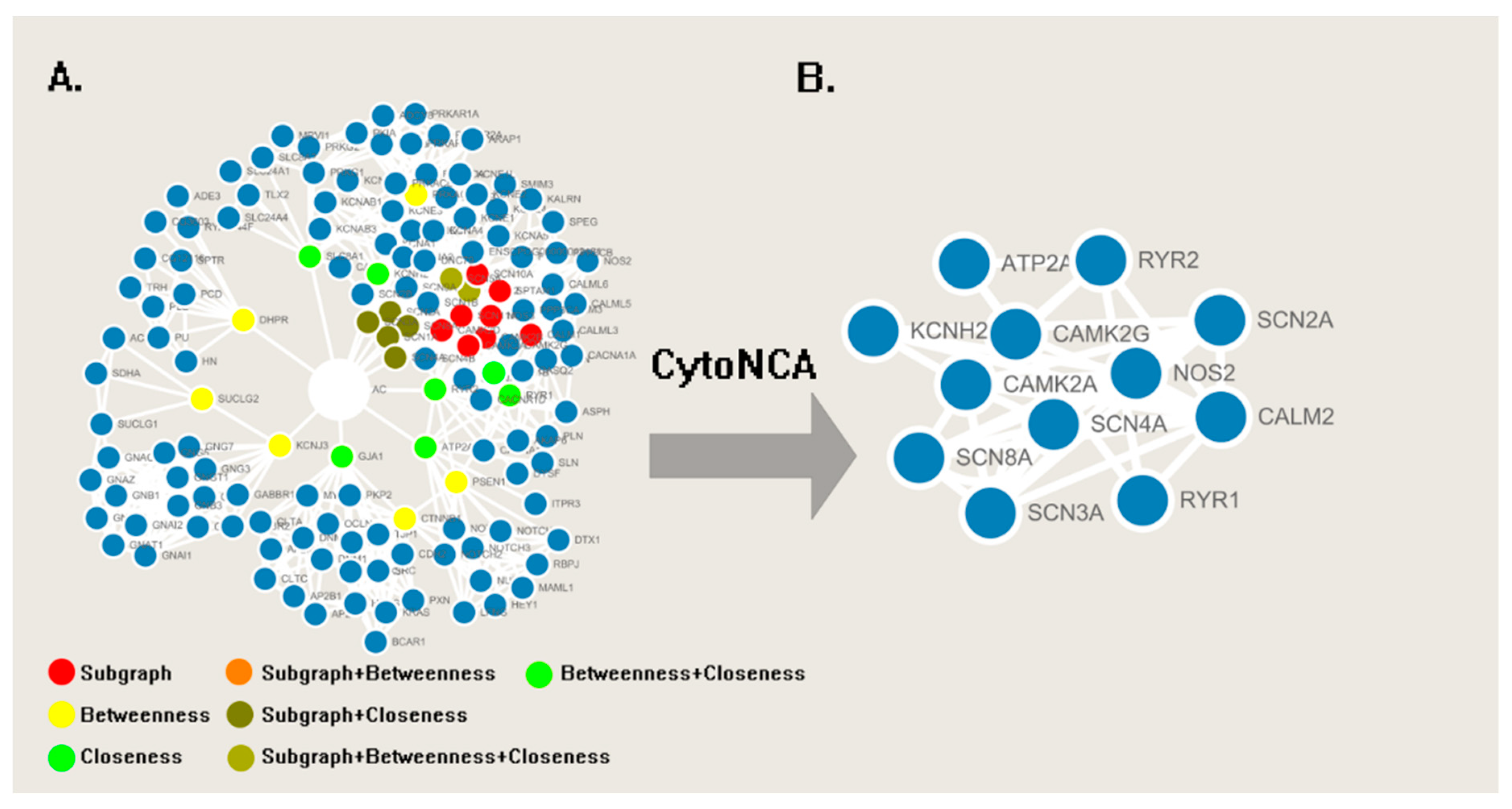
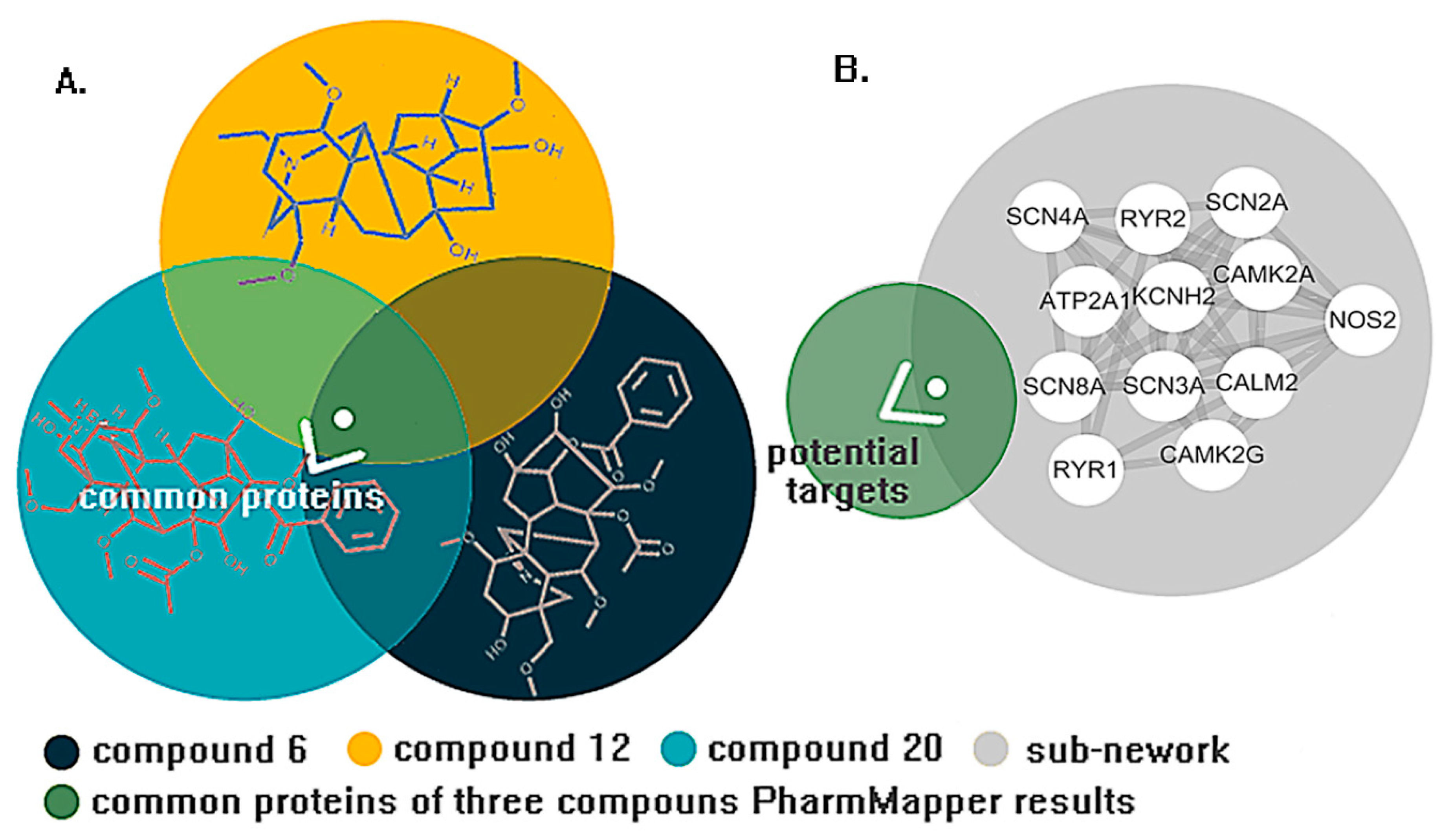
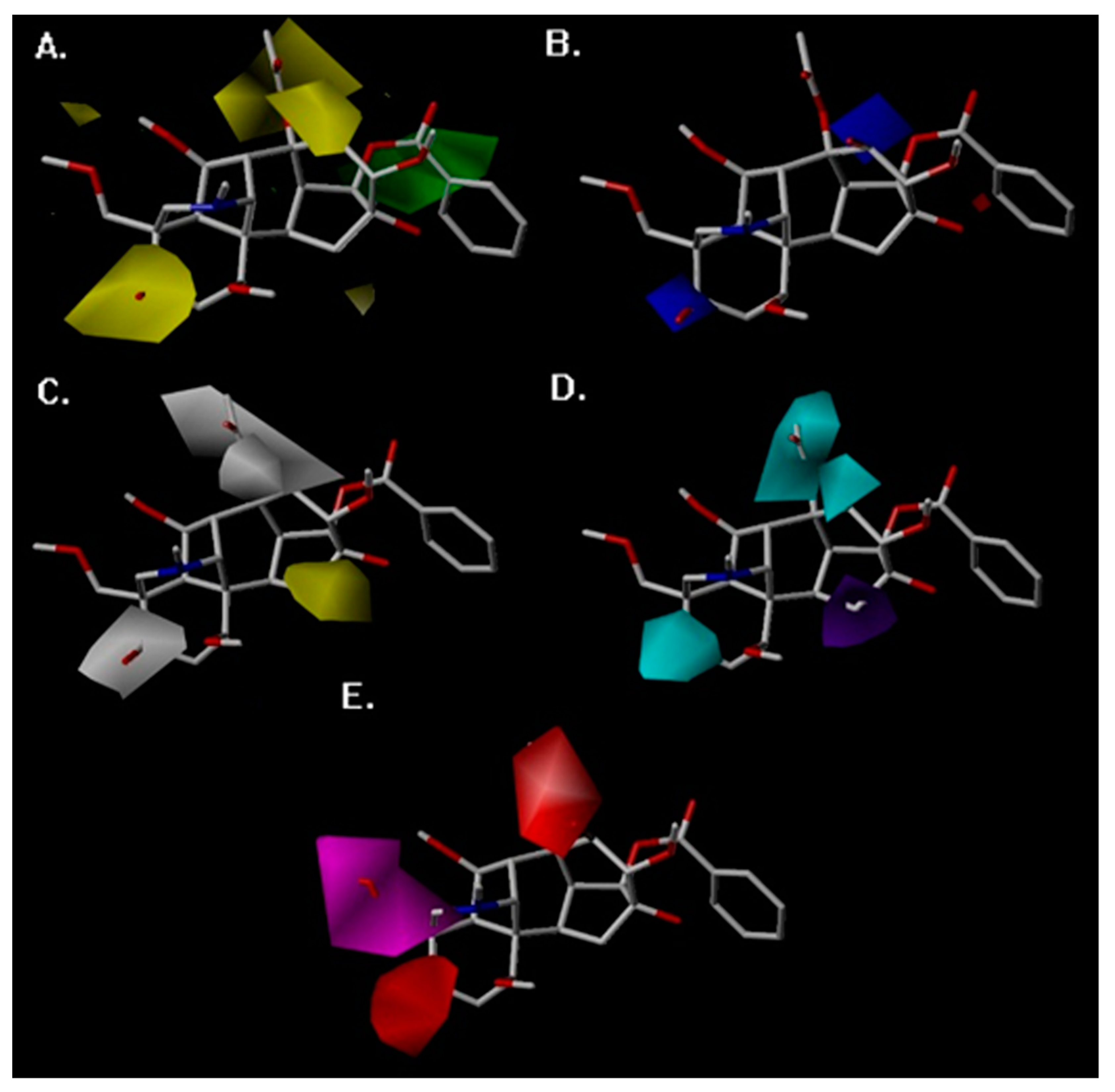
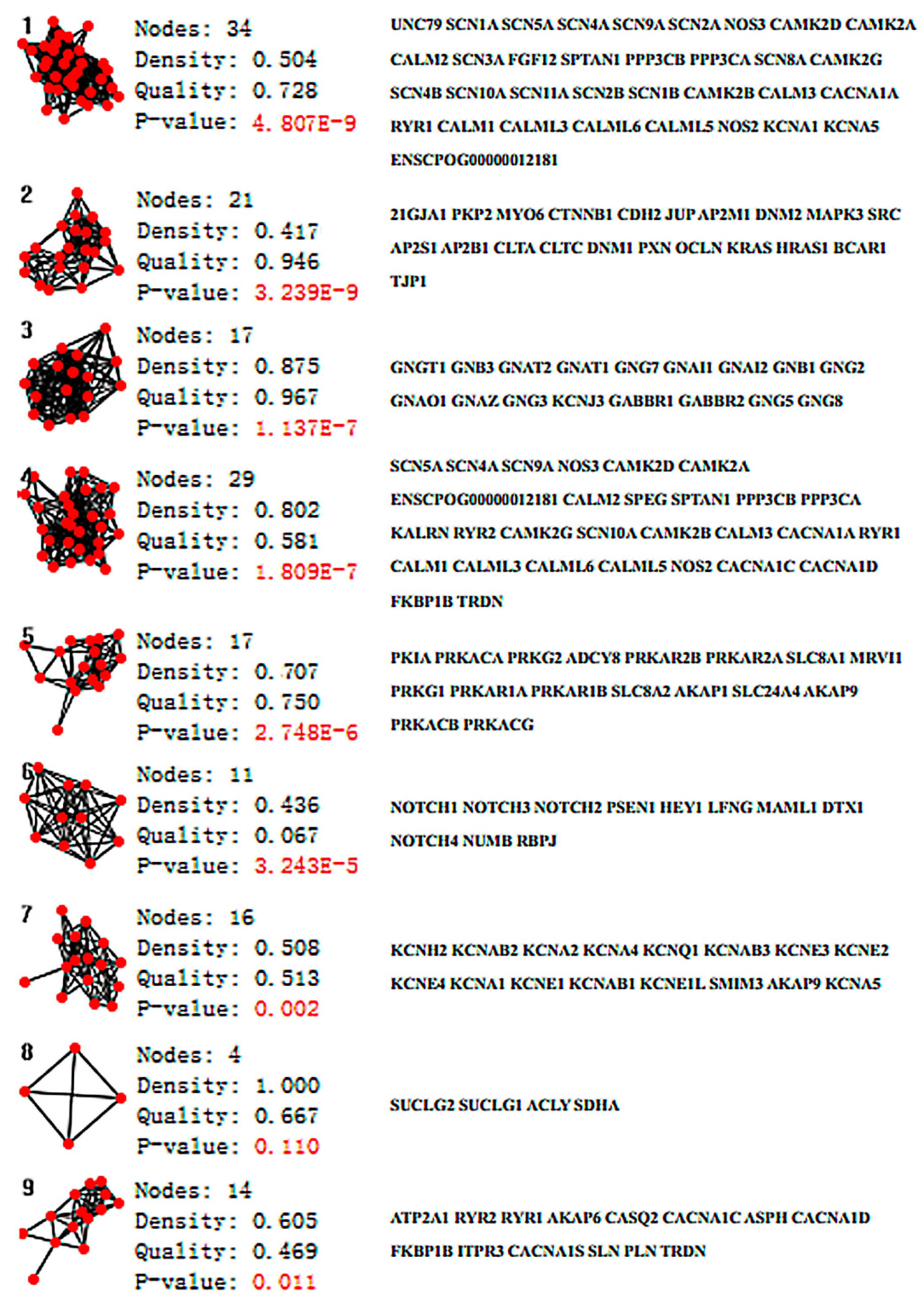
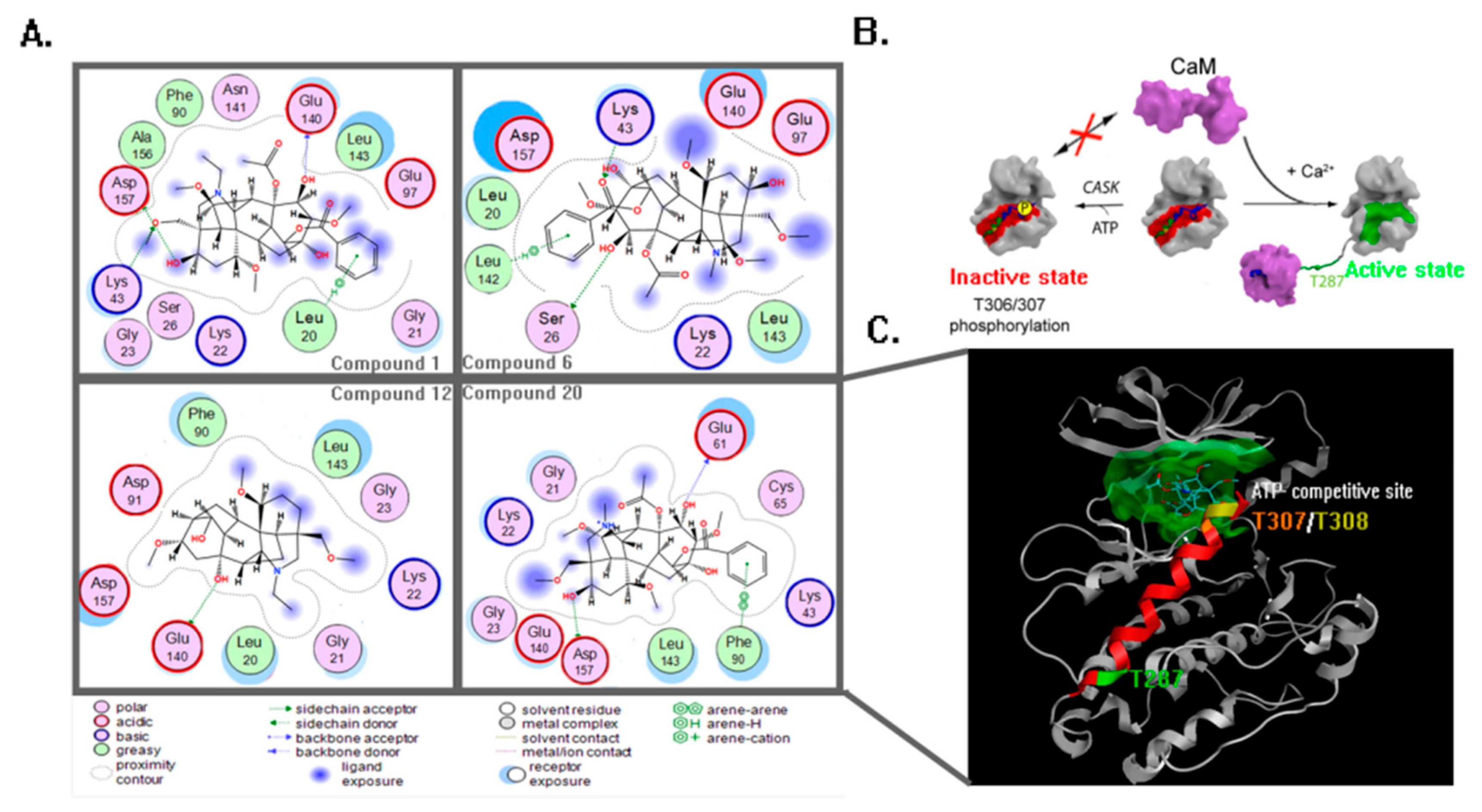
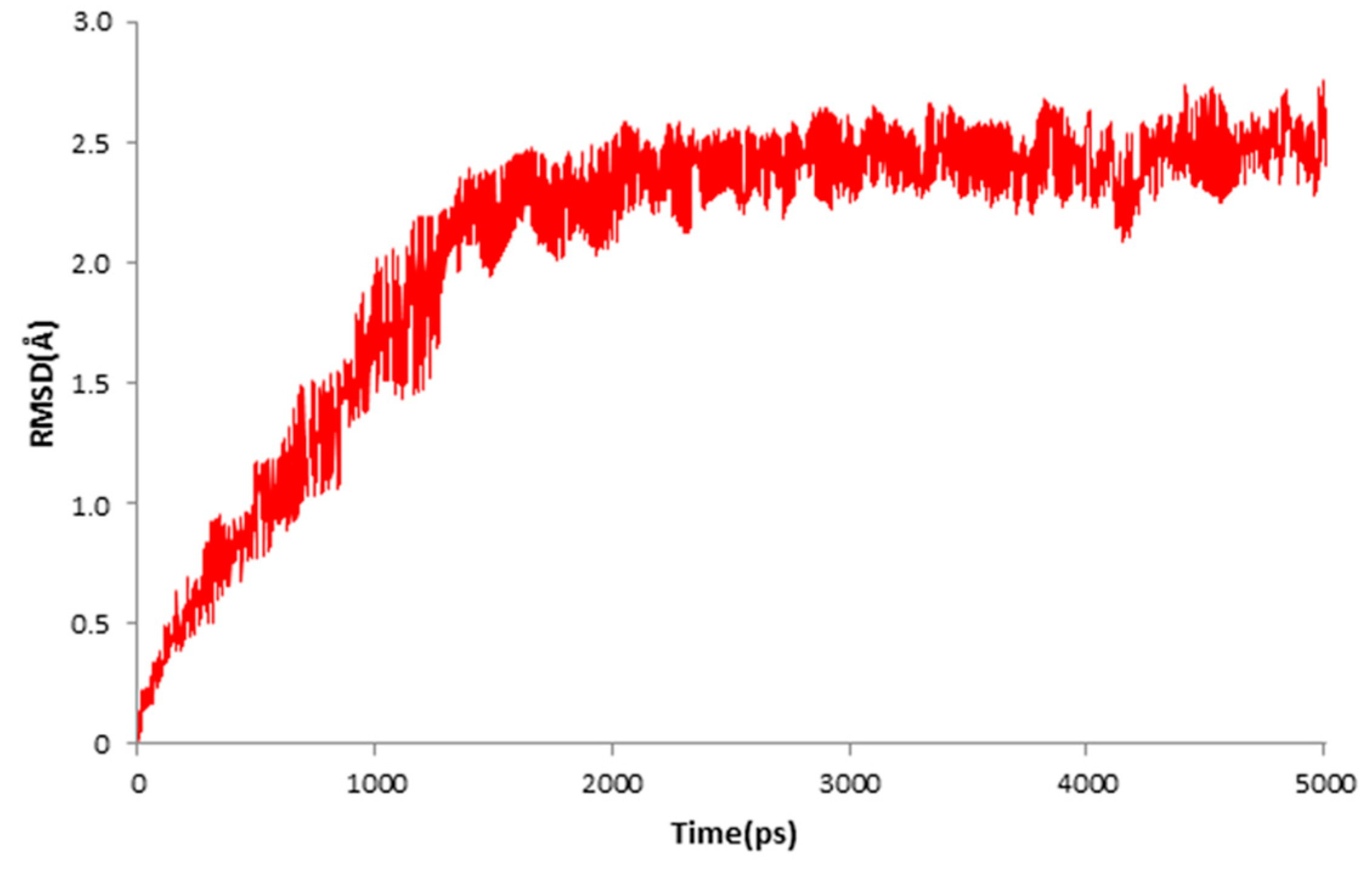
2. Results
3. Discussion
4. Materials and Methods
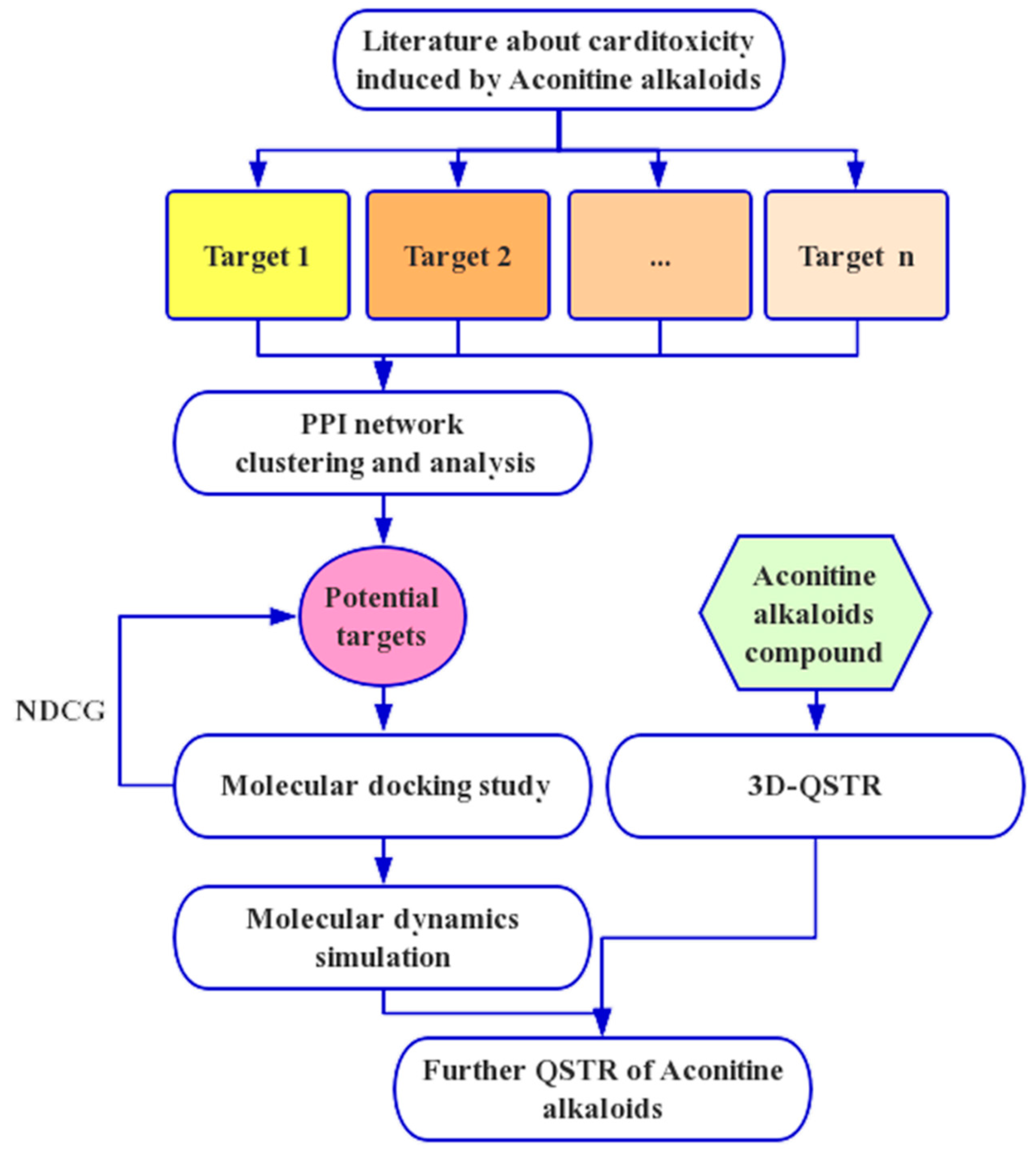
4.1. Network Analysis
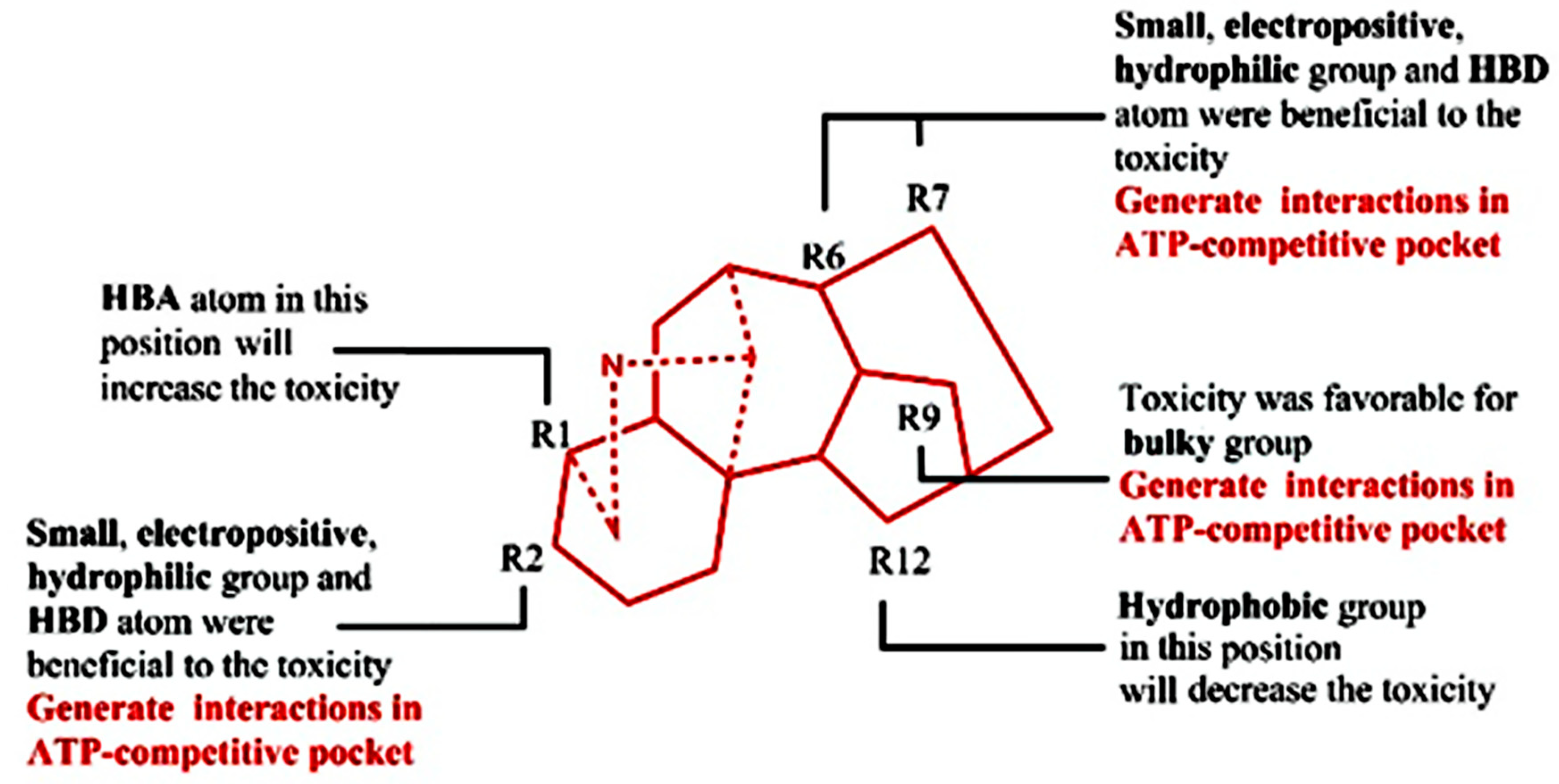
4.2. QSTR Research
4.3. Molecular Docking and Dynamics Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Atal, C.K.; Sharma, M.L.; Kaul, A.; Khajuria, A. Immunomodulating agents of plant origin. I: Preliminary screening. J. Ethnopharmacol. 1986, 18, 133–141. [Google Scholar] [CrossRef]
- Tang, W.; Eisenbrand, G. Chinese Drugs Plant Origin; Springer: Berlin/Heidelberg, Germany, 1992; pp. 997–1002. [Google Scholar]
- Chan, T.Y. Aconitine poisoning: A global perspective. Vet. Hum. Toxicol. 1994, 36, 326–328. [Google Scholar] [PubMed]
- Dhesi, P.; Ng, R.; Shehata, M.M.; Shah, P.K. Ventricular tachycardia after ingestion of ayurveda herbal antidiarrheal medication containing aconitum. Arch. Internal Med. 2010, 170, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Dickens, P.; Tai, Y.T.; But, P.P.; Tomlinson, B.; Ng, H.K.; Yan, K.W. Fatal accidental aconitine poisoning following ingestion of Chinese herbal medicine: A report of two cases. Forensic Sci. Int. 1994, 67, 55–58. [Google Scholar] [CrossRef]
- Fujita, Y.; Terui, K.; Fujita, M.; Kakizaki, A.; Sato, N.; Oikawa, K.; Aoki, H.; Takahashi, K.; Endo, S. Five cases of aconite poisoning: Toxicokinetics of aconitines. J. Anal. Toxicol. 2007, 31, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Pullela, R.; Young, L.; Gallagher, B.; Avis, S.P.; Randell, E.W. A case of fatal aconitine poisoning by monkshood ingestion. J. Forensic Sci. 2010, 53, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, Y. Determination of aconitine and hypaconitine in gucixiaotong ye by capillary electrophoresis with field-amplified sample injection. China J. Chin. Mater. Med. 2010, 35, 3287–3290. [Google Scholar]
- Chen, M.G.; Wang, Q.H.; Lin, W.; Lin, Y.B.; Chen, K.L.; Liu, F.; Lin, Q.; Lin, H.Y.; Cai, H.D. A clinical study in epidural injection with lappaconitine compound for post-operative analgesia. Chin. J. Integr. Tradit. West. Med. 1997, 3, 257–260. [Google Scholar]
- Kobayashi, M.; Takahashi, H.; Herndon, D.N.; Pollard, R.B.; Suzuki, F. Therapeutic effects of il-12 combined with benzoylmesaconine, a non-toxic aconitine-hydrolysate, against herpes simplex virus type 1 infection in mice following thermal injury. Burns 2003, 29, 37–42. [Google Scholar] [CrossRef]
- Li, X.; Gu, L.; Yang, L.; Zhang, D.; Shen, J. Aconitine: A potential novel treatment for systemic lupus erythematosus. J. Pharmacol. Sci. 2017, 133, 115. [Google Scholar] [CrossRef] [PubMed]
- Pyaskovskaya, O.N.; Boychuk, I.V.; Fedorchuk, A.G.; Kolesnik, D.L.; Dasyukevich, O.I.; Solyanik, G.I. Aconitine-containing agent enhances antitumor activity of dichloroacetate against ehrlich carcinoma. Exp. Oncol. 2015, 37, 192–196. [Google Scholar] [PubMed]
- Liu, G.; Wong, L.; Chua, H.N. Complex discovery from weighted ppi networks. Bioinformatics 2009, 25, 1891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Su, S.; Bhatnagar, R.K.; Hassett, D.J.; Lu, L.J. Prediction and analysis of the protein interactome in pseudomonas aeruginosa to enable network-based drug target selection. PLoS ONE 2012, 7, e41202. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The string database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ning, X.; Zhang, X.S. Identification of functional modules in a ppi network by clique percolation clustering. Comput. Biol. Chem. 2006, 30, 445. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, Y.; Niu, Z.; Wu, F.X. United complex centrality for identification of essential proteins from ppi networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.C.; Cui, Y.P.; Wu, J.T.; Wang, J.M.; Liu, Q.Z.; Gao, Z.L. The ppi network and cluster one analysis to explain the mechanism of bladder cancer. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 618–623. [Google Scholar] [PubMed]
- Le, G.H.; Guo, J.X. The progress of novel drug delivery systems. Acta Pharm. Sin. 2017, 52, 181–188. [Google Scholar]
- Berardi, M.J.; Shih, W.M.; Harrison, S.C.; Chou, J.J. Mitochondrial uncoupling protein 2 structure determined by nmr molecular fragment searching. Nature 2011, 476, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G. Structural basis for membrane anchoring of hiv-1 envelope spike. Science 2016, 353, 172. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Xie, S.; Berardi, M.J.; Zhao, X.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis c virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the m2 proton channel of influenza a virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.C.; Scotti, L.; Feitosa, A.M.; Mf, F.M.D.; Scotti, M.T. Computer-aided drug design using sesquiterpene lactones as sources of new structures with potential activity against infectious neglected diseases. Molecules 2017, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Shi, Z.; Fallahi, M.; Hammond, G.L. Successful in silico discovery of novel nonsteroidal ligands for human sex hormone binding globulin. J. Med. Chem. 2005, 48, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Jujjavarapu, S.E.; Dhagat, S. In silico discovery of novel ligands for antimicrobial lipopeptides for computer-aided drug design. Probiotics Antimicrob. Proteins 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem. 2004, 11, 2105–2134. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Coupling interaction between thromboxane a2 receptor and alpha-13 subunit of guanine nucleotide-binding protein. J. Proteome Res. 2005, 4, 1681. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Jones, D.; Heinrikson, R.L. Prediction of the tertiary structure and substrate binding site of caspase-8. FEBS Lett. 1997, 419, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Q.; Du, Q.S.; Chou, K.C. Study of drug resistance of chicken influenza a virus (h5n1) from homology-modeled 3d structures of neuraminidases. Biochem. Biophys. Res. Commun. 2007, 354, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Du, Q.S.; Huang, R.B.; Zhang, D.W.; Chou, K.C. Insights from investigating the interaction of oseltamivir (tamiflu)with neuraminidase of the 2009 h1 n1 swine flu virus. Biochem. Biophys. Res. Commun. 2009, 386, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Tomasselli, A.G.; Heinrikson, R.L. Prediction of the tertiary structure of a caspase-9/inhibitor complex. FEBS Lett. 2000, 470, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS ONE 2012, 7, e38546. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Mezey, P.G.; Chou, K.C. Heuristic molecular lipophilicity potential (hmlp): A 2d-qsar study to ladh of molecular family pyrazole and derivatives. J. Comput. Chem. 2005, 26, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Huang, R.B.; Wei, Y.T.; Pang, Z.W.; Du, L.Q.; Chou, K.C. Fragment-based quantitative structure & ndash; activity relationship (fb-qsar) for fragment-based drug design. J. Comput. Chem. 2009, 30, 295–304. [Google Scholar] [PubMed]
- Wei, H.; Wang, C.H.; Du, Q.S.; Meng, J.; Chou, K.C. Investigation into adamantane-based m2 inhibitors with fb-qsar. Med. Chem. 2009, 5, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Deaayuela, M.A.; Pérezcastillo, Y.; Menesesmarcel, A.; Ubeira, F.M.; Bolasfernández, F.; Chou, K.C.; Gonzálezdíaz, H. Hp-lattice qsar for dynein proteins: Experimental proteomics (2d-electrophoresis, mass spectrometry) and theoretic study of a leishmania infantum sequence. Bioorg. Med. Chem. 2008, 16, 7770–7776. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.Y.; Forsen, S. The biological functions of low-frequency phonons: 2. Cooperative effects. Chem. Scr. 1981, 18, 126–132. [Google Scholar]
- Chou, K.C. Low-frequency collective motion in biomacromolecules and its biological functions. Biophys. Chem. 1988, 30, 3–48. [Google Scholar] [CrossRef]
- Chou, K.C.; Maggiora, G.M.; Mao, B. Quasi-continuum models of twist-like and accordion-like low-frequency motions in DNA. Biophys. J. 1989, 56, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Kuo-Chen, C.; Mao, B. Collective motion in DNA and its role in drug intercalation. Biopolymers 2010, 27, 1795–1815. [Google Scholar]
- Martel, P. Biophysical aspects of neutron scattering from vibrational modes of proteins. Progr. Biophys. Mol. Biol. 1992, 57, 129–179. [Google Scholar] [CrossRef]
- Zhou, G.P. Biological functions of soliton and extra electron motion in DNA structure. Phys. Scr. 1989, 40, 698. [Google Scholar] [CrossRef]
- Chou, K.C. Low-frequency resonance and cooperativity of hemoglobin. Trends Biochem. Sci. 1989, 14, 212. [Google Scholar] [CrossRef]
- Chou, K.C.; Zhang, C.T.; Maggiora, G.M. Solitary wave dynamics as a mechanism for explaining the internal motion during microtubule growth. Biopolymers 2010, 34, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.A. Designed electromagnetic pulsed therapy: Clinical applications. J. Cell. Physiol. 2007, 216, 851. [Google Scholar] [CrossRef]
- Madkan, A.; Blank, M.; Elson, E.; Chou, K.C.; Geddis, M.S.; Goodman, R. Steps to the clinic with elf emf. Nat. Sci. 2009, 1, 157–165. [Google Scholar] [CrossRef]
- Zhu, Z.; Schuster, D.I.; Tuckerman, M.E. Molecular dynamics study of the connection between flap closing and binding of fullerene-based inhibitors of the hiv-1 protease. Biochemistry 2003, 42, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Gong, K.; Wei, D.Q.; Li, Y.X.; Chou, K.C. Molecular dynamics studies on the interactions of ptp1b with inhibitors: From the first phosphate-binding site to the second one. Protein Eng. Des. Sel. 2009, 22, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H. The cambridge structural database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (comsia) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, C.; Cai, B.C.; Lu, Y.M.; Wu, H. Single channel analysis of aconitine blockade of calcium channels in rat myocardiocytes. Acta Pharm. Sin. 1995, 30, 168–171. [Google Scholar]
- Hardick, D.J.; Cooper, G.; Scottward, T.; Blagbrough, I.S.; Potter, B.V.; Wonnacott, S. Conversion of the sodium channel activator aconitine into a potent alpha 7-selective nicotinic ligand. FEBS Lett. 1995, 365, 79–82. [Google Scholar] [CrossRef]
- Li, Y.; Tu, D.; Xiao, H.; Du, Y.; Zou, A.; Liao, Y.; Dong, S. Aconitine blocks herg and kv1.5 potassium channels. J. Ethnopharmacol. 2010, 131, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Laver, D.; Lamb, G. Inactivation of Ca2+ release channels (ryanodine receptors ryr1 and ryr2) with rapid steps in [Ca2+] and voltage. Biophys. J. 1998, 74, 2352–2364. [Google Scholar] [CrossRef]
- Pan, Y.; Zvaritch, E.; Tupling, A.R.; Rice, W.J.; De, L.S.; Rudnicki, M.; Mckerlie, C.; Banwell, B.L.; Maclennan, D.H. Targeted disruption of the atp2a1 gene encoding the sarco(endo)plasmic reticulum Ca2+ atpase isoform 1 (serca1) impairs diaphragm function and is lethal in neonatal mice. J. Biol. Chem. 2003, 278, 13367–13375. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.H.; Thompson, W.J.; Lee, A.K.; Strada, S.J. Cyclic gmp-dependent protein kinase activity in rat pulmonary microvascular endothelial cells. Biochem. Biophys. Res. Commun. 1994, 202, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Yasufukutakano, J.; Kozasa, T.; Nakajima, S.; Nakajima, Y. Different g proteins mediate somatostatin-induced inward rectifier K+ currents in murine brain and endocrine cells. J. Physiol. 1997, 502, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chu, G.; Kranias, E.G.; Bers, D.M. Cardiac myocyte calcium transport in phospholamban knockout mouse: Relaxation and endogenous camkii effects. Am. J. Physiol. 1998, 274, H1335. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, T.; Wang, W.; Culter, M.J.; Wang, Q.; Voigt, N.; Rosenbaum, D.S.; Dobrev, D.; Wehrens, X.H.T. Inhibition of camkii phosphorylation of ryr2 prevents induction of atrial fibrillation in fkbp12.6 knock-out mice. Circ. Res. 2012, 110, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Livshitz, L.M.; Rudy, Y. Regulation of Ca2+ and electrical alternans in cardiac myocytes: Role of camkii and repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2854. [Google Scholar] [CrossRef] [PubMed]
- Couchonnal, L.F.; Anderson, M.E. The role of calmodulin kinase ii in myocardial physiology and disease. Physiology 2008, 23, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ashpole, N.M.; Hudmon, A. Excitotoxic neuroprotection and vulnerability with camkii inhibition. Mol. Cell. Neurosci. 2011, 46, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Rellos, P.; Pike, A.C.; Niesen, F.H.; Salah, E.; Lee, W.H.; Von, D.F.; Knapp, S. Structure of the camkiiδ/calmodulin complex reveals the molecular mechanism of camkii kinase activation. PLoS Biol. 2010, 8, e1000426. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Watenpaugh, K.D.; Heinrikson, R.L. A model of the complex between cyclin-dependent kinase 5 and the activation domain of neuronal cdk5 activator. Biochem. Biophys. Res. Commun. 1999, 259, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Wei, D.Q.; Zhong, W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against sars. Biochem. Biophys. Res. Commun. 2003, 308, 148–151. [Google Scholar] [CrossRef]
- Huang, R.B.; Du, Q.S.; Wang, C.H.; Chou, K.C. An in-depth analysis of the biological functional studies based on the nmr m2 channel structure of influenza a virus. Biochem. Biophys. Res. Commun. 2008, 377, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Chou, K. Molecular therapeutic target for type-2 diabetes. J. Proteome Res. 2004, 3, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Novel inhibitor design for hemagglutinin against h1n1 influenza virus by core hopping method. PLoS ONE 2011, 6, e28111. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P. The string database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2010, 39, D561. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. J. Gen. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein–protein interaction networks. Nat. Methods 2012, 9, 471. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. Cytonca: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E.; Rodríguez-Velázquez, J.A. Subgraph centrality and clustering in complex hyper-networks. Phys. A Stat. Mech. Appl. 2006, 364, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Prountzos, D.; Pingali, K. Betweenness centrality. ACM Sigplan. Not. 2013, 48. [Google Scholar] [CrossRef]
- Okamoto, K.; Chen, W.; Li, X.Y. Ranking closeness centrality for large-scale social networks. In International Workshop on Rontiers in Algorithmics; Springer: Berlin/Heidelberg, Germany, 2008; pp. 186–195. [Google Scholar]
- Wang, X.; Pan, C.; Gong, J.; Liu, X.; Li, H. Enhancing the enrichment of pharmacophore-based target prediction for the polypharmacological profiles of drugs. J. Chem. Inf. Model. 2016, 56, 1175. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (comfa). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Bush, B.L.; Nachbar, R.B., Jr. Sample-distance partial least squares: Pls optimized for many variables, with application to comfa. J. Comput. Aided Mol. Des. 1993, 7, 587–619. [Google Scholar] [CrossRef] [PubMed]
- Bello Ramirez, A.M.; Nava Ocampo, A.A. A qsar analysis of toxicity of aconitum alkaloids. Fundam. Clin. Pharmacol. 2004, 18, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Huang, R.B.; Chou, K.C. Recent advances in qsar and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr. Protein Peptide Sci. 2008, 9, 248–259. [Google Scholar] [CrossRef]
- Pradoprado, F.J.; de la Vega, O.M.; Uriarte, E.; Ubeira, F.M.; Chou, K.C.; Gonzálezdíaz, H. Unified qsar approach to antimicrobials. 4. Multi-target qsar modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg. Med. Chem. 2009, 17, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Virupaksha, B.; Alpana, G. Comfa qsar models of camptothecin analogues based on the distinctive sar features of combined abc, cd and e ring substitutions. Comput. Biol. Med. 2012, 42, 890. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, D.; Mangiatordi, G.F.; Catto, M.; Carotti, A.; Nicolotti, O. Applicability domain for qsar models: Where theory meets reality. Int. J. Quant. Struct. Prop. Relat. 2016. [Google Scholar] [CrossRef]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of qsar models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, B.; Rasouli, Z.; Hassanzadeh, Z.; Ghavami, R. Molecular docking and qsar analysis of naphthyridone derivatives as atad2 bromodomain inhibitors: Application of comfa, ls-svm, and rbf neural network. Med. Chem. Res. 2016, 25, 1–11. [Google Scholar] [CrossRef]
- Qiao, Y.Y.; Guo, S. Concise applications of molecular modeling software-moe. Comput. Appl. Chem. 2005, 2, 157–160. [Google Scholar]
- Vilar, S.; Cozza, G.S. Medicinal chemistry and the molecular operating environment (moe): Application of qsar and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Foroozesh, M.; Stevens, C.L.K. Qsar models of cytochrome p450 enzyme 1a2 inhibitors using comfa, comsia and hqsar. SAR QSAR Environ. Res. 2011, 22, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.Z.; Aono, M.; Seddiqui, M.H. Estimating a Ranked List of Human Hereditary Diseases for Clinical Phenotypes by Using Weighted Bipartite Network. In Proceedings of the 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Osaka, Japan, 3–7 July 2013. [Google Scholar] [CrossRef]
- Phillips, J.C.; Zheng, G.; Kumar, S.; Kalé, L.V. Namd: Biomolecular simulation on thousands processors. In Proceedings of the 2002 ACM/IEEE Conference on Supercomputing, Baltimore, MD, USA, 16–22 November 2002. [Google Scholar] [CrossRef]
- Farrokhnia, M.; Mahnam, K. Molecular dynamics and docking investigations of several zoanthamine-type marine alkaloids as matrix metaloproteinase-1 inhibitors. Iran. J. Pharm. Res. 2017, 16, 173–186. [Google Scholar] [PubMed]
- Laudadio, E.; Mobbili, G.; Minnelli, C.; Massaccesi, L.; Galeazzi, R. Salts influence cathechins and flavonoids encapsulation in liposomes: A molecular dynamics investigation. Mol. Inf. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Review: Recent advances in developing web-servers for predicting protein attributes. Nat. Sci. 2009, 1, 63–92. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. Irna-ai: Identifying the adenosine to inosine editing sites in rna sequences. Oncotarget 2016, 8, 4208. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. Iss-psednc: Identifying splicing sites using pseudo dinucleotide composition. Biomed. Res. Int. 2014, 2014, 623149. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.C. Irna-pseu: Identifying rna pseudouridine sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar] [PubMed]
- Cheng, X.; Xiao, X.; Chou, K.C. Ploc-mplant: Predict subcellular localization of multi-location plant proteins by incorporating the optimal go information into general pseaac. Gene 2017, 13, 1722–1727. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xiao, X.; Chou, K.C. Ploc-mhum: Predict subcellular localization of multi-location human proteins via general pseaac to winnow out the crucial go information. Bioinformatics 2018, 34, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhao, S.G.; Xiao, X.; Chou, K.C. Iatc-misf: A multi-label classifier for predicting the classes of anatomical therapeutic chemicals. Bioinformatics 2017, 33, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. Psuc-lys: Predict lysine succinylation sites in proteins with pseaac and ensemble random forest approach. J. Theor. Biol. 2016, 394, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Jiang, S.Y.; Xu, Z.C.; Xiao, X.; Chou, K.C. Irnam5c-psednc: Identifying rna 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget 2017, 8, 41178–41188. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Sun, B.Q.; Xiao, X.; Xu, Z.C.; Jia, J.H.; Chou, K.C. Ikcr-pseens: Identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics 2017, 110, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Hui, D.; Feng, P.; Hao, L.; Chou, K.C. Iacp: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar]
- Xiang, C.; Xuan, X.; Chou, K.C. Ploc-meuk: Predict subcellular localization of multi-label eukaryotic proteins by extracting the key go information into general pseaac. Genomics 2017, 110, 50. [Google Scholar]
- Xiang, C.; Zhao, S.G.; Xuan, X.; Chou, K.C. Iatc-mhyb: A hybrid multi-label classifier for predicting the classification of anatomical therapeutic chemicals. Oncotarget 2017, 8, 58494–58503. [Google Scholar]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. Ihsp-pseraaac: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. Irna-psecoll: Identifying the occurrence sites of different rna modifications by incorporating collective effects of nucleotides into pseknc. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015, 11, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr. Top. Med. Chem. 2017, 17, 2337–2358. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistical Parameters | CoMFA | CoMSIA |
|---|---|---|
| q2 a | 0.624 | 0.719 |
| r2 b | 0.966 | 0.901 |
| ONC c | 6 | 4 |
| SEE d | 0.043 | 0.116 |
| F e | 124.127 | 157.458 |
| rpred2 f | 0.903 | 0.894 |
| Fraction of Field contribution g | ||
| steric | 0.621 | 0.120 |
| Electrostatic | 0.379 | 0.204 |
| Hydrophobic | - | 0.327 |
| H-bond acceptor | - | 0.216 |
| H-bond donor | - | 0.133 |
| Name | Classification | Frequency |
|---|---|---|
| RYR2 | Ryanodine receptor 2 | 19 |
| RYR1 | Ryanodine receptor 1 | 15 |
| GJA1 | Gap junction α-1 protein (connexin43) | 13 |
| SLC8A1 | Sodium/calcium exchanger 1 | 11 |
| ATP2A1 | Calcium transporting ATPase fast twitch 1 | 9 |
| KCNH2 | Potassium voltage-gated channel H2 | 7 |
| SCN3A | Sodium voltage-gated channel type 3, | 3 |
| SCN2A | Sodium voltage-gated channel type 2 | 3 |
| SCN8A | Sodium voltage-gated channel type 8 | 2 |
| SCN1A | Sodium voltage-gated channel type 1 | 2 |
| SCN4A | Sodium voltage-gated channel type 4 | 1 |
| KCNJ3 | Potassium inwardly-rectifying channel J3 | 1 |
| Compounds | Experimental pLD50 | Fit Score (2V7O) | Fit Score (2VZ6) |
|---|---|---|---|
| 6 | 1 | 3 | 3 |
| 20 | 2 | 1 | 12 |
| 12 | 3 | 4 | 9 |
| 1 | 4 | 2 | 4 |
| 11 | 5 | 7 | 2 |
| 14 | 6 | 8 | 13 |
| 16 | 7 | 5 | 6 |
| 7 | 8 | 17 | 15 |
| 8 | 9 | 10 | 11 |
| 27 | 10 | 23 | 17 |
| 13 | 11 | 12 | 19 |
| 15 | 12 | 11 | 5 |
| 32 | 13 | 18 | 18 |
| 5 | 14 | 22 | 8 |
| 33 | 15 | 13 | 29 |
| 21 | 16 | 15 | 1 |
| 25 | 17 | 9 | 20 |
| 22 | 18 | 25 | 25 |
| 17 | 19 | 20 | 16 |
| 28 | 20 | 24 | 30 |
| 9 | 21 | 16 | 32 |
| 29 | 22 | 32 | 14 |
| 2 | 23 | 30 | 24 |
| 30 | 24 | 31 | 26 |
| 18 | 25 | 21 | 27 |
| 10 | 26 | 26 | 21 |
| 23 | 27 | 29 | 31 |
| 31 | 28 | 33 | 7 |
| 26 | 29 | 14 | 23 |
| 4 | 30 | 28 | 33 |
| 3 | 31 | 6 | 10 |
| 19 | 32 | 27 | 28 |
| 24 | 33 | 19 | 22 |
| NDCG | 1 | 0.9122 | 0.8503 |

| No. | CAS. NO | Substituent in R1 to R13 | pLD50 |
|---|---|---|---|
| 1 | 302-27-2 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-ethyl | 4.92 |
| 2* | 545-56-2 | methoxymethyl-H-hydroxy-methoxy-hydroxy-hydroxy-H-methoxy-hydroxy-H-H-H-ethyl | 1.96 |
| 3 | 127-29-7 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-H-methoxy-methyl 2,3-dimethoxybenzoate-H-hydroxy-H-ethyl | 1.44 |
| 4 | 509-18-2 | methoxymethyl-H-hydroxy-methoxy-hydroxy-hydroxy-H-methoxy-methoxy-H-H-H-ethyl | 1.76 |
| 5* | 466-24-0 | methoxymethyl-hydroxy-methoxy-methoxy-H-hydroxy-hydroxy-methoxy-benzoxy-H-hydroxy-H-ethyl | 3.00 |
| 6 | 2752-64-9 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-methy | 5.00 |
| 7 | 4491-19-4 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-H-methoxy-benzoxy-H-hydroxy-H-ethyl | 4.33 |
| 8* | 6900-87-4 | methoxymethyl-H-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-methy | 4.33 |
| 9 | 1356-52-1 | H-H-methoxy-H-H-hydroxy-H-methoxy-benzoxy-H-H-hydroxy-H-ethyl | 2.55 |
| 10 | 6836-11-9 | methy-H-methoxy-acetoxyl-dioxolane-H-H-methoxy-methoxy-H-H-hydroxy-ethyl | 1.88 |
| 11 | 8006-38-0 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-ethyl | 4.78 |
| 12* | 20501-56-8 | methoxymethyl-H-methoxy-H-H-hydroxy-H-methoxy-hydroxy-H-H-H-ethyl | 4.94 |
| 13 | 21019-30-7 | 2-(3-methyl-2,5-dioxopyrrolidin-1-yl)benzoate ethyl-H-methoxy-methoxy-hydroxy-hydroxy-H-methoxy-methoxy-H-H-H-ethyl | 3.52 |
| 14 | 41849-35-8 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-hydroxy-ethyl | 4.66 |
| 15 | 26000-16-8 | 2-(3-methyl-2,5-dioxopyrrolidin-1-yl)benzoate ethyl-H-methoxy-methoxy-a-H-H-methoxy-methoxy-H-H-H-ethyl | 3.3 |
| 16 | 77181-26-1 | methoxymethyl-acetoxyl-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-ethyl | 4.4 |
| 17 | 71402-60-3 | methoxymethyl-hydroxy-methoxy-methoxy-H-hydroxy-hydroxy-methoxy-benzoxy-H-hydroxy-H-trimethylethanaminium | 2.59 |
| 18 | 67806-02-4 | methoxymethyl-acetoxyl-methoxy-methoxy-H-acetoxyl-acetoxyl-methoxy-benzoxy-H-acetoxyl-H-ethyl | 1.9 |
| 19 | 85031-25-0 | methoxymethyl-acetoxyl-methoxy-methoxy-H-acetoxyl-acetoxyl-methoxy-acetoxyl-H-acetoxyl-H-ethyl | 1.17 |
| 20 | 71425-64-4 | methoxymethyl-hydroxy-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-trimethylethanaminium | 4.95 |
| 21* | 63238-67-5 | methoxymethyl-hydroxy-methoxy-methoxy-H-hydroxy-hydroxy-methoxy-benzoxy-H-hydroxy-H-methy | 2.68 |
| 22 | 71402-61-4 | methoxymethyl-hydroxy-methoxy-methoxy-H-hydroxy-hydroxy-methoxy-benzoxy-hydroxy-H-H-trimethylethanaminium | 2.62 |
| 23 | 38146-89-3 | methoxymethyl-hydroxy-methoxy-methoxy-H-hydroxy-H-methoxy-hydroxy-H-hydroxy-H-ethyl | 1.85 |
| 24 | 82144-73-8 | methoxymethyl-acetoxyl-methoxy-methoxy-H-acetoxyl-H-methoxy-benzoxy-H-hydroxy-H-ethyl | 0.84 |
| 25 | 82144-74-9 | methoxymethyl-acetoxyl-methoxy-methoxy-H-acetoxyl-H-methoxy-benzoxy-H-acetoxyl-H-ethyl | 2.66 |
| 26 | 38146-91-7 | methoxymethyl-acetoxyl-methoxy-methoxy-H-acetoxyl-H-methoxy-acetoxyl-H-acetoxyl-H-ethyl | 1.82 |
| 27* | 71402-59-0 | methoxymethyl-H-methoxy-methoxy-H-acetoxyl-hydroxy-methoxy-benzoxy-H-hydroxy-H-ethyl | 4.27 |
| 28 | 71402-62-5 | methoxymethyl-H-hydroxy-methoxy-H-hydroxy-hydroxy-methoxy-benzoxy-H-hydroxy-H-methy | 2.59 |
| 29 | 39089-30-0 | methy-H-hydroxy-H-H-hydroxy-H-methoxy-hydroxy-H-H-H-ethyl | 2.29 |
| 30 | 58111-33-4 | methoxymethyl-H-hydroxy-methoxy-hydroxy-hydroxy-H-methoxy-H-methoxy-H-H-trimethylethanaminium | 1.93 |
| 31* | 23943-93-3 | hydroxy-H-methoxy-hydroxy-H-hydroxy-H-methoxy-methoxy-H-H-H-ethyl | 1.85 |
| 32 | 32854-75-4 | 2-acetamidobenzoate ethyl-H-methoxy-H-H-hydroxy-H-methoxy-methoxy-hydroxy-H-H-ethyl | 3.16 |
| 33 | 138729-51-8 | 2-acetamidobenzoate ethyl-H-methoxy-H-H-acetoxyl-H-methoxy-methoxy-acetoxyl-H-H-ethyl | 2.84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.-Y.; Liang, J.-W.; Olounfeh, K.M.; Sun, Q.; Zhao, N.; Meng, F.-H. A Comprehensive In Silico Method to Study the QSTR of the Aconitine Alkaloids for Designing Novel Drugs. Molecules 2018, 23, 2385. https://doi.org/10.3390/molecules23092385
Wang M-Y, Liang J-W, Olounfeh KM, Sun Q, Zhao N, Meng F-H. A Comprehensive In Silico Method to Study the QSTR of the Aconitine Alkaloids for Designing Novel Drugs. Molecules. 2018; 23(9):2385. https://doi.org/10.3390/molecules23092385
Chicago/Turabian StyleWang, Ming-Yang, Jing-Wei Liang, Kamara Mohamed Olounfeh, Qi Sun, Nan Zhao, and Fan-Hao Meng. 2018. "A Comprehensive In Silico Method to Study the QSTR of the Aconitine Alkaloids for Designing Novel Drugs" Molecules 23, no. 9: 2385. https://doi.org/10.3390/molecules23092385