Novel Protein Kinase Inhibitors Related to Tau Pathology Modulate Tau Protein-Self Interaction Using a Luciferase Complementation Assay

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of the Benzofuropyridines
2.2. Protein Kinase Inhibition
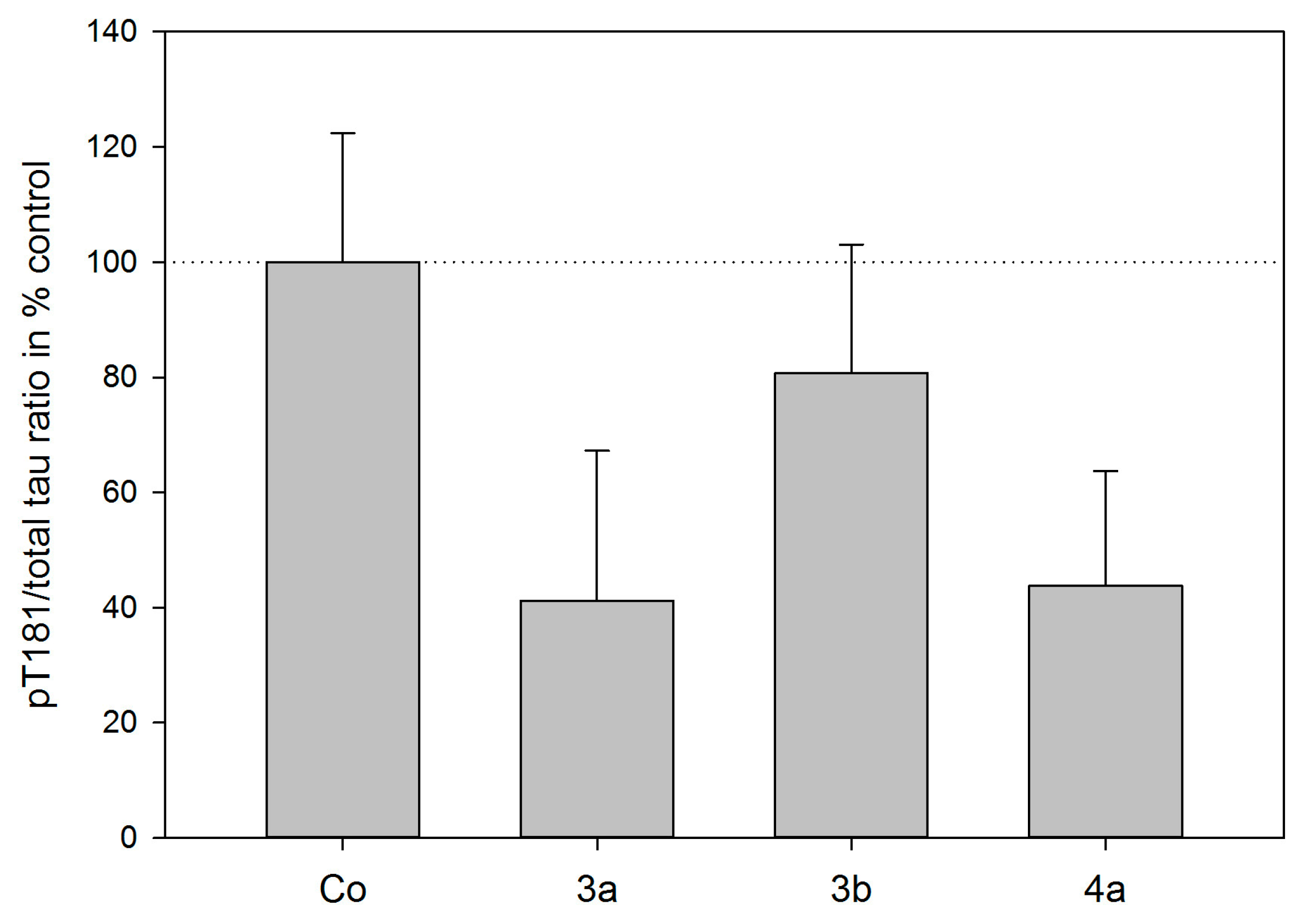
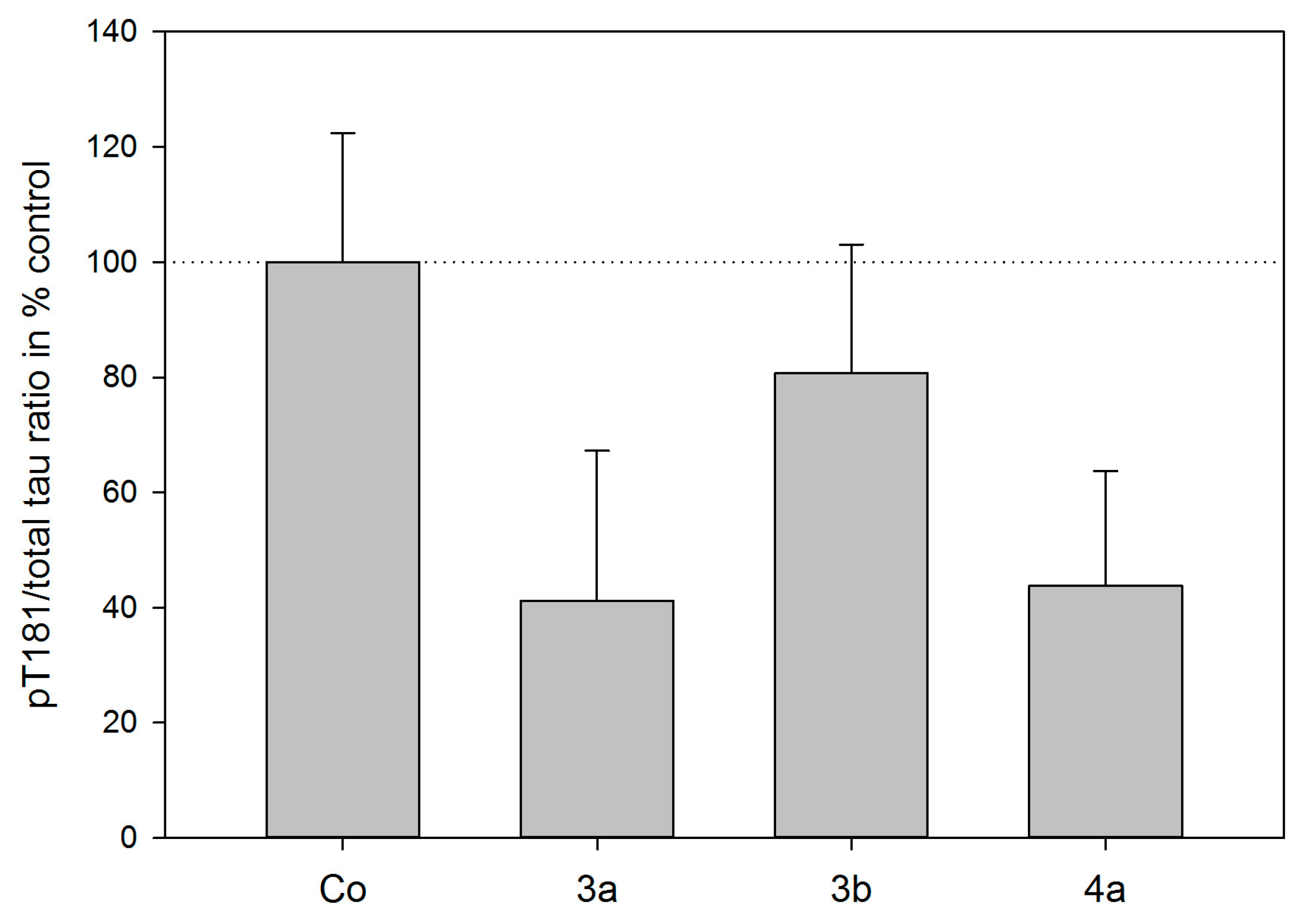
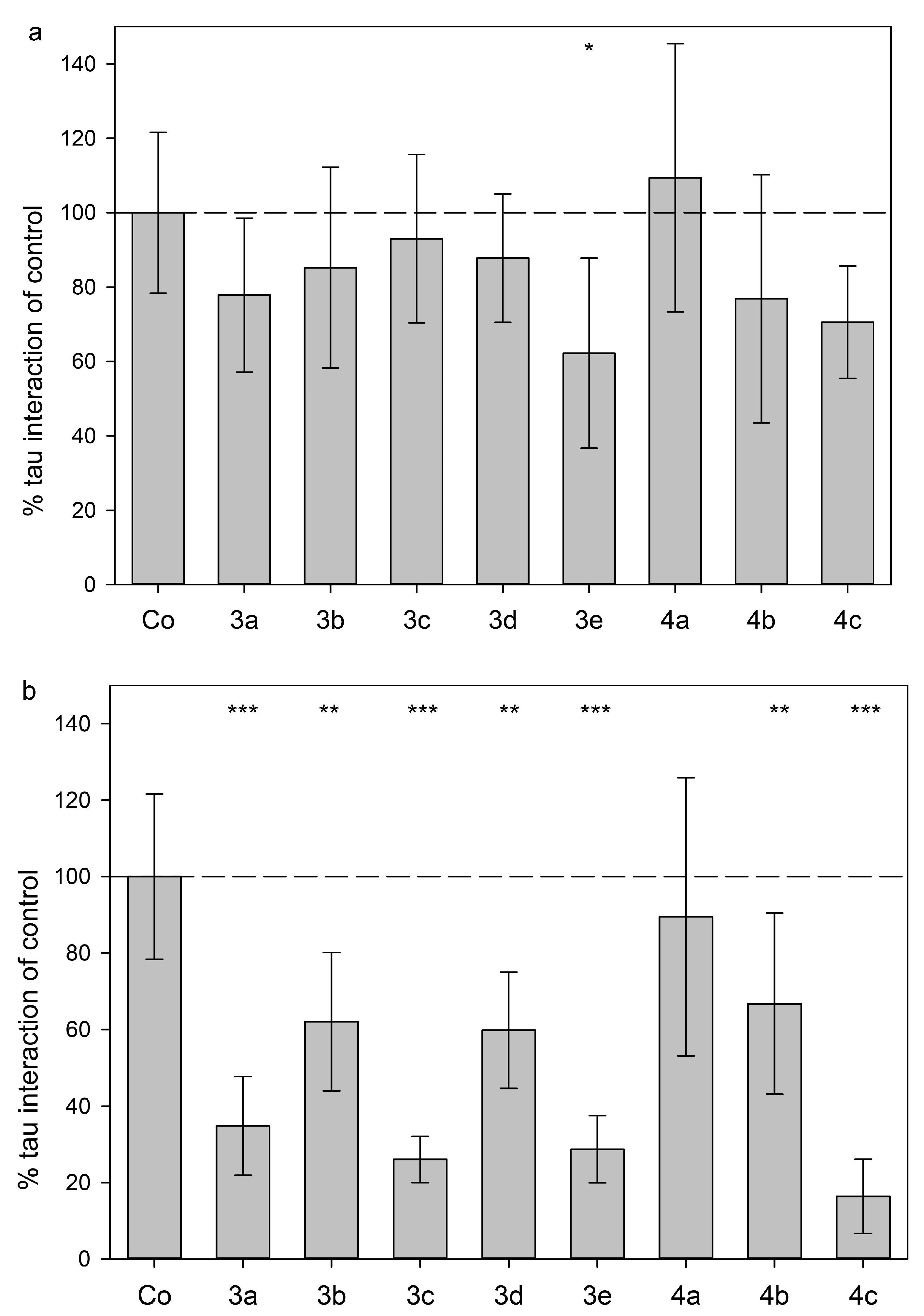
2.3. Tau Protein Interaction Assay
3. Material and Methods
3.1. Chemical Reagents and Instruments
3.2. General Procedure for the Synthesis of Compounds 1
3.3. General Procedure for the Synthesis of Compounds 2
3.4. General Procedure for the Synthesis of Compounds 3 and 4
3.5. General Procedure for the Synthesis of 3-Hydroxy Compounds 3e and 4c
3.6. Protein Kinase Assay
3.7. Tau Protein Phosphorylation Assay
3.8. Tau Protein Interaction Assay
Cell Culture and Transfection
NanoBiT® Luciferase Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, A.; Nisha, C.M.; Silakari, C.; Sharma, I.; Anusha, K.; Gupta, N.; Nai, P.; Tripathi, T.; Kumar, A. Current and novel therapeutic molecules and targets in Alzheimer’s disease. J. Formos. Med. Assoc. 2016, 115, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, L.; Rosset, I.; Roriz-Cruz, M. Global Epidemiology of Dementia: Alzheimer’s and Vascular Types. BioMed Res. Int. 2014, 2014, 908915. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrigi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedowicz, D.M.; Nelson, P.T.; Murphy, M.P. Alzheimer’s disease: Pathological mechanisms and recent insights. Curr. Neuropharmacol. 2011, 9, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Bäckmann, L.; Jones, S.; Berger, A.K.; Laukka, E.J.; Smail, B.J. Multiple cognitive deficits during the transition to Alzheimer’s disease. J. Intern. Med. 2004, 256, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Coman, H.; Nemes, B. New Therapeutic Targets in Alzheimer’s Disease. Int. J. Gerontol. 2017, 11, 2–6. [Google Scholar] [CrossRef]
- Allsop, D.; Mayes, J. Amyloid β-peptides and Alzheimer’s disease. Essays Biochem. 2014, 56, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Mikulca, J.A.; Nguyen, V.; Gajdosik, D.A. Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J. Clin. Pharm. Ther. 2014, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Giardina, G.A. γ-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: Disappointments and hopes. Curr. Top. Med. Chem. 2011, 11, 1555–1570. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.; Rits, S. Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid Sporadic Alzheimer’s Disease. Med. Sci. 2018, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Delrieu, J.; Ousset, P.J.; Caillaud, C. Clinical trials in Alzheimer’s disease: Immunotherapy approaches. J. Neurochem. 2012, 120 (Suppl. S1), 186–193. [Google Scholar] [CrossRef] [PubMed]
- Delrieu, J.; Ousset, P.J.; Vellas, B. Gantenerumab for the treatment of Alzheimer’s disease. Expert Opin. Biol. Ther. 2012, 12, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Hanger, D.P.; Miller, C.C.J.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Leko, M.B.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Delacourte, A.; Sergeant, N.; Wattez, A. Tau aggregation in the hippocampal formation: An ageing or a pathological process? Exp. Gerontol. 2002, 37, 1291–1296. [Google Scholar] [CrossRef]
- Wang, D.; Fu, Q.; Zhou, Y. β2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J. Biol. Chem. 2013, 21, 394–402. [Google Scholar]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Meijer, L. Indirubins inhibit glycogen synthase kinase -3β and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Roxbee, C.L.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.C.; Jerning, E.; Markgren, P.O.; Borgegard, T.; et al. Structural insights and biological effects of glycogen synthase kinase specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Martinez, A. GSK3-inhibitors: Preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Hernández, F. GSK-3 inhibitors for Alzheimer’s disease. Exp. Rev. Neurotherap. 2007, 7, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen Synthase Kinase 3 Inhibitors in the Next Horizon for Alzheimer’s Disease Treatment. Int. J. Alzheimer’s Dis. 2011, 2011, 280502. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kudrimoti, S.; Prasanna, S.; Odde, S.; Doerksen, R.J.; Pennaka, H.K.; Choo, Y.-M.; Rao, K.V.; Tekwani, B.L.; Madgula, V.; et al. Structure-activity relationship and mechanism of action studies of manzamine analogues for the control of neuroinflammation and cerebral infections. J. Med. Chem. 2010, 53, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.-J.; Calero, M.; Andrés, M.V.; Gomez-Carillo, B.; Leon, T. A Phase II Trial of Tideglusib in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 1, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci. Signal. 2016, 9, ra110. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, A.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Ramos, M.T.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congdon, E.E.; Kim, S.; Bonchak, J.; Songrug, T.; Matzavinos, A.; Kuret, J. Nucleation-dependent tau filament formation: The importance of dimerization and an estimation of elementary rate constants. J. Biol. Chem. 2008, 283, 13806–13816. [Google Scholar] [CrossRef] [PubMed]
- Brachwitz, K.; Voigt, B.; Meijer, L.; Schächtele, C.; Molnár, J.; Hilgeroth, A. Evaluation of the first cytostatically active 1-aza-9-oxafluorenes as novel selective CDK1 inhibitors with P-glycoprotein modulating properties. J. Med. Chem. 2003, 46, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Ishiguro, K.; Hisanaga, S.-I. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Vincent, F.; Shah, K. Deregulated Cdk5 Triggers Aberrant Activation of Cell Cycle Kinases and Phosphatases Inducing Neuronal Death. J. Cell Sci. 2012, 125, 5124–5137. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.; Myers, A.; Hardy, J. The law of mass action applied to neurodegenerative disease: A hypothesis concerning the etiology and pathogenesis of complex diseases. Hum. Mol. Genet. 2004, 13, R123–R126. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hashi, K.; Hirata, Y.; Kiuchi, K. SOD1 dimerization monitoring using a novel split NanoLuc, NanoBit. Cell Biochem. Funct. 2016, 34, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberstadt, M.; Stieler, J.; Simpong, D.L.; Römuß, U.; Urban, N.; Schaefer, M.; Arendt, T.; Holzer, M. TDP-43 self-interaction is modulated by redox-active compounds Auranofin, Chelerythrine and Riluzole. Sci. Rep. 2018, 8, 2248. [Google Scholar] [CrossRef] [PubMed]
- Von Einem, B.; Eschbach, J.; Kiechle, M.; Wahler, A.; Thal, D.R.; McLean, P.J.; Weishaupt, J.H.; Ludolph, A.C.; von Arnim, C.A.F.; Danzer, K.M. The Golgi-localized, gamma ear-containing, ARF-binding (GGA) protein family alters alpha synuclein (α-syn) oligomerization and secretion. Aging (Albany NY) 2017, 9, 1677–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Cpd. | R | Protein Kinase Affinities (µM) a | ||
|---|---|---|---|---|
| cdk1 | cdk5/p25 | gsk-3β | ||
| 3a | OEt | 0.17 ± 0.031 | 0.46 ± 0.022 | 0.083 ± 0.034 |
| 3b | OBn | 2.30 ± 0.33 | n.a. b | 5.80 ± 1.20 |
| 3c | OMe | 0.14 ± 0.023 | 0.51 ± 0.012 | 0.062 ± 0.023 |
| 3d | F | 1.40 ± 0.24 | n.a. b | 0.38 ± 0.93 |
| 3e | OH | 0.013 ± 0.0031 | 0.11 ± 0.043 | 0.024 ± 0.0021 |
| 4a | OBn | 0.091 ± 0.012 | 2.10 ± 0.24 | 1.60 ± 0.23 |
| 4b | F | 1.06 ± 0.63 | 160 ± 34 | 190 ± 37 |
| 4c | OH | 0.77 ± 0.062 | 0.073 ± 0.012 | 0.012 ± 0.013 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holzer, M.; Schade, N.; Opitz, A.; Hilbrich, I.; Stieler, J.; Vogel, T.; Neukel, V.; Oberstadt, M.; Totzke, F.; Schächtele, C.; et al. Novel Protein Kinase Inhibitors Related to Tau Pathology Modulate Tau Protein-Self Interaction Using a Luciferase Complementation Assay. Molecules 2018, 23, 2335. https://doi.org/10.3390/molecules23092335
Holzer M, Schade N, Opitz A, Hilbrich I, Stieler J, Vogel T, Neukel V, Oberstadt M, Totzke F, Schächtele C, et al. Novel Protein Kinase Inhibitors Related to Tau Pathology Modulate Tau Protein-Self Interaction Using a Luciferase Complementation Assay. Molecules. 2018; 23(9):2335. https://doi.org/10.3390/molecules23092335
Chicago/Turabian StyleHolzer, Max, Nico Schade, Ansgar Opitz, Isabel Hilbrich, Jens Stieler, Tim Vogel, Valentina Neukel, Moritz Oberstadt, Frank Totzke, Christoph Schächtele, and et al. 2018. "Novel Protein Kinase Inhibitors Related to Tau Pathology Modulate Tau Protein-Self Interaction Using a Luciferase Complementation Assay" Molecules 23, no. 9: 2335. https://doi.org/10.3390/molecules23092335