Structural Characterization of the Lactobacillus Plantarum FlmC Protein Involved in Biofilm Formation

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
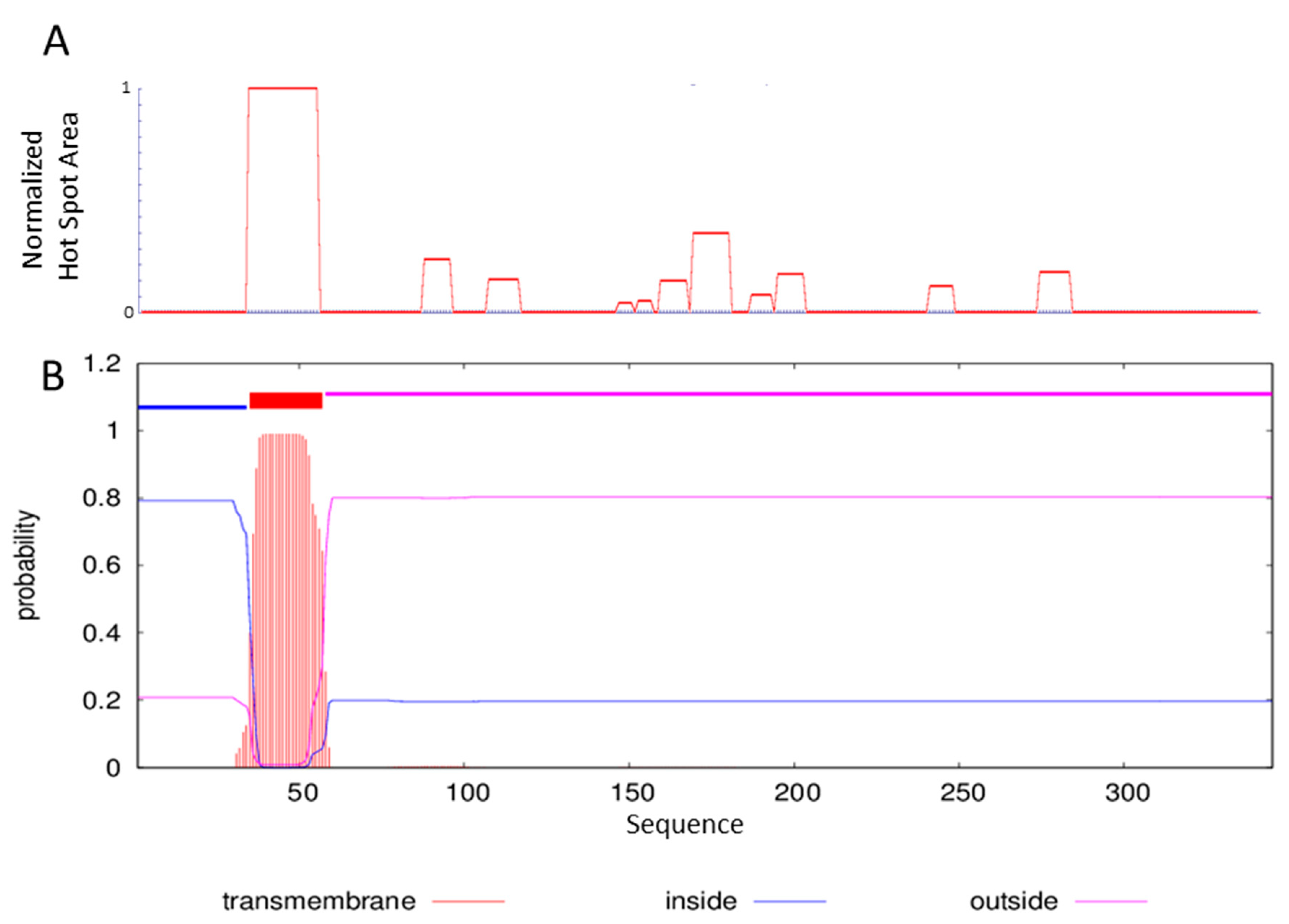
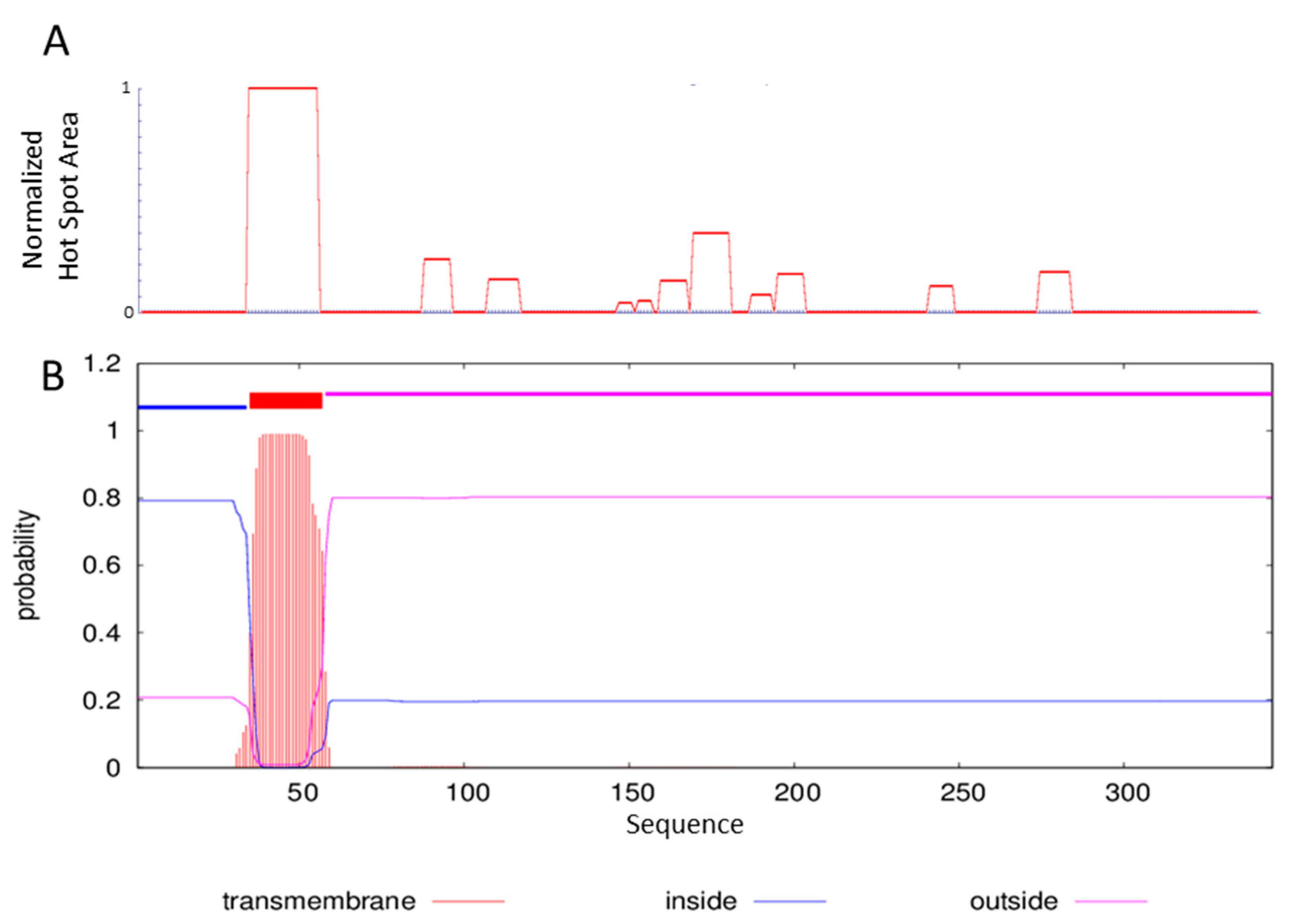
2.1. Sequence Analysis and ΔTM-FlmC Expression
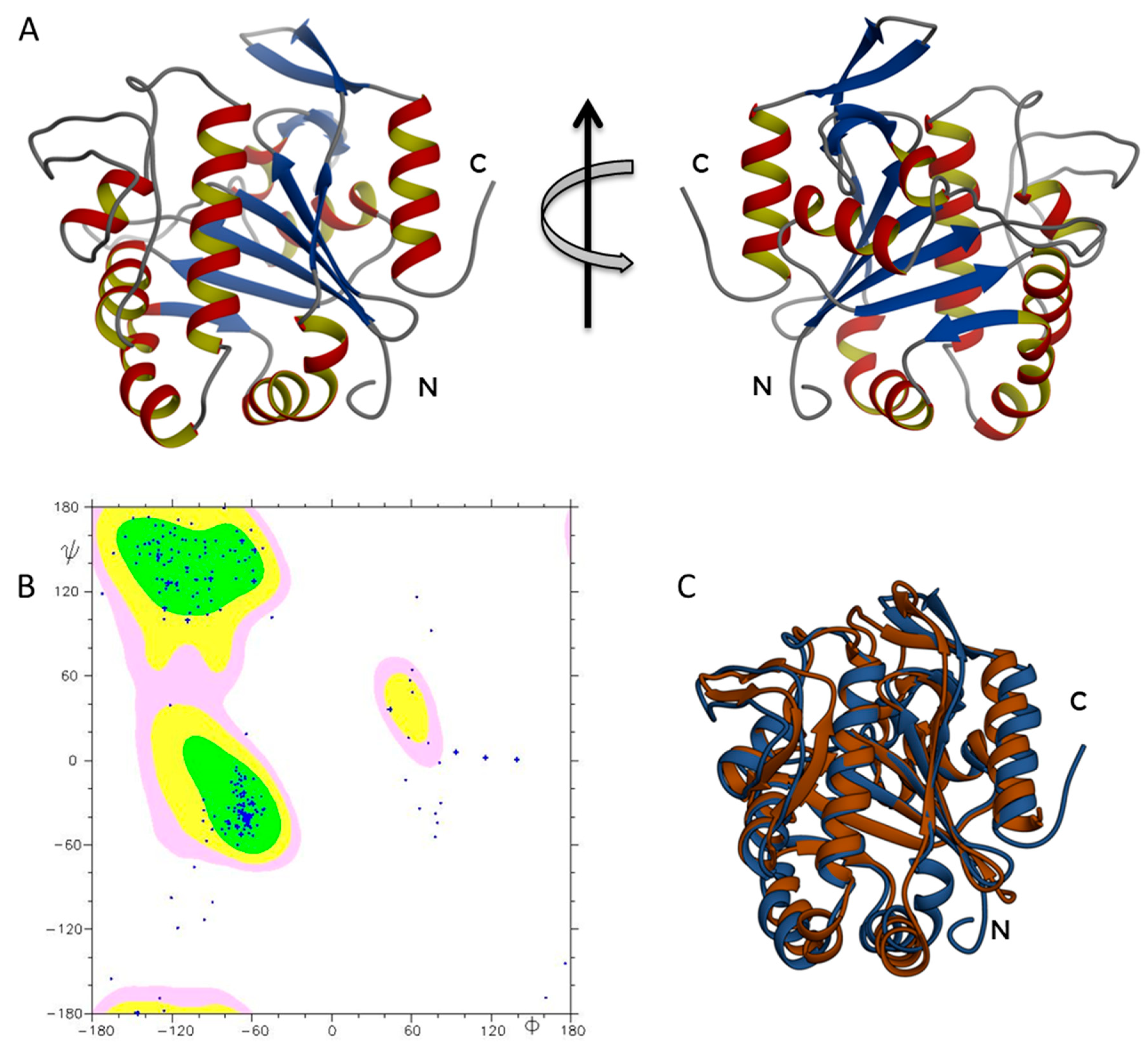
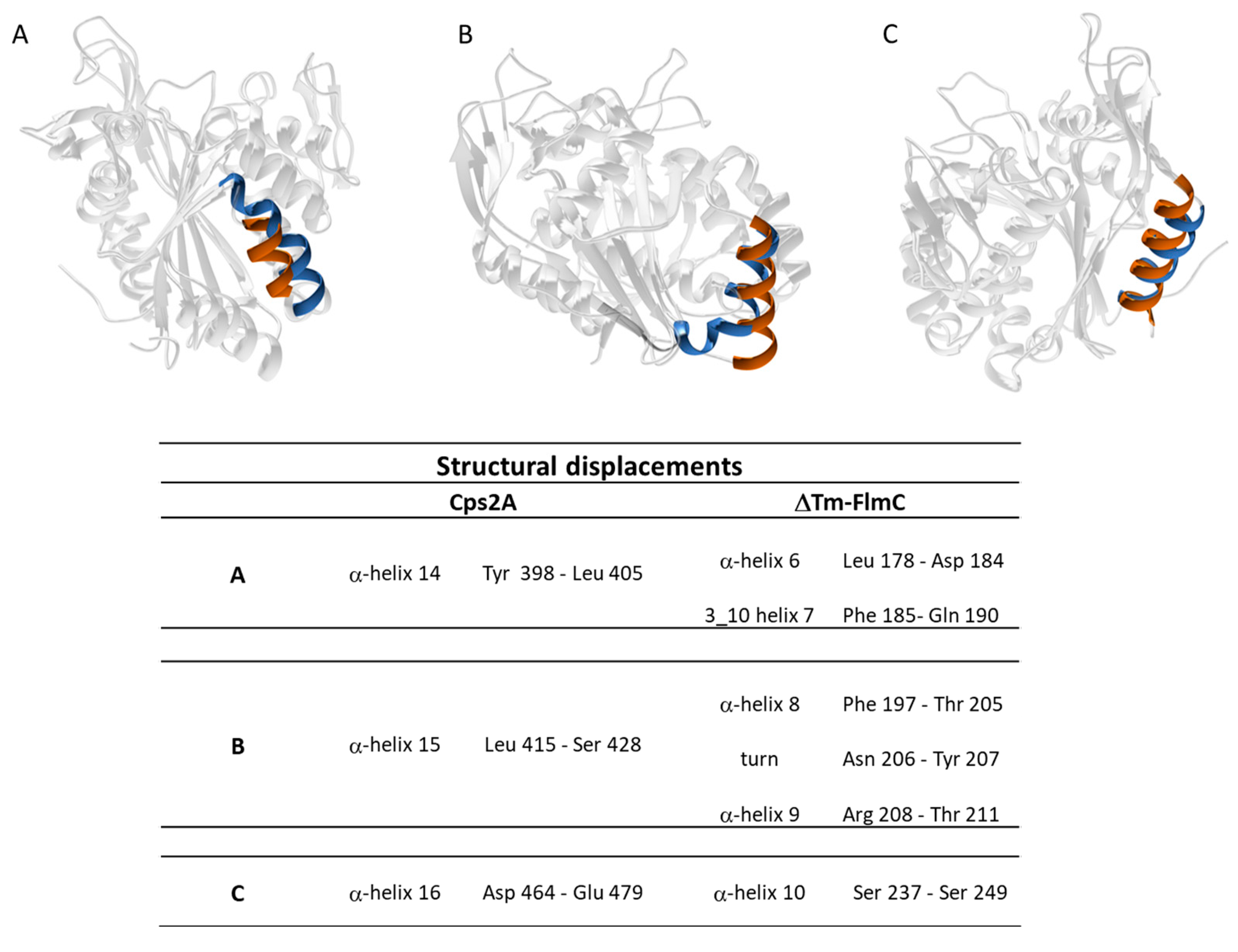
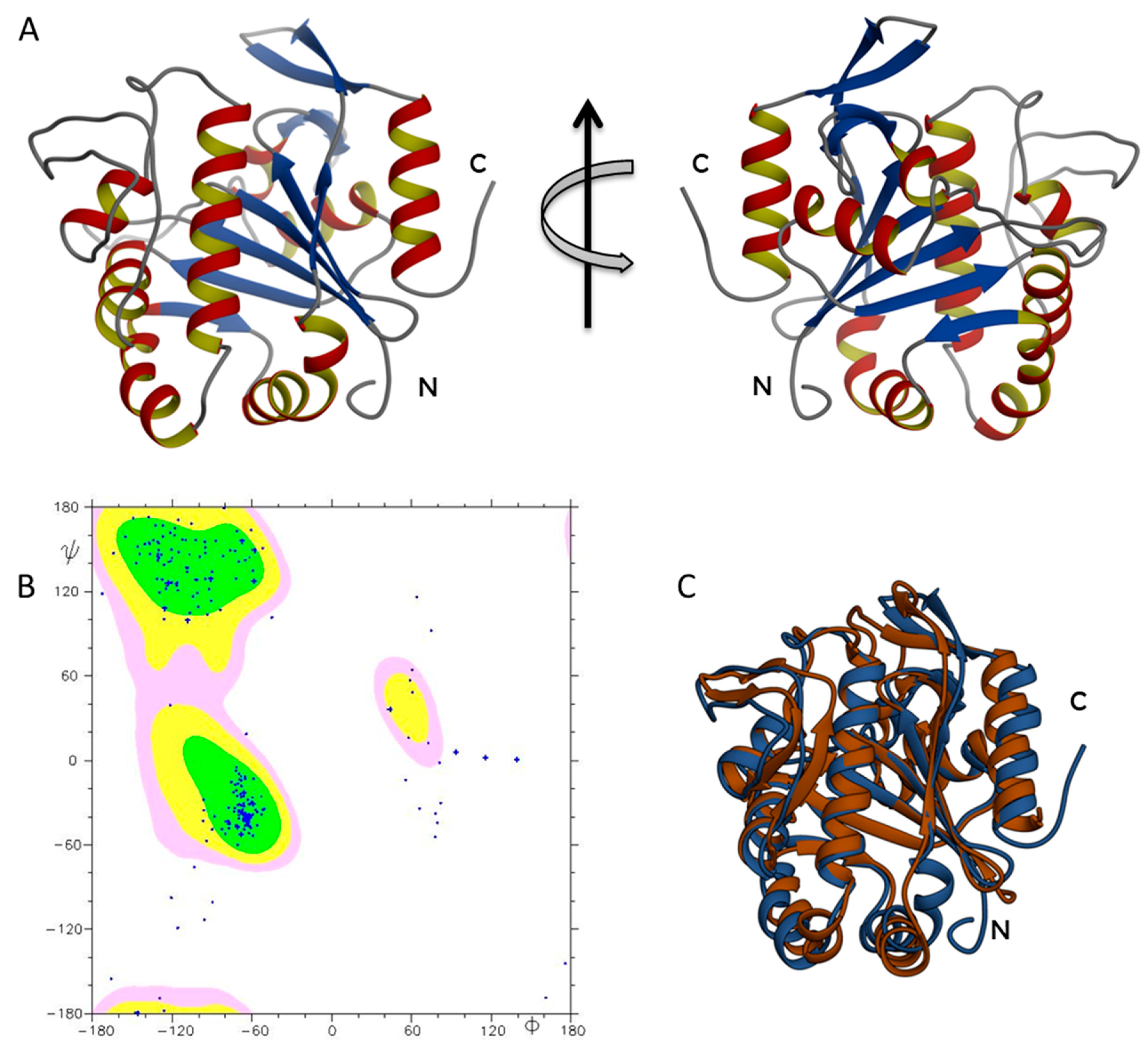
2.2. ΔTM-FlmC Structural Model
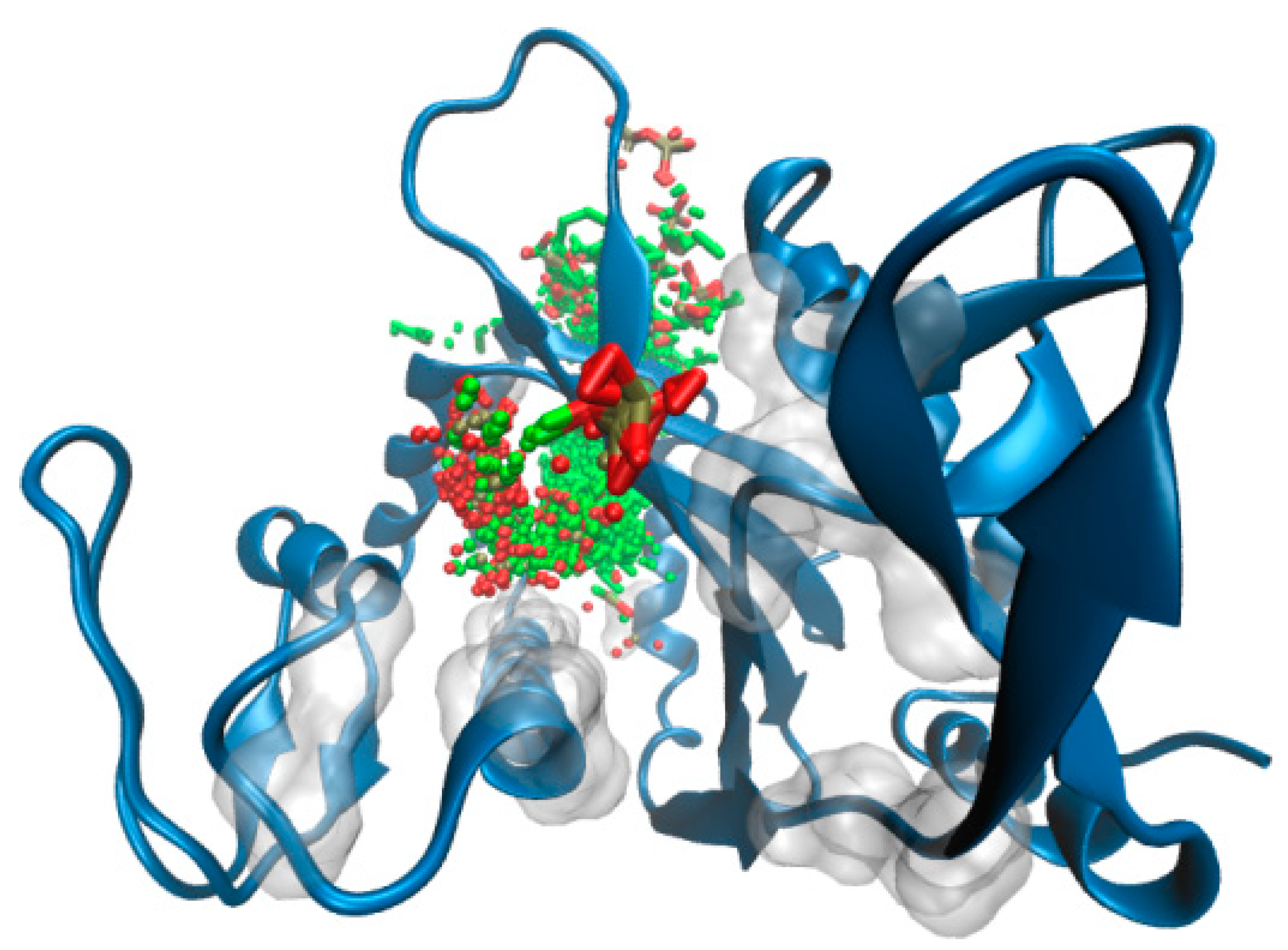
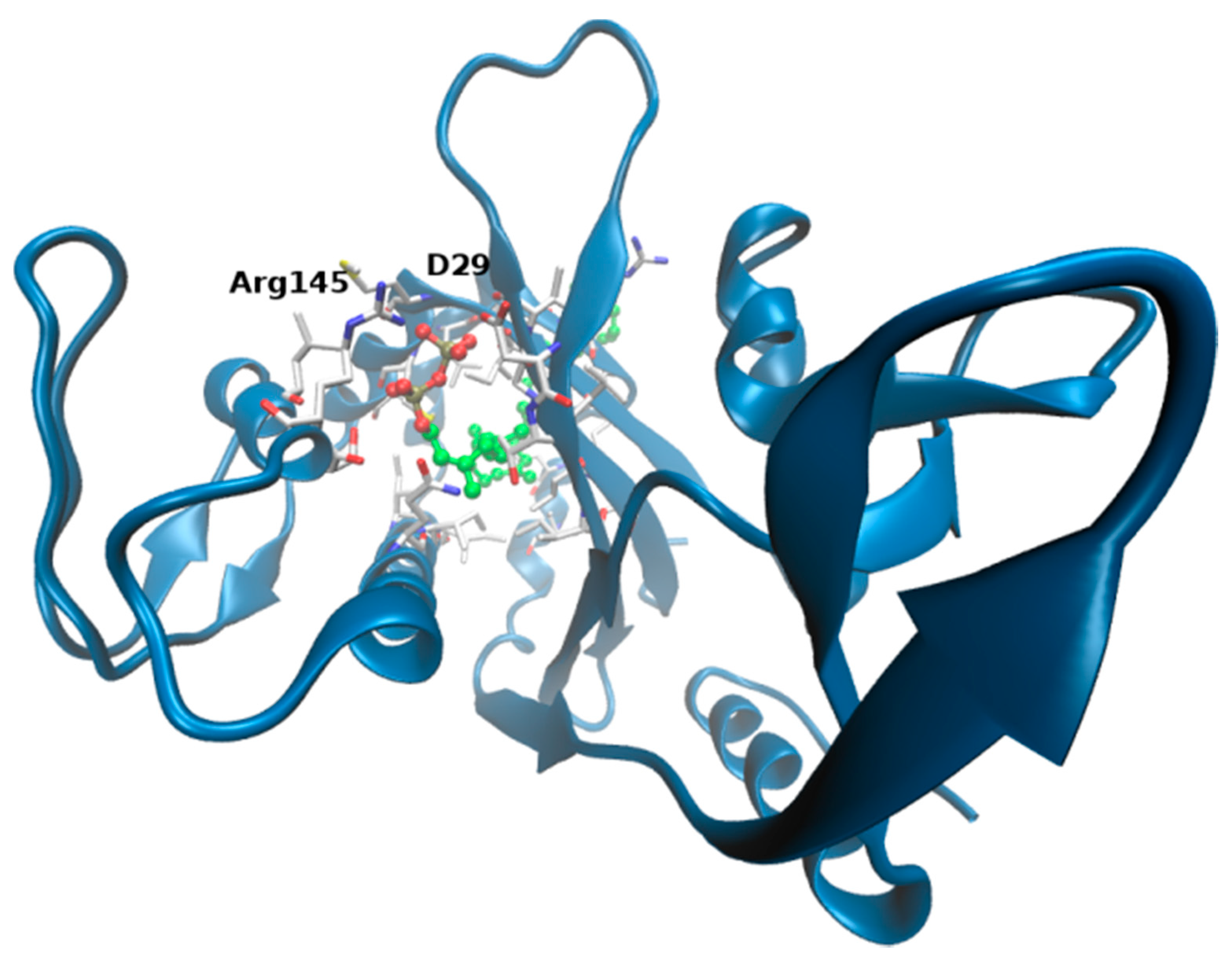
2.3. ΔTM-Flmc Binding Pocket
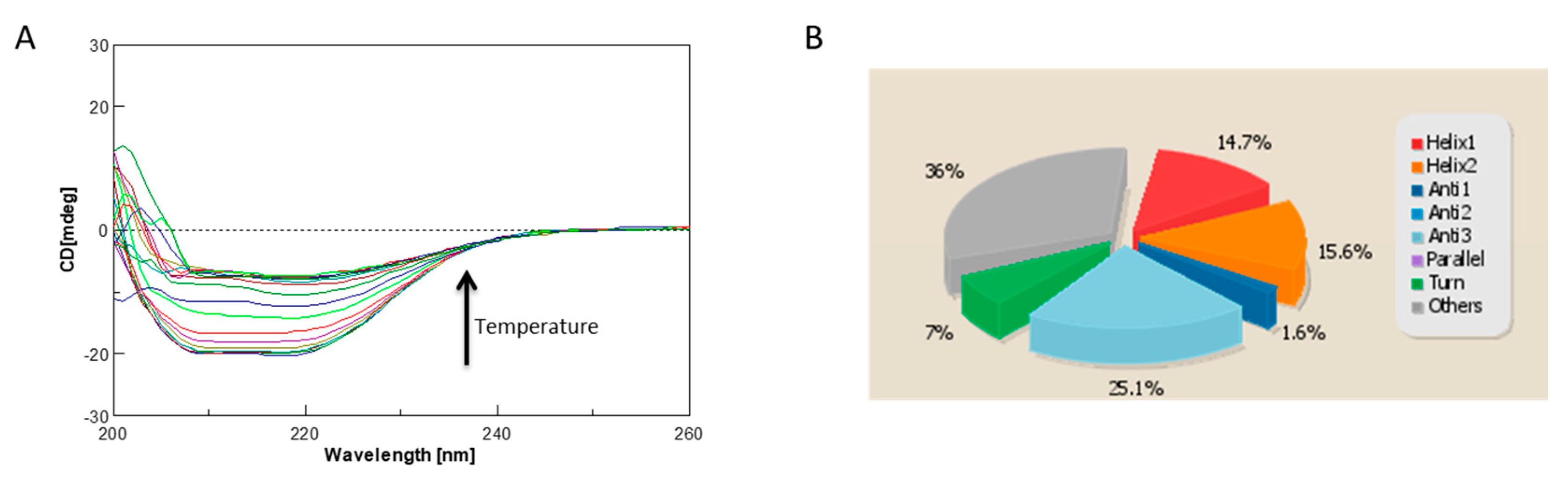
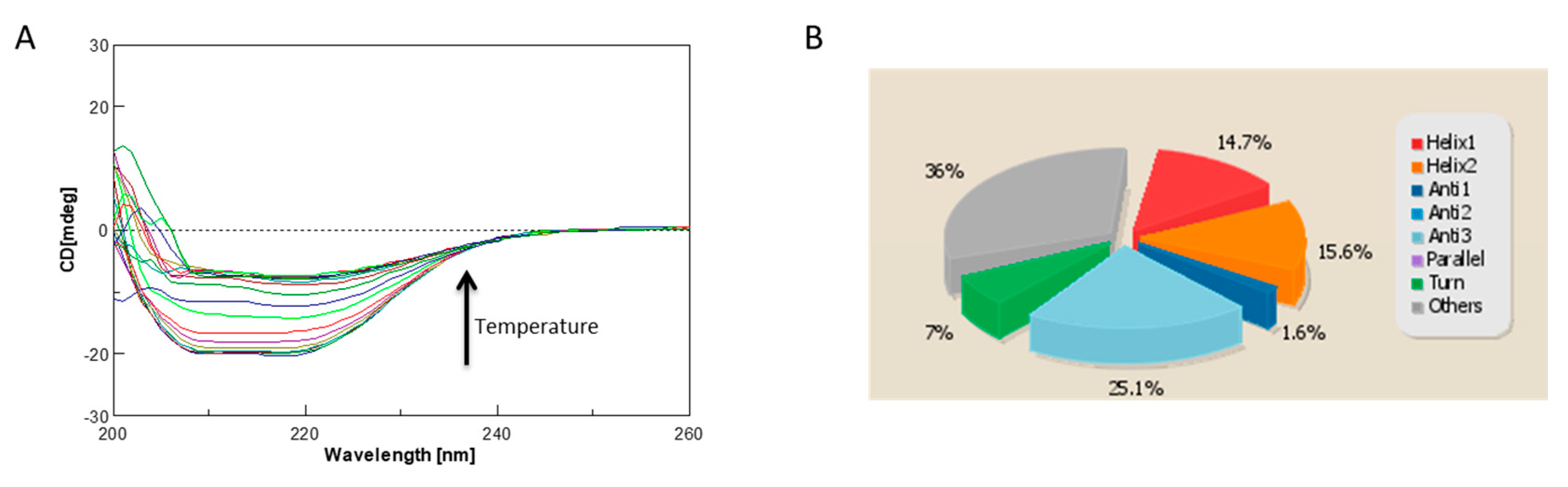
2.4. Circular Dichroism Characterization and Validation of the Obtained Model
2.5. ΔTM-FlmC Domain Binds a Lipid Molecule and a Magnesium Ion
3. Materials and Methods
3.1. Bioinformatics
3.2. Expression and Purification of ΔTM-FlmC from L. plantarum
3.3. Circular Dichroism
3.4. 3D Structural Models
3.5. Molecular Dynamics
3.6. ICP-MS
3.7. Lipid Extraction
3.8. UHPLC-HRMS Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mihai, M.M.; Holban, A.M.; Giurcaneanu, C.; Popa, L.G.; Oanea, R.M.; Lazar, V.; Chifiriuc, M.C.; Popa, M.; Popa, M.I. Microbial biofilms: Impact on the pathogenesis of periodontitis, cystic fibrosis, chronic wounds and medical device-related infections. Curr. Top. Med. Chem. 2015, 15, 1552–1576. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Jabra-Rizk, M.A.; O’May, G.A.; Costerton, J.W.; Shirtliff, M.E. Polymicrobial interactions: Impact on pathogenesis and human disease. Clin. Microbiol. Rev. 2012, 25, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Mangwani, N.; Kumari, S.; Das, S. Bacterial biofilms and quorum sensing: Fidelity in bioremediation technology. Biotechnol. Genet. Eng. Rev. 2016, 32, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Caggianiello, G.; Kleerebezem, M.; Spano, G. Exopolysaccharides produced by lactic acid bacteria: From health-promoting benefits to stress tolerance mechanisms. Appl. Microbiol. Biotechnol. 2016, 100, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Jagmann, N.; Philipp, B. Reprint of design of synthetic microbial communities for biotechnological production processes. J. Biotechnol. 2014, 192, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Nancharaiah, Y.V.; Venkata Mohan, S.; Lens, P.N. Metals removal and recovery in bioelectrochemical systems: A review. Bioresour. Technol. 2015, 195, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Lagrafeuille, R.; Souweine, B.; Forestier, C. Anti-biofilm activity as a health issue. Front. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Malgieri, G.; Avitabile, C.; Palmieri, M.; D’Andrea, L.D.; Isernia, C.; Romanelli, A.; Fattorusso, R. Structural basis of a temporin 1b analogue antimicrobial activity against gram negative bacteria determined by cd and nmr techniques in cellular environment. ACS Chem. Biol. 2015, 10, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Iacovino, R.; Caso, J.V.; Rapuano, F.; Russo, A.; Isidori, M.; Lavorgna, M.; Malgieri, G.; Isernia, C. Physicochemical characterization and cytotoxic activity evaluation of hydroxymethylferrocene: β-cyclodextrin inclusion complex. Molecules 2012, 17, 6056–6070. [Google Scholar] [CrossRef] [PubMed]
- Iacovino, R.; Rapuano, F.; Caso, J.V.; Russo, A.; Lavorgna, M.; Russo, C.; Isidori, M.; Russo, L.; Malgieri, G.; Isernia, C. β-cyclodextrin inclusion complex to improve physicochemical properties of pipemidic acid: Characterization and bioactivity evaluation. Int. J. Mol. Sci. 2013, 14, 13022–13041. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, C.; Netti, F.; Orefice, G.; Palmieri, M.; Nocerino, N.; Malgieri, G.; D’Andrea, L.D.; Capparelli, R.; Fattorusso, R.; Romanelli, A. Design, structural and functional characterization of a temporin-1b analog active against gram-negative bacteria. Biochim. Biophys. Acta 2013, 1830, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Bayramov, D.F.; Neff, J.A. Beyond conventional antibiotics—New directions for combination products to combat biofilm. Adv. Drug Deliv. Rev. 2017, 112, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Coleman, S.R.; Hancock, R.E. Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr. Opin. Microbiol. 2016, 33, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlane, S.; Dillon, J.F. Microbial biofilms in the human gastrointestinal tract. J. Appl. Microbiol. 2007, 102, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watters, C.; Fleming, D.; Bishop, D.; Rumbaugh, K.P. Host responses to biofilm. Prog. Mol. Biol Transl. Sci。 2016, 142, 193–239. [Google Scholar] [PubMed]
- Santos, C.M.; Pires, M.C.; Leão, T.L.; Hernández, Z.P.; Rodriguez, M.L.; Martins, A.K.; Miranda, L.S.; Martins, F.S.; Nicoli, J.R. Selection of Lactobacillus strains as potential probiotics for vaginitis treatment. Microbiology 2016, 162, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, E.; Adrain, V.; Diana, P. Microbial biofilm formation under the influence of various physical-chemical factors. Biotechnol. Biotechnol. Equip. 2010, 24, 1993–1996. [Google Scholar] [CrossRef]
- Leccese Terraf, M.C.; Juárez Tomás, M.S.; Rault, L.; Le Loir, Y.; Even, S.; Nader-Macías, M.E. Biofilms of vaginal Lactobacillus reuteri CRL 1324 and Lactobacillus rhamnosus CRL 1332: Kinetics of formation and matrix characterization. Arch. Microbiol. 2016, 198, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Aoudia, N.; Rieu, A.; Briandet, R.; Deschamps, J.; Chluba, J.; Jego, G.; Garrido, C.; Guzzo, J. Biofilms of Lactobacillus plantarum and Lactobacillus fermentum: Effect on stress responses, antagonistic effects on pathogen growth and immunomodulatory properties. Food Microbiol. 2016, 53, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Lönnermark, E.; Friman, V.; Lappas, G.; Sandberg, T.; Berggren, A.; Adlerberth, I. Intake of Lactobacillus plantarum reduces certain gastrointestinal symptoms during treatment with antibiotics. J. Clin. Gastroenterol. 2010, 44, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Caggianiello, G.; van Swam, I.I.; Taverne, N.; Meijerink, M.; Bron, P.A.; Spano, G.; Kleerebezem, M. Strain-specific features of extracellular polysaccharides and their impact on Lactobacillus plantarum-host interactions. Appl. Environ. Microbiol. 2016, 82, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Vicariotto, F.; Mogna, L.; Del Piano, M. Effectiveness of the two microorganisms Lactobacillus fermentum LF15 and Lactobacillus plantarum LP01, formulated in slow-release vaginal tablets, in women affected by bacterial vaginosis: A pilot study. J. Clin. Gastroenterol. 2014, 48, S106–S112. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; Wilson, G. Probiotics genomics. Microb. Biotechnol. 2010, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siezen, R.J.; van Hylckama Vlieg, J.E. Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microb. Cell Fact. 2011. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Arena, M.P.; Fiocco, D.; Capozzi, V.; Drider, D.; Spano, G. Lactobacillus plantarum with broad antifungal activity: A promising approach to increase safety and shelf-life of cereal-based products. Int. J. Food Microbiol. 2017, 247, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, C.; Vastano, V.; Siciliano, R.A.; Candela, M.; Vici, M.; Muscariello, L.; Marasco, R.; Sacco, M. Surface displaced alfa-enolase of Lactobacillus plantarum is a fibronectin binding protein. Microb. Cell Factor. 2009. [Google Scholar] [CrossRef] [PubMed]
- Vastano, V.; Capri, U.; Candela, M.; Siciliano, R.A.; Russo, L.; Renda, M.; Sacco, M. Identification of binding sites of Lactobacillus plantarum enolase involved in the interaction with human plasminogen. Microbiol. Res. 2013, 168, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Salzillo, M.; Vastano, V.; Capri, U.; Muscariello, L.; Sacco, M.; Marasco, R. Identification and characterization of enolase as a collagen-binding protein in Lactobacillus plantarum. J. Basic Microbiol. 2015, 55, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Vastano, V.; Pagano, A.; Fusco, A.; Merola, G.; Sacco, M.; Donnarumma, G. The Lactobacillus plantarum enoA1 enolase is involved in immunostimulation of caco-2 cells and in biofilm development. Adv. Exp. Med. Biol. 2016, 897, 33–44. [Google Scholar] [PubMed]
- Muscariello, L.; Marino, C.; Capri, U.; Vastano, V.; Marasco, R.; Sacco, M. CcpA and three newly identified proteins are involved in biofilm development in Lactobacillus plantarum. J. Basic Microbiol. 2013, 53, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, C.H.; Koo, H.; Quivey, R.G. The putative autolysin regulator LytR in Streptococcus mutans plays a role in cell division and is growth-phase regulated. Microbiology 2005, 151, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Patras, K.A.; Derieux, J.; Al-Bassam, M.M.; Adiletta, N.; Vrbanac, A.; Lapek, J.D.; Zengler, K.; Gonzalez, D.J.; Nizet, V. Group b Streptococcus biofilm regulatory protein a contributes to bacterial physiology and innate immune resistance. J. Infect. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.T.; Scott-Anne, K.; Liao, S.; De, A.; Luo, M.; Kovacs, C.; Narvaez, B.S.; Faustoferri, R.C.; Yu, Q.; Taylor, C.M.; et al. Deficiency of BrpA in Streptococcus mutans reduces virulence in rat caries model. Mol. Oral Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.T.; Baker, H.V.; Burne, R.A. Influence of BrpA on critical virulence attributes of Streptococcus mutans. J. Bacteriol. 2006, 188, 2983–2992. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, J.P.; Liao, S.; Yao, X.; Ahn, S.J.; Isoda, R.; Nguyen, A.H.; Brady, L.J.; Burne, R.A.; Abranches, J.; Wen, Z.T. BrpA is involved in regulation of cell envelope stress responses in Streptococcus mutans. Appl. Environ. Microbiol. 2012, 78, 2914–2922. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, J.P.; Liao, S.; McKey, B.A.; Yao, X.; Fan, Y.; Abranches, J.; Beatty, W.L.; Wen, Z.T. Psr is involved in regulation of glucan production and double deficiency of BrpA and Psr is lethal in Streptococcus mutans. Microbiology 2013, 159, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Marles-Wright, J.; Cleverley, R.M.; Emmins, R.; Ishikawa, S.; Kuwano, M.; Heinz, N.; Bui, N.K.; Hoyland, C.N.; Ogasawara, N.; et al. A widespread family of bacterial cell wall assembly proteins. EMBO J. 2011, 30, 4931–4941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gräslund, S.; Nordlund, P.; Weigelt, J.; Hallberg, B.M.; Bray, J.; Gileadi, O.; Knapp, S.; Oppermann, U.; Arrowsmith, C.; Hui, R.; et al. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. Aggrescan: A server for the prediction and evaluation of “Hot spots” of aggregation in polypeptides. BMC Bioinform. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-tasser: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Kellis, J.T.; Nyberg, K.; Fersht, A.R. Energetics of complementary side-chain packing in a protein hydrophobic core. Biochemistry 1989, 28, 4914–4922. [Google Scholar] [CrossRef] [PubMed]
- Shortle, D.; Stites, W.E.; Meeker, A.K. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry 1990, 29, 8033–8041. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Stigter, D. Modeling protein stability as heteropolymer collapse. Adv. Protein. Chem. 1995, 46, 59–104. [Google Scholar] [PubMed]
- Baglivo, I.; Palmieri, M.; Rivellino, A.; Netti, F.; Russo, L.; Esposito, S.; Iacovino, R.; Farina, B.; Isernia, C.; Fattorusso, R.; et al. Molecular strategies to replace the structural metal site in the prokaryotic zinc finger domain. Biochim. Biophys. Acta 2014, 1844, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Malgieri, G.; Eliezer, D. Structural effects of parkinson’s disease linked DJ-1 mutations. Protein Sci. 2008, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Hendlich, M.; Rippmann, F.; Barnickel, G. Ligsite: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J. 3dligandsite: Predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010, 38, W469–W473. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. Swissdock, a protein-small molecule docking web service based on eadock dss. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Park, H.; Heo, L.; Seok, C. Galaxyweb server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- De Paola, I.; Pirone, L.; Palmieri, M.; Balasco, N.; Esposito, L.; Russo, L.; Mazzà, D.; Di Marcotullio, L.; Di Gaetano, S.; Malgieri, G.; et al. Cullin3-btb interface: A novel target for stapled peptides. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Spaziano, G.; Sorrentino, R.; Matteis, M.; Malgieri, G.; Sgambato, M.; Russo, T.P.; Terlizzi, M.; Roviezzo, F.; Rossi, F.; Pinto, A.; et al. Nociceptin reduces the inflammatory immune microenvironment in a conventional murine model of airway hyperresponsiveness. Clin. Exp. Allergy 2017, 47, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Malgieri, G.; Russo, L.; Baglivo, I.; Esposito, S.; Netti, F.; Del Gatto, A.; de Paola, I.; Zaccaro, L.; Pedone, P.V.; et al. Structural Zn(ii) implies a switch from fully cooperative to partly downhill folding in highly homologous proteins. J. Am. Chem. Soc. 2013, 135, 5220–5228. [Google Scholar] [CrossRef] [PubMed]
- D′Abrosca, G.; Russo, L.; Palmieri, M.; Baglivo, I.; Netti, F.; de Paola, I.; Zaccaro, L.; Farina, B.; Iacovino, R.; Pedone, P.V.; et al. The (unusual) aspartic acid in the metal coordination sphere of the prokaryotic zinc finger domain. J. Inorg. Biochem. 2016, 161, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.; Lloyd, G.; Joe, M.; Lowary, T.L.; Reynolds, E.; Walters-Morgan, H.; Bhatt, A.; Lovering, A.; Besra, G.S.; Alderwick, L.J. Lcp1 is a phosphotransferase responsible for ligating arabinogalactan to peptidoglycan in Mycobacterium tuberculosis. Am. Soc. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Paladino, A.; Zangi, R. Ribose 2′-hydroxyl groups stabilize RNA hairpin structures containing GCUAA pentaloop. J. Chem. Theory Comput. 2013, 9, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad Sci. USA 2015, 112, 3095–3103. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. Aqua and procheck-nmr: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool, 2nd ed.; CCP4 Newsletter on Protein Crystallography: San Carlos, CA, USA, 2002. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wüthrich, K. Molmol: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Joosten, R.P.; te Beek, T.A.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.; Schneider, R.; Sander, C.; Vriend, G. A series of pdb related databases for everyday needs. Nucleic. Acids Res. 2011, 39, D411–D419. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- York, D.M.; Wlodawer, A.; Pedersen, L.G.; Darden, T.A. Atomic-level accuracy in simulations of large protein crystals. Proc. Natl. Acad. Sci. USA 1994, 91, 8715–8718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. P-lincs: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Long, G.L.; Winefordner, J.D. Limit of detection a closer look at the iupac definition. Anal. Chem. 1983, 55, 712A–724A. [Google Scholar]
- Hübscher, J.; Lüthy, L.; Berger-Bächi, B.; Stutzmann Meier, P. Phylogenetic distribution and membrane topology of the LytR-CpsA-Psr protein family. BMC Genet. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, S.D.; Liu, J.; Ton-That, H. Biogenesis of the gram-positive bacterial cell envelope. Curr. Opin. Microbiol. 2016, 34, 31–37. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Abrosca, G.; Paladino, A.; Cuoco, E.; Marasco, R.; Pacifico, S.; Piccolella, S.; Vastano, V.; Sacco, M.; Isernia, C.; Muscariello, L.; et al. Structural Characterization of the Lactobacillus Plantarum FlmC Protein Involved in Biofilm Formation. Molecules 2018, 23, 2252. https://doi.org/10.3390/molecules23092252
D’Abrosca G, Paladino A, Cuoco E, Marasco R, Pacifico S, Piccolella S, Vastano V, Sacco M, Isernia C, Muscariello L, et al. Structural Characterization of the Lactobacillus Plantarum FlmC Protein Involved in Biofilm Formation. Molecules. 2018; 23(9):2252. https://doi.org/10.3390/molecules23092252
Chicago/Turabian StyleD’Abrosca, Gianluca, Antonella Paladino, Emilio Cuoco, Rosangela Marasco, Severina Pacifico, Simona Piccolella, Valeria Vastano, Margherita Sacco, Carla Isernia, Lidia Muscariello, and et al. 2018. "Structural Characterization of the Lactobacillus Plantarum FlmC Protein Involved in Biofilm Formation" Molecules 23, no. 9: 2252. https://doi.org/10.3390/molecules23092252