DSC, FTIR and Raman Spectroscopy Coupled with Multivariate Analysis in a Study of Co-Crystals of Pharmaceutical Interest


Abstract
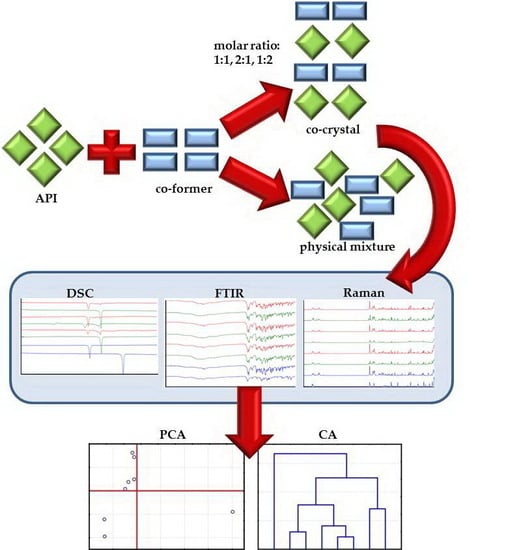
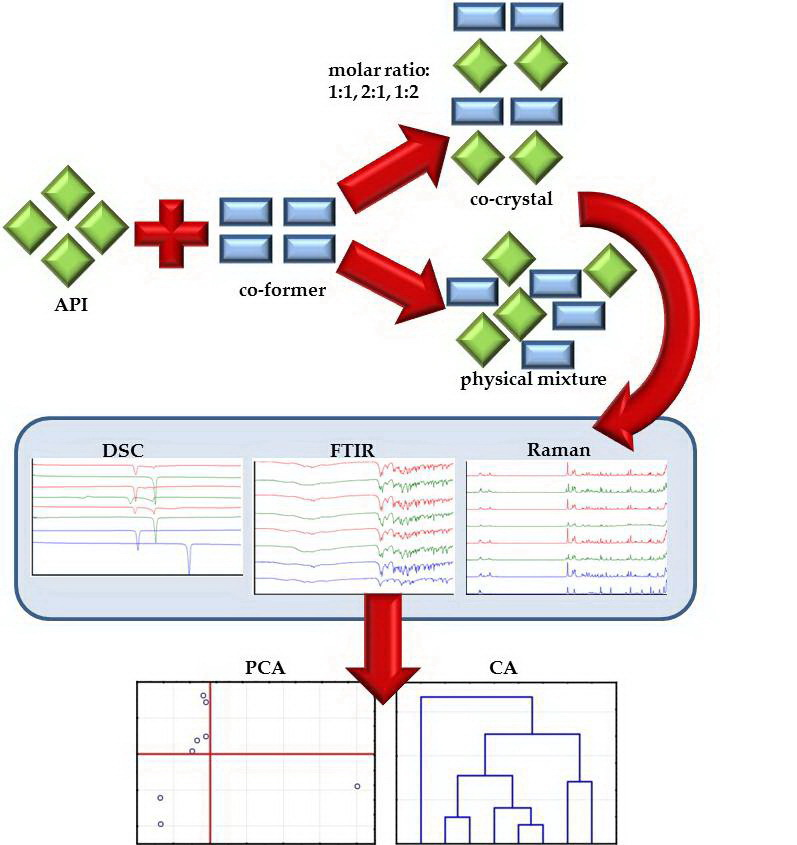
:
1. Introduction
2. Results and Discussion
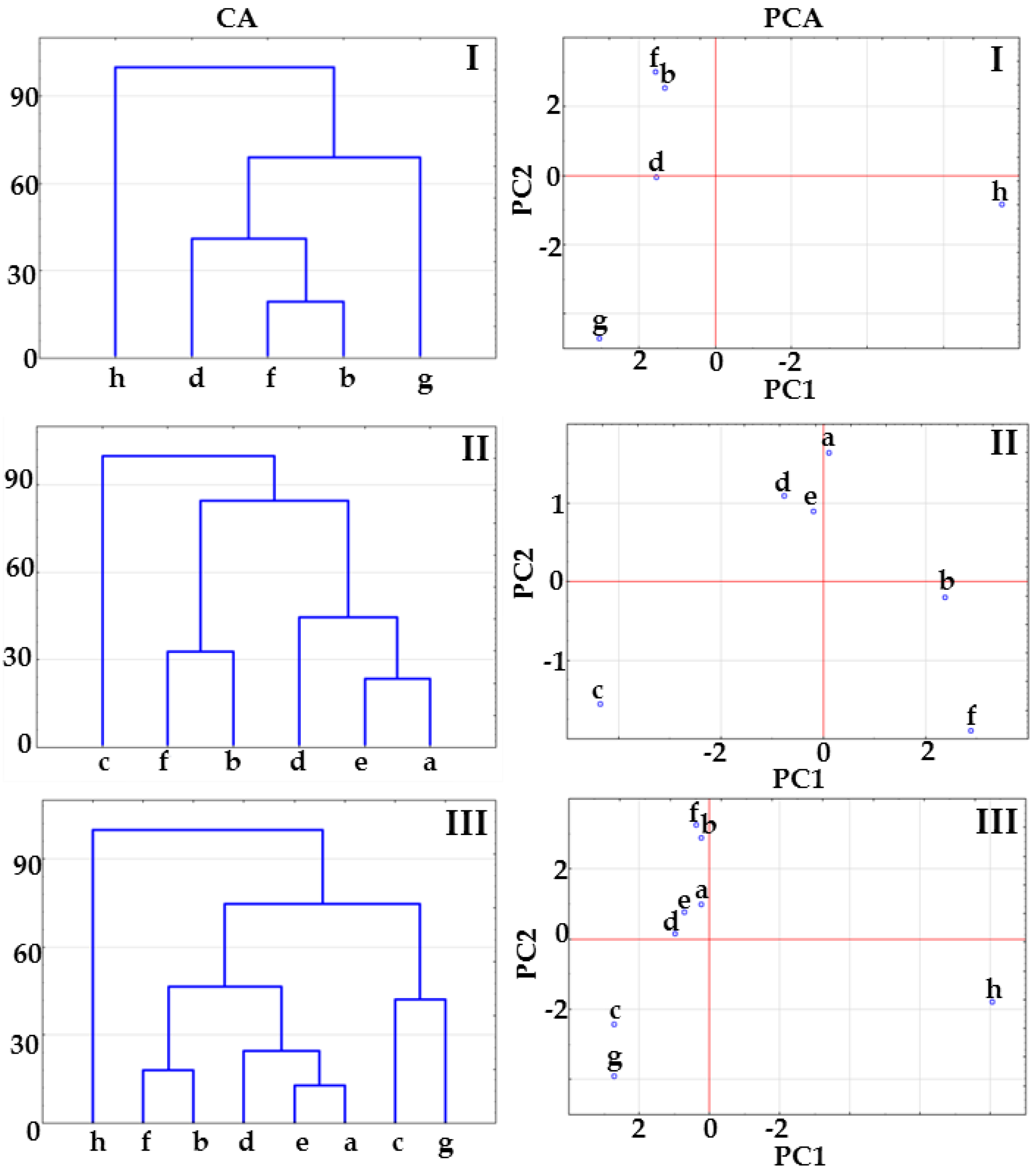
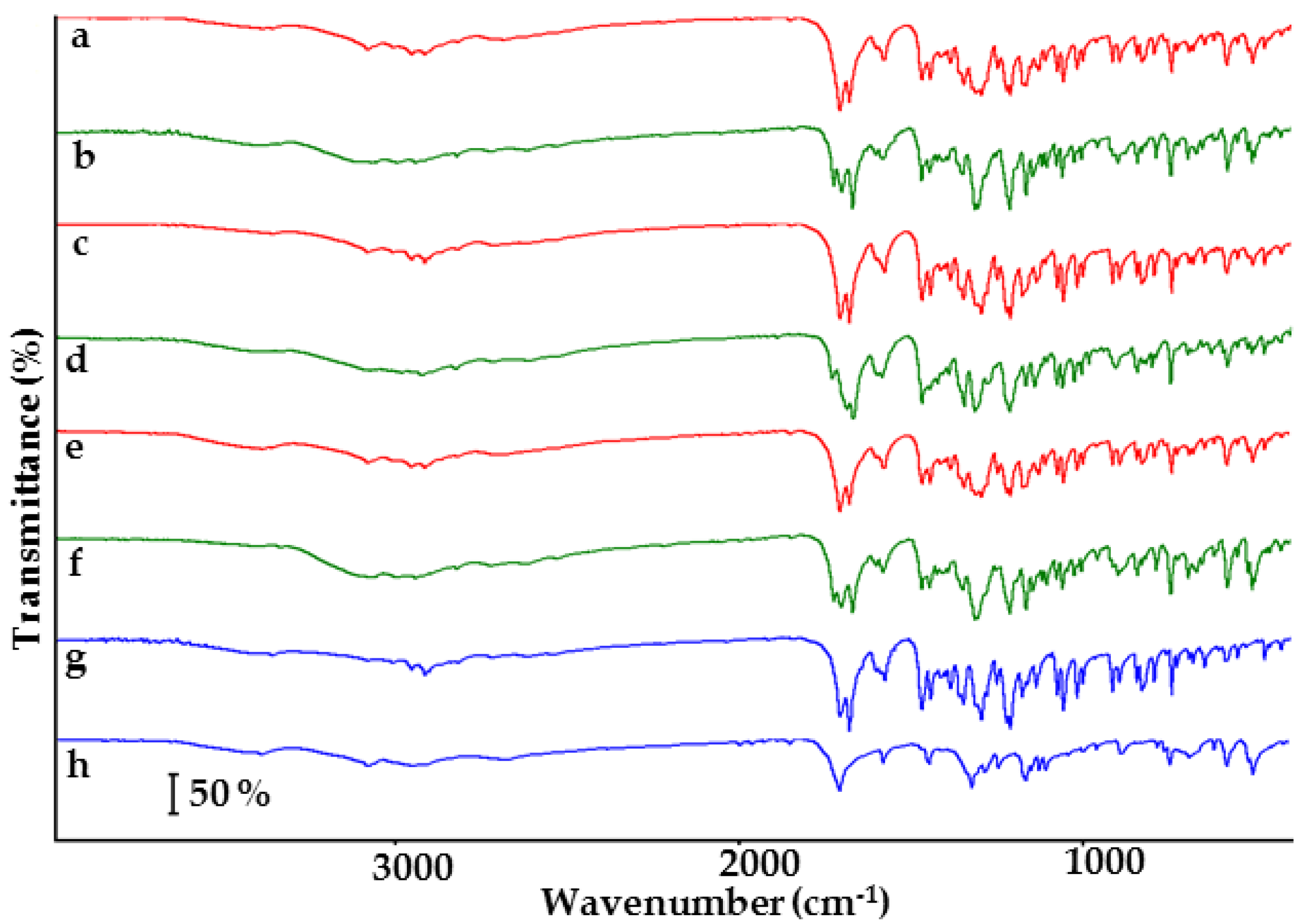
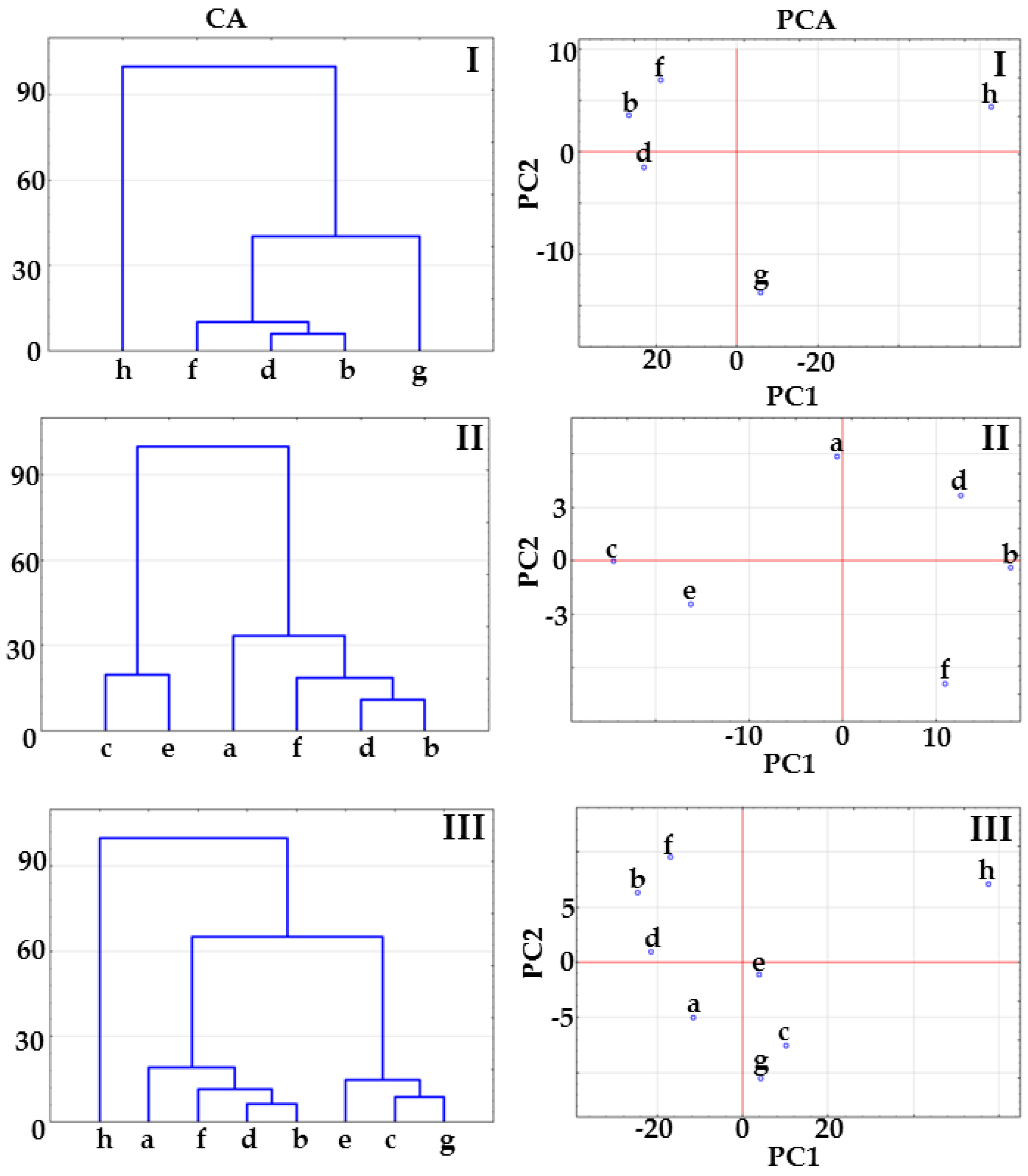
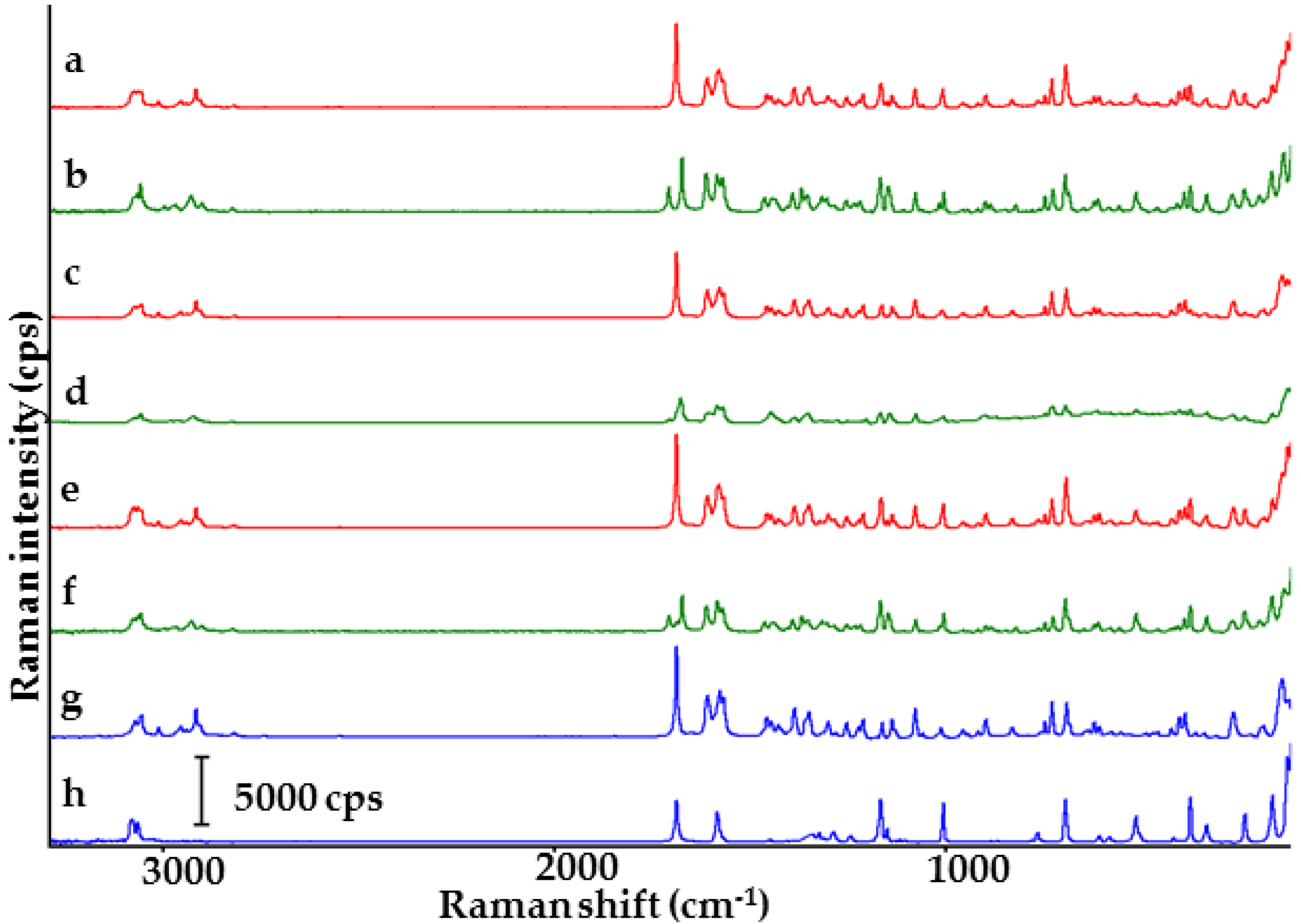
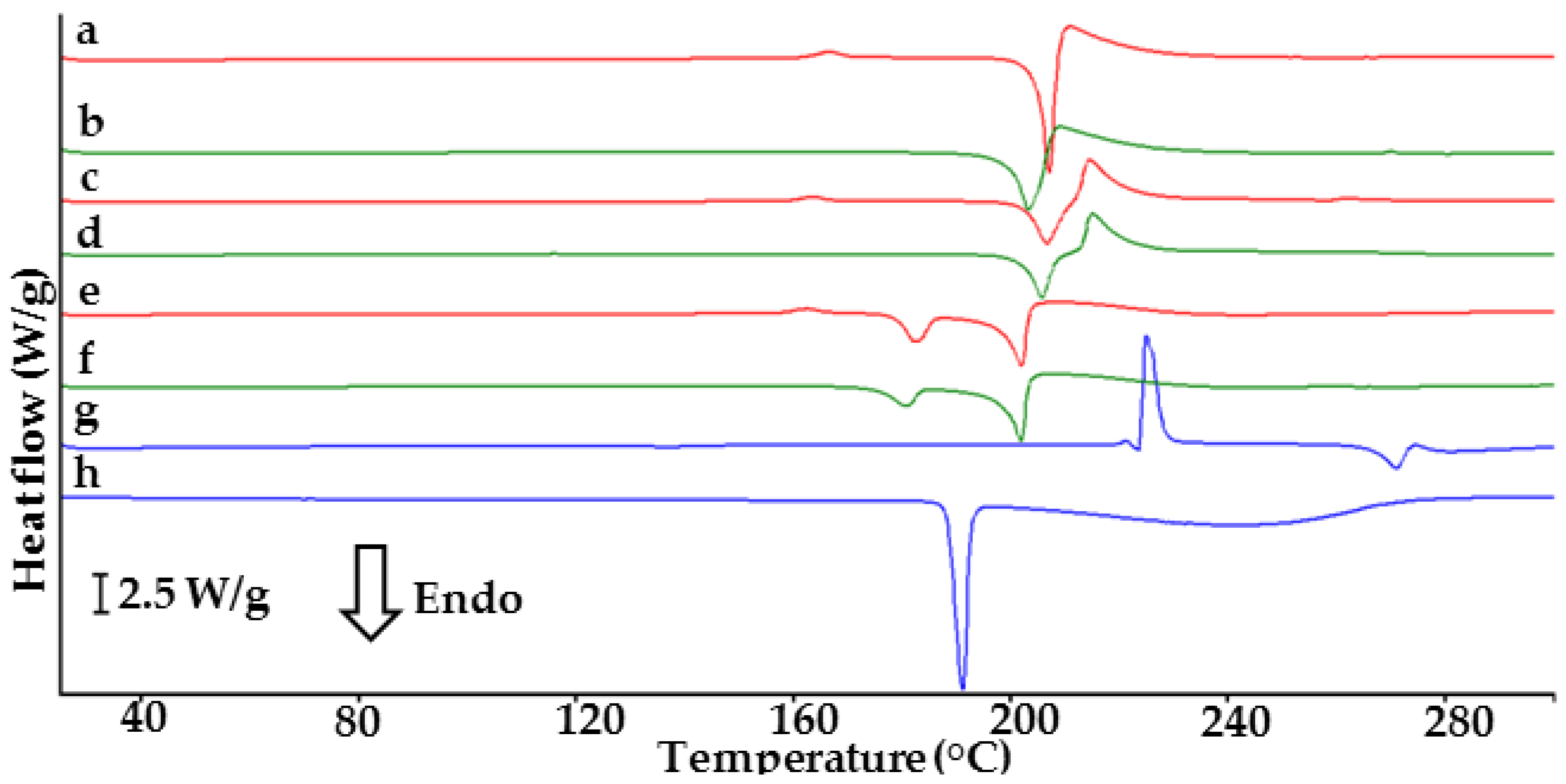
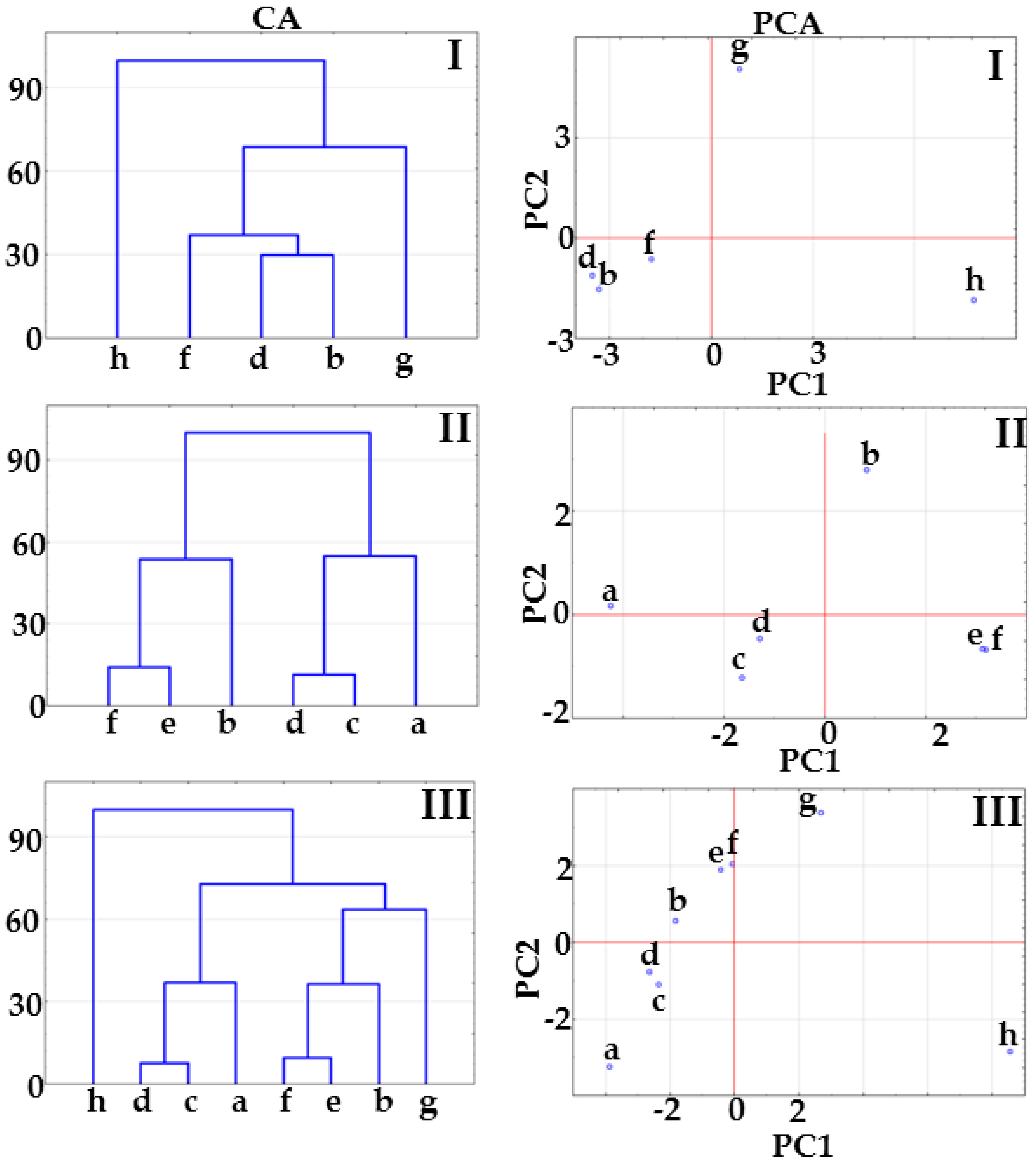
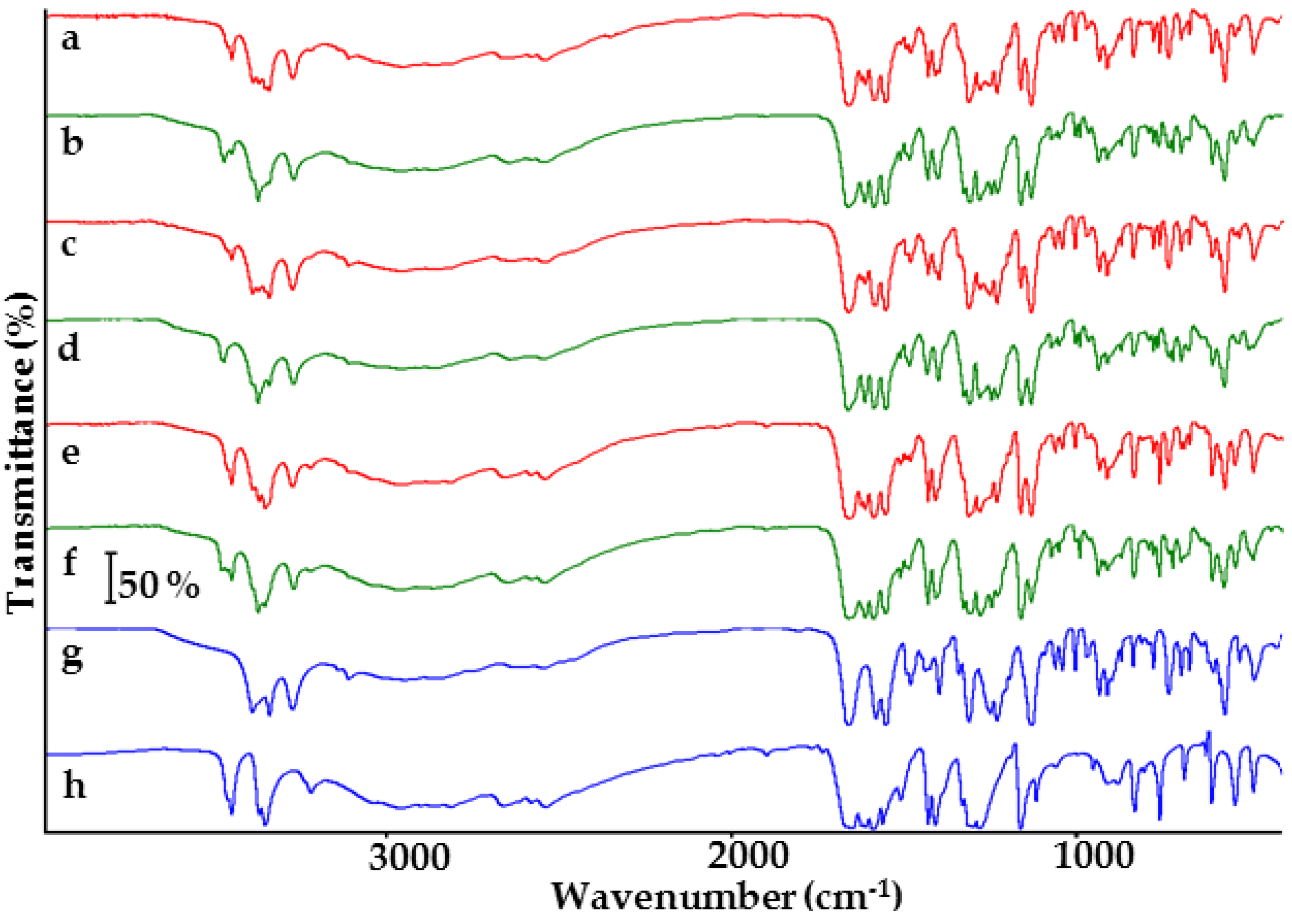
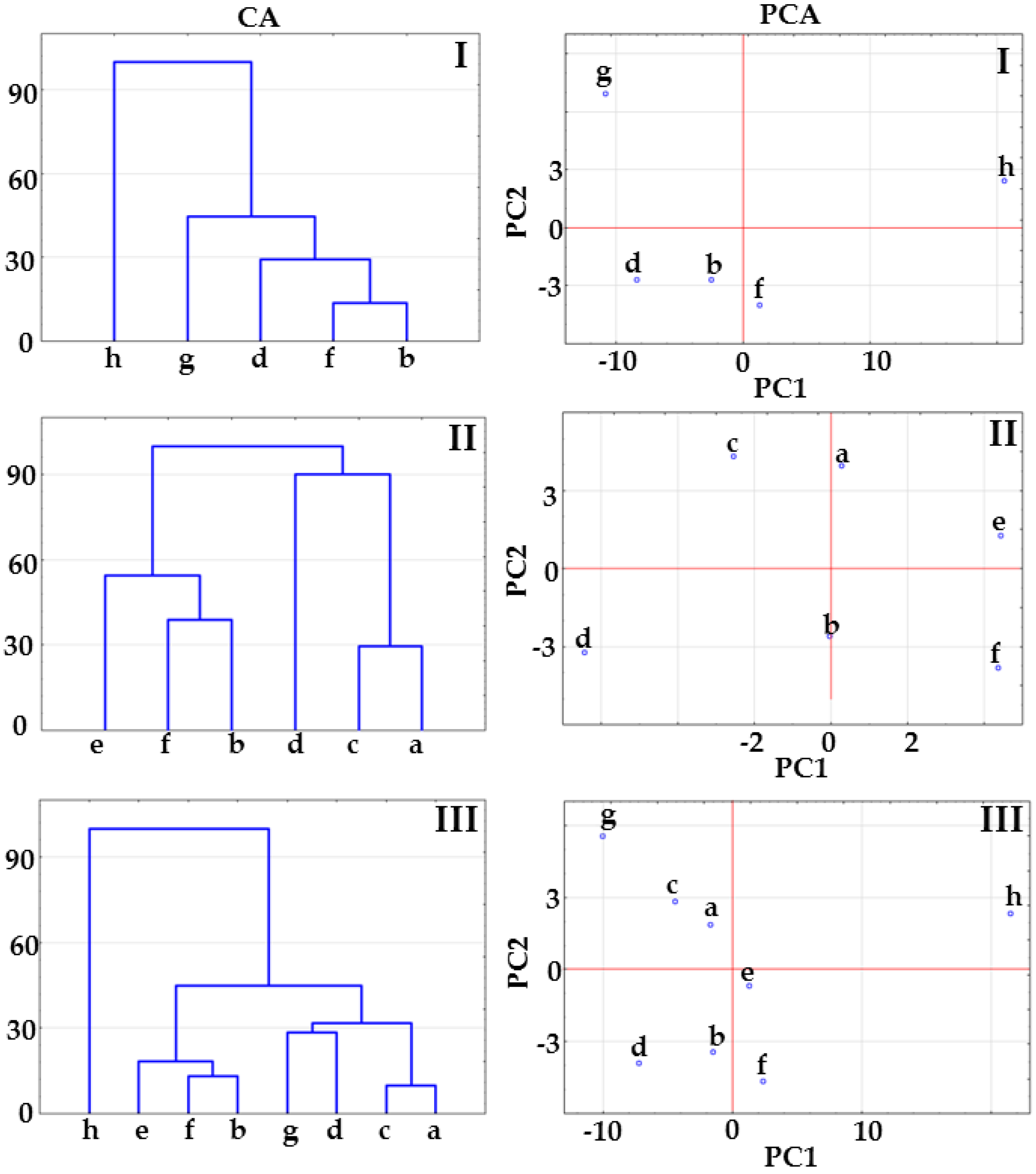
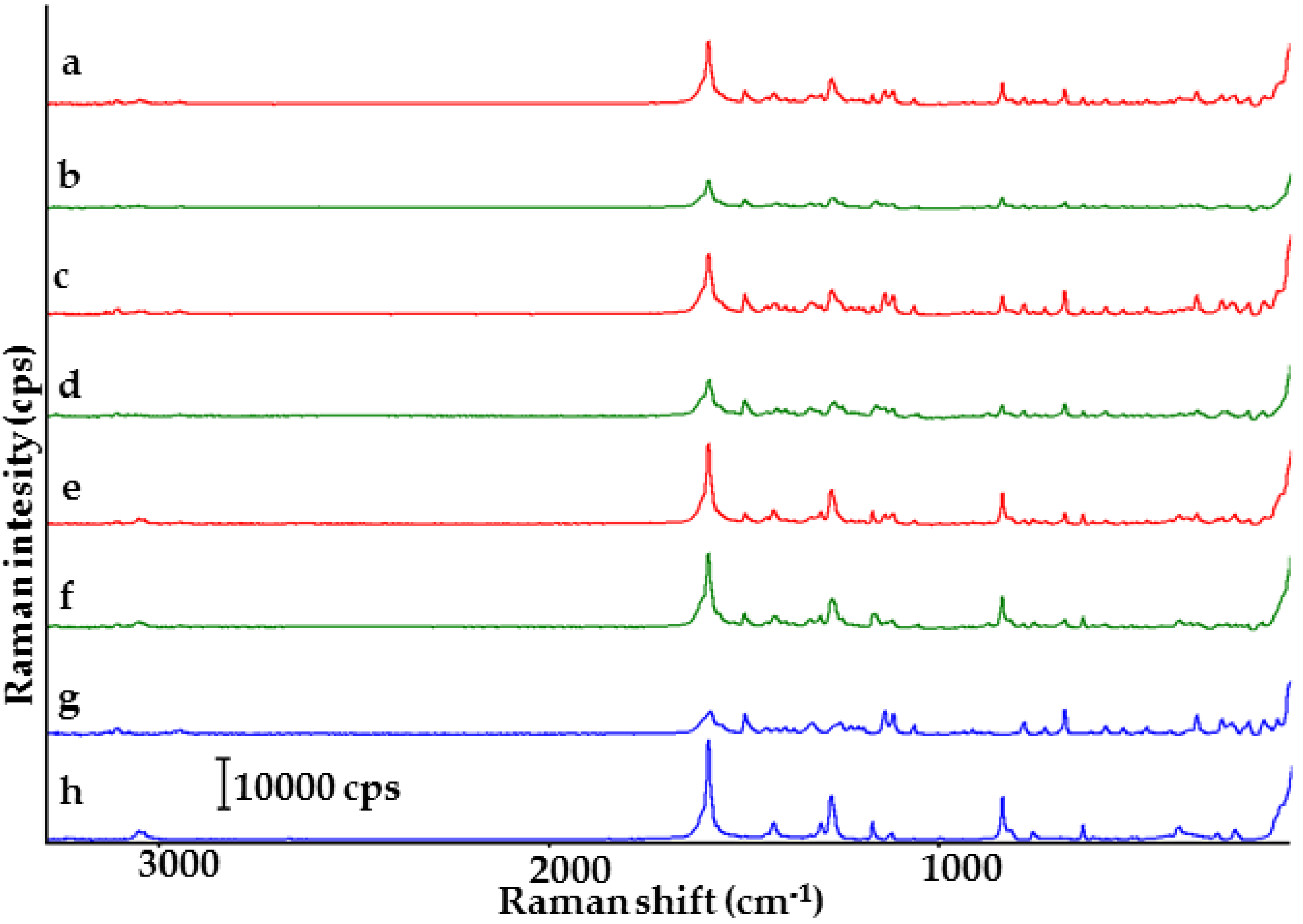
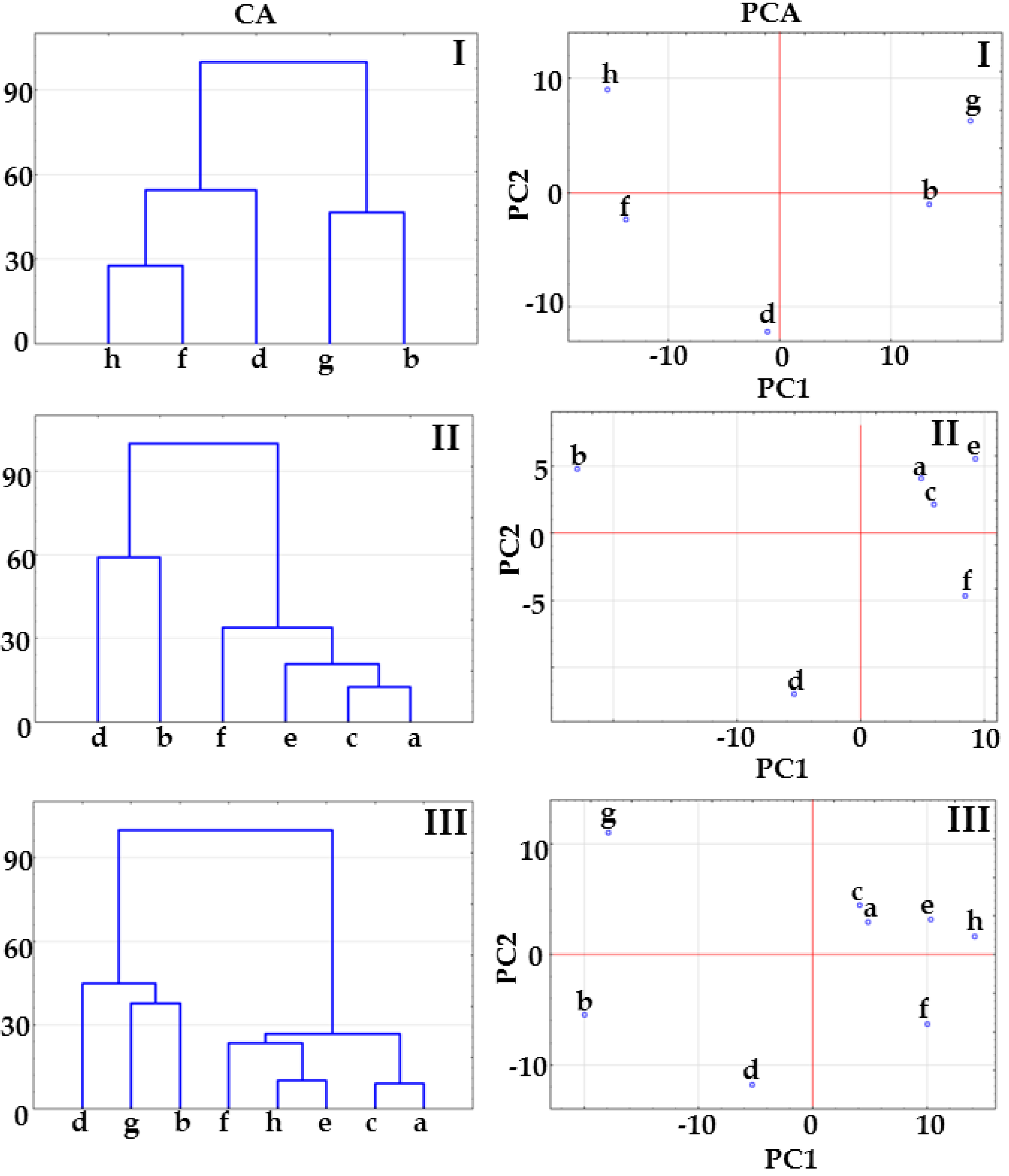
2.1. Co-Crystals of Indomethacin with Saccharin
2.2. Co-Crystals of Furosemide with p-Aminobenzoic Acid
3. Materials and Methods
3.1. Reagents
3.2. Preparation of Mixtures and Co-Crystals
3.3. DSC Conditions
3.4. FTIR Conditions
3.5. Raman Conditions
3.6. Multivariate Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hörter, D.; Dressman, J.B. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv. Drug Deliv. Rev. 1997, 25, 3–14. [Google Scholar] [CrossRef]
- Steed, J.W. The role of co-crystals in pharmaceutical design. Trends Pharmacol. Sci. 2013, 34, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regulatory Classification of Pharmaceutical Co-Crystals. Guidance for Industry. 2013; pp. 1–5. Available online: http://www.fda.gov/downloads/Drugs/Guidances/UCM281764.pdf (accessed on 30 June 2018).
- Huang, Y.; Zhang, B.; Gao, Y.; Zhang, J.; Shi, L. Baicalein-nicotinamide cocrystal with enhanced solubility, dissolution, and oral bioavailability. J. Pharm. Sci. 2014, 103, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Hiendrawan, S.; Veriansyah, B.; Widjojokusumo, E.; Soewandhi, S.N.; Wikarsa, S.; Tjandrawinata, R.R. Physicochemical and mechanical properties of paracetamol cocrystal with 5-nitroisophthalic acid. Int. J. Pharm. 2016, 497, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Huang, Y.T.; Lin, S.Y. Spectroscopic and thermal approaches to investigate the formation mechanism of piroxicam-saccharin co-crystal induced by liquid-assisted grinding or thermal stress. J. Therm. Anal. Calorim. 2016, 123, 2345–2356. [Google Scholar] [CrossRef]
- Hong, C.; Xie, Y.; Yao, Y.; Li, G.; Yuan, X.; Shen, H. A Novel strategy for pharmaceutical cocrystal generation without knowledge of stoichiometric ratio: Myricetin cocrystals and a ternary phase diagram. Pharm. Res. 2015, 32, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Chun, N.H.; Wang, I.C.; Liu, J.J.; Jeong, M.Y.; Choi, G.J. Understanding the formation of indomethacin-saccharin cocrystals by anti-solvent crystallization. Cryst. Growth Des. 2013, 13, 2067–2074. [Google Scholar] [CrossRef]
- Ervasti, T.; Aaltonen, J.; Ketolainen, J. Theophylline-nicotinamide cocrystal formation in physical mixture during storage. Int. J. Pharm. 2015, 486, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Zhang, G.C.; Hsu, P.C.; Lin, S.Y. A portable fiber-optic Raman analyzer for fast real-time screening and identifying cocrystal formation of drug-coformer via grinding process. Microchem. J. 2013, 110, 15–20. [Google Scholar] [CrossRef]
- Sarraguça, M.C.; Ribeiro, P.R.; Santos, A.O.; Silva, M.C.; Lopes, J.A. A PAT approach for the on-line monitoring of pharmaceutical co-crystals formation with near infrared spectroscopy. Int. J. Pharm. 2014, 471, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Sarraguça, M.C.; Paisana, M.; Pinto, J.; Lopes, J.A. Real-time monitoring of cocrystallization processes by solvent evaporation: A near infrared study. Eur. J. Pharm. Sci. 2016, 90, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.K.; Lin, S.Y.; Lin, H.L.; Huang, Y.T. Simultaneous DSC-FTIR microspectroscopy used to screen and detect the co-crystal formation in real time. Bioorg. Med. Chem. Lett. 2011, 21, 3148–3151. [Google Scholar] [CrossRef] [PubMed]
- Harriss, B.I.; Vella-Zarb, L.; Wilson, C.; Evans, I.R. Furosemide cocrystals: Structures, hydrogen bonding, and implications for properties. Cryst. Growth Des. 2014, 14, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Sowa, M.; Ślepokura, K.; Matczak-Jon, E. Solid-state characterization and solubility of a genistein-caffeine cocrystal. J. Mol. Struct. 2014, 1076, 80–88. [Google Scholar] [CrossRef]
- Chun, N.H.; Lee, M.J.; Song, G.H.; Chang, K.Y.; Kim, C.S.; Choi, G.J. Combined anti-solvent and cooling method of manufacturing indomethacin-saccharin (IMC-SAC) co-crystal powders. J. Cryst. Growth. 2014, 408, 112–118. [Google Scholar] [CrossRef]
- Rehder, S.; Klukkert, M.; Löbmann, K.A.M.; Strachan, C.J.; Sakmann, A.; Gordon, K.; Rades, T.; Leopold, C.S. Investigation of the formation process of two piracetam cocrystals during grinding. Pharmaceutics 2011, 3, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Basavoju, S.; Boström, D.; Velaga, S.P. Indomethacin-saccharin cocrystal: Design, synthesis and preliminary pharmaceutical characterization. Pharm. Res. 2008, 25, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Chun, N.H.; Wang, I.C.; Lee, M.J.; Jung, Y.T.; Lee, S.; Kim, W.S.; Choi, G.J. Characteristics of indomethacin-saccharin (IMC-SAC) co-crystals prepared by an anti-solvent crystallization process. Eur. J. Pharm. Biopharm. 2013, 85, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Padrela, L.; Rodrigues, M.A.; Velaga, S.P.; Matos, H.A.; de Azevedo, E.G. Formation of indomethacin-saccharin cocrystals using supercritical fluid technology. Eur. J. Pharm. Sci. 2009, 38, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Zhang, G.C.; Lin, S.Y. Real-time co-crystal screening and formation between indomethacin and saccharin via DSC analytical technique or DSC-FTIR microspectroscopy. J. Therm. Anal. Calorim. 2015, 120, 679–687. [Google Scholar] [CrossRef]
- Goud, R.N.; Gangavaram, S.; Suresh, K.; Pal, S.; Manjunatha, S.G.; Nambiar, S.; Nangia, A. Novel furosemide cocrystals and selection of high solubility drugs forms. J. Pharm. Sci. 2012, 101, 664–680. [Google Scholar] [CrossRef] [PubMed]
- Khomane, K.S.; More, P.K.; Raghavendra, G.; Bansal, A.K. Molecular understanding of the compaction behavior of indomethacin polymorphs. Mol. Pharm. 2013, 10, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Roopa, B.N.; Abu, K.; Manjunath, S.G.; Nambiar, S. Thermal studies of furosemide-caffeine binary system that forms a cocrystal. J. Therm. Anal. Calorim. 2014, 115, 2261–2268. [Google Scholar] [CrossRef]
- Zhang, G.C.; Lin, H.L.; Lin, S.Y. Thermal analysis and FTIR spectral curve-fitting investigation of formation mechanism and stability of indomethacin-saccharin cocrystals via solid-state grinding process. J. Pharm. Biomed. Anal. 2012, 66, 162–169. [Google Scholar] [CrossRef] [PubMed]
- De Cássia da Silva, R.; Semaan, F.S.; Novák, C.; Cavalheiro, E.T.G. Thermal behavior of furosemide. J. Therm. Anal. Calorim. 2013, 111, 1933–1937. [Google Scholar] [CrossRef]
- Teixeira, J.A.; Nunes, W.D.G.; Colman, T.A.D.; do Nascimento, A.L.C.S.; Caires, F.J. Thermal and spectroscopic study to investigate p-aminobenzoic acid, sodium p-aminobenzoate and its compounds with some lighter trivalent lanthanides. Thermochim. Acta 2016, 624, 59–68. [Google Scholar] [CrossRef]
- Garg, R.K.; Sarkar, D. Polymorphism control of p-aminobenzoic acid by isothermal anti-solvent crystallization. J. Cryst. Growth 2016, 454, 180–185. [Google Scholar] [CrossRef]
- Ali, H.; Singh, S.K.; Verma, P.R.P. Preformulation and physicochemical interaction study of furosemide with different solid lipids. J. Pharm. Investig. 2015, 45, 385–398. [Google Scholar] [CrossRef]
- Świsłocka, R.; Samsonowicz, M.; Regulska, E.; Lewandowski, W. Molecular structure of 4-aminobenzoic acid salts with alkali metals. J. Mol. Struct. 2006, 792–793, 227–238. [Google Scholar] [CrossRef]
- Rinnan, Å.; van den Berg, F.; Engelsen, S.B. Review of the most common pre-processing techniques for near-infrared spectra. Trends Anal. Chem. 2009, 10, 1201–1222. [Google Scholar] [CrossRef]
- Caliandro, R.; Di Profio, G.; Nicolotti, O. Multivariate analysis of quaternary carbamazepine-saccharin mixtures by X-ray diffraction and infrared spectroscopy. J. Pharm. Biomed. Anal. 2013, 78–79, 269–279. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Principal Component | Indomethacin and Saccharin | Furosemide and p-Aminobenzoic Acid | |||||
|---|---|---|---|---|---|---|---|
| Matrix I | Matrix II | Matrix III | Matrix I | Matrix II | Matrix III | ||
| Calculation for DSC Data | |||||||
| PC1 | Eigenvalue | 18.23 | 6.70 | 11.58 | 21.80 | 8.59 | 15.88 |
| Variance (%) | 59.04 | 63.90 | 50.04 | 62.94 | 67.49 | 54.78 | |
| PC2 | Eigenvalue | 9.64 | 2.14 | 6.40 | 8.25 | 2.10 | 5.70 |
| Variance (%) | 31.20 (90.24) | 20.41 (84.31) | 27.65 (77.69) | 23.81 (86.76) | 16.47 (83.96) | 19.66 (74.44) | |
| PC3 | Eigenvalue | 2.46 | 1.12 | 2.62 | 2.77 | 1.77 | 4.57 |
| Variance (%) | 7.97 (98.21) | 10.67 (94.98) | 11.30 (88.99) | 8.01 (94.77) | 13.87 (97.83) | 15.77 (90.21) | |
| Calculation for FTIR Data | |||||||
| PC1 | Eigenvalue | 1396.59 | 293.44 | 707.85 | 154.68 | 17.30 | 92.34 |
| Variance (%) | 95.00 | 90.42 | 90.91 | 85.63 | 51.93 | 79.14 | |
| PC2 | Eigenvalue | 68.03 | 20.14 | 53.41 | 21.10 | 13.44 | 13.84 |
| Variance (%) | 4.63 (99.63) | 6.21 (96.63) | 6.86 (97.77) | 11.68 (97.31) | 40.36 (92.29) | 11.87 (91.06) | |
| PC3 | Eigenvalue | 4.77 | 8.77 | 13.27 | 4.28 | 1.66 | 6.21 |
| Variance (%) | 0.32 (99.95) | 2.70 (99.33) | 1.70 (99.47) | 2.37 (99.68) | 4.97 (97.26) | 5.32 (96.38) | |
| Calculation for Raman Data | |||||||
| PC1 | Eigenvalue | 157.99 | 305.50 | 101.11 | 226.53 | 153.49 | 171.22 |
| Variance (%) | 48.52 | 80.41 | 41.47 | 65.47 | 71.19 | 65.69 | |
| PC2 | Eigenvalue | 106.56 | 58.91 | 94.05 | 68.41 | 47.80 | 53.35 |
| Variance (%) | 32.73 (81.25) | 15.51 (95.92) | 38.58 (80.05) | 19.77 (85.24) | 22.17 (93.36) | 20.47 (86.16) | |
| PC3 | Eigenvalue | 58.68 | 9.56 | 41.39 | 50.25 | 13.53 | 34.98 |
| Variance (%) | 18.02 (99.27) | 2.52 (98.43) | 16.98 (97.02) | 14.52 (99.76) | 6.28 (99.64) | 13.42 (99.58) | |
| Principal Component | Indomethacin and Saccharin | Furosemide and p-Aminobenzoic Acid | |||||
|---|---|---|---|---|---|---|---|
| Matrix I | Matrix II | Matrix III | Matrix I | Matrix II | Matrix III | ||
| Calculation for FTIR Data | |||||||
| PC1 | Wavenumber (cm−1) | 1240–1216 1363–1305 1717–1679 | 1683–1679 1328–1317 1224, 1737 | 1232–1220 1683–1679 1324–1313 1714–1710 | 773–769 553–549 1425–1421 842, 3461 | 1425–1421 773–769, 3461 | 773–769 1425–1421, 3461 |
| PC2 | Wavenumber (cm−1) | 1691, 1336 1737, 518–511 | 1695–1691, 1066 838–804, 1027 | 1691, 518 1737, 804 | 1178–1170 1340, 1629–1621 | 1344–1340 1166, 730 | 1178–1170 1340, 1629–1621 |
| PC3 | Wavenumber (cm−1) | 1726–1718 1186–1182 1232–1224 | 526–507 1232–1220 1683–1679 | 518–507 1224, 1683–1679 | 1425–1421 1309, 730 | 769, 410–403 3492–3488 | 1425–1421 1309, 730 |
| Calculation for Raman Data | |||||||
| PC1 | Wavenumber (cm−1) | 1622–1614 1583–1575, 735 | 1703–1695 1683–1680, 1155 | 1703–1695 1683–1680 1622–1618, 1579 | 1606–1595, 843 1286–1278 | 1610–1591 1286–1278, 843 | 1614–1595 846–843 1286–1278 |
| PC2 | Wavenumber (cm−1) | 1683–1680 1699–1695 1174, 704 | 1699–1695 704–700, 1174 | 1699–1695, 1174 1618–1614 | 1174–1143 1602–1595 1286–1282 | 1170–1163 1506, 1159, 1255 | 1147–1124 1174–1163 850–846 |
| PC3 | Wavenumber (cm−1) | 1699–1695 1174, 704–700 | 384, 1178–1174 735, 1618 | 1699–1695, 1174 704–700 | 684–681 1147–1120 1506–1502 | 1151–1143 684–681 1506–1502 | 683–681 1151–1143 1506–1502 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbacz, P.; Wesolowski, M. DSC, FTIR and Raman Spectroscopy Coupled with Multivariate Analysis in a Study of Co-Crystals of Pharmaceutical Interest. Molecules 2018, 23, 2136. https://doi.org/10.3390/molecules23092136
Garbacz P, Wesolowski M. DSC, FTIR and Raman Spectroscopy Coupled with Multivariate Analysis in a Study of Co-Crystals of Pharmaceutical Interest. Molecules. 2018; 23(9):2136. https://doi.org/10.3390/molecules23092136
Chicago/Turabian StyleGarbacz, Patrycja, and Marek Wesolowski. 2018. "DSC, FTIR and Raman Spectroscopy Coupled with Multivariate Analysis in a Study of Co-Crystals of Pharmaceutical Interest" Molecules 23, no. 9: 2136. https://doi.org/10.3390/molecules23092136
APA StyleGarbacz, P., & Wesolowski, M. (2018). DSC, FTIR and Raman Spectroscopy Coupled with Multivariate Analysis in a Study of Co-Crystals of Pharmaceutical Interest. Molecules, 23(9), 2136. https://doi.org/10.3390/molecules23092136