
Evaluation of the Antioxidant and Neuroprotectant Activities of New Asymmetrical 1,3-Diketones

 , , ,
, , ,  ,
,
Abstract
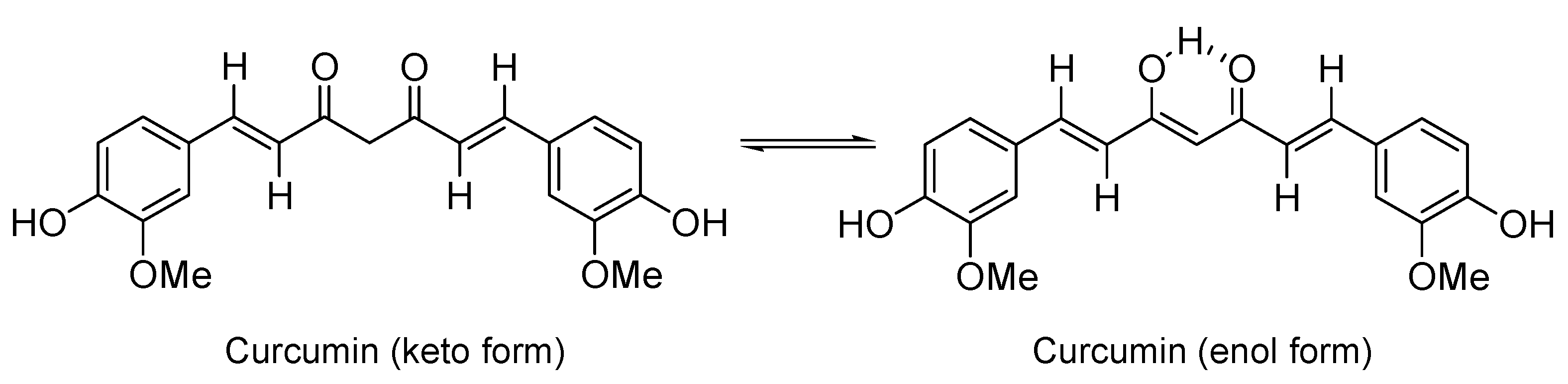
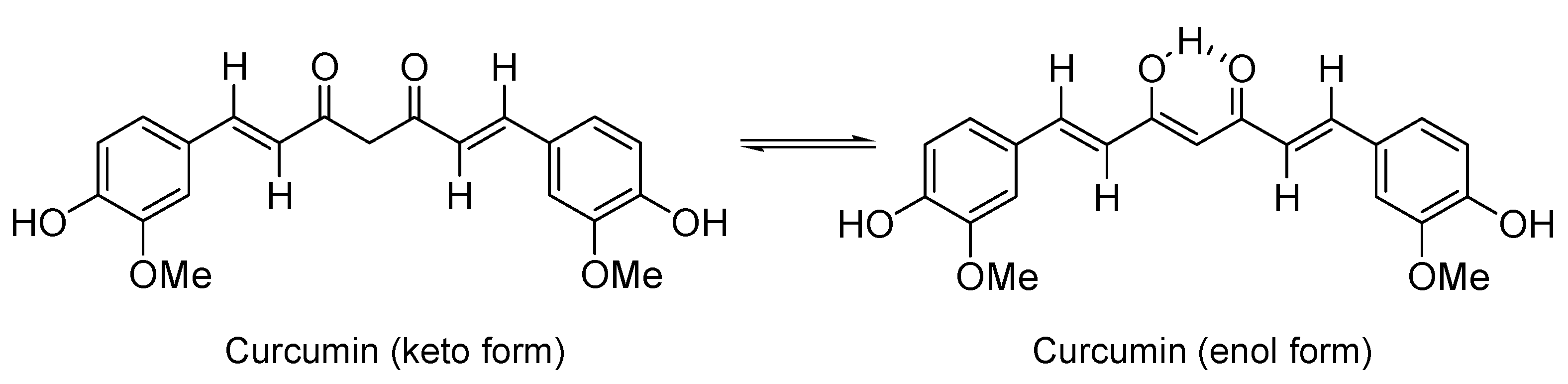
:1. Introduction
2. Results and Discussion
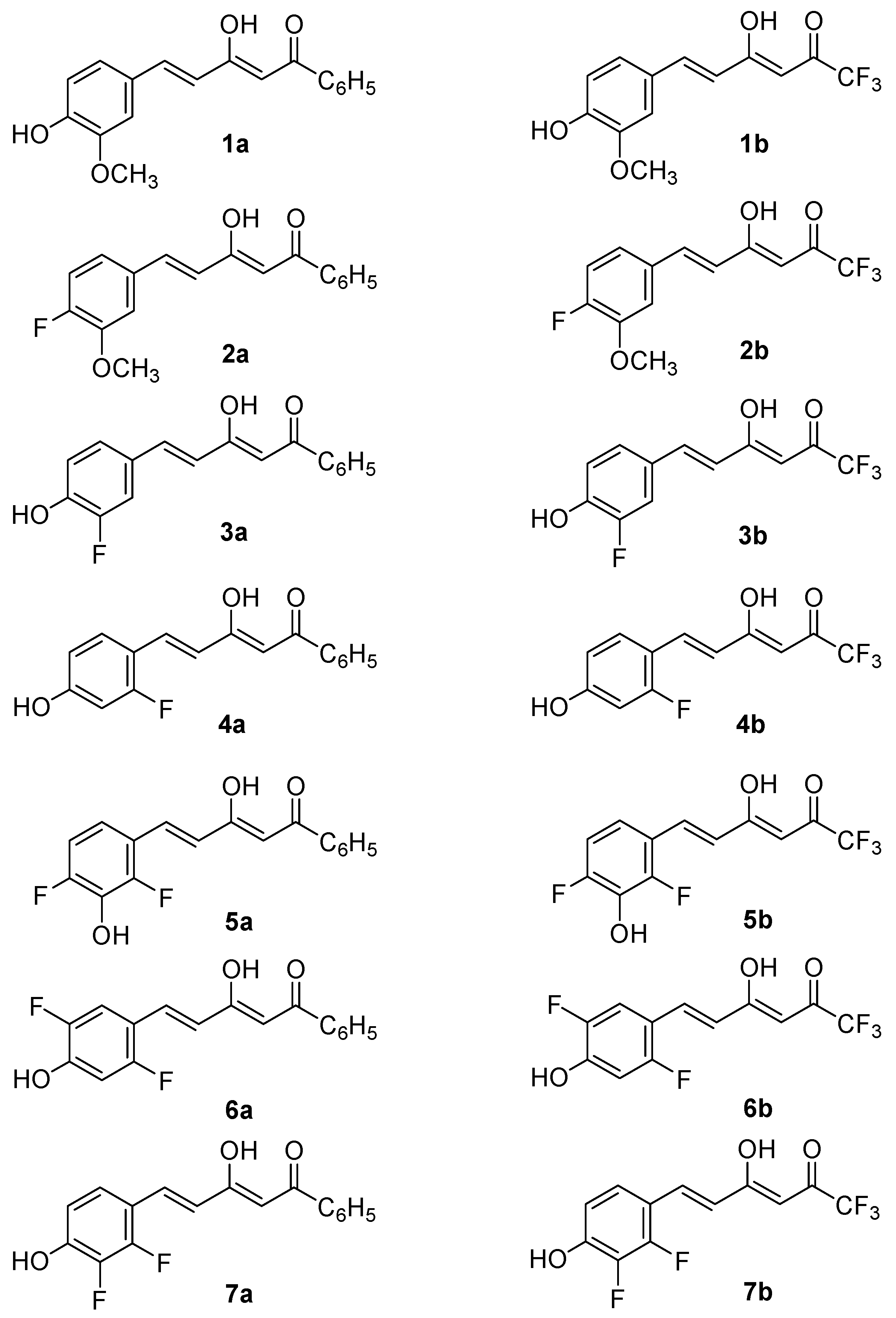
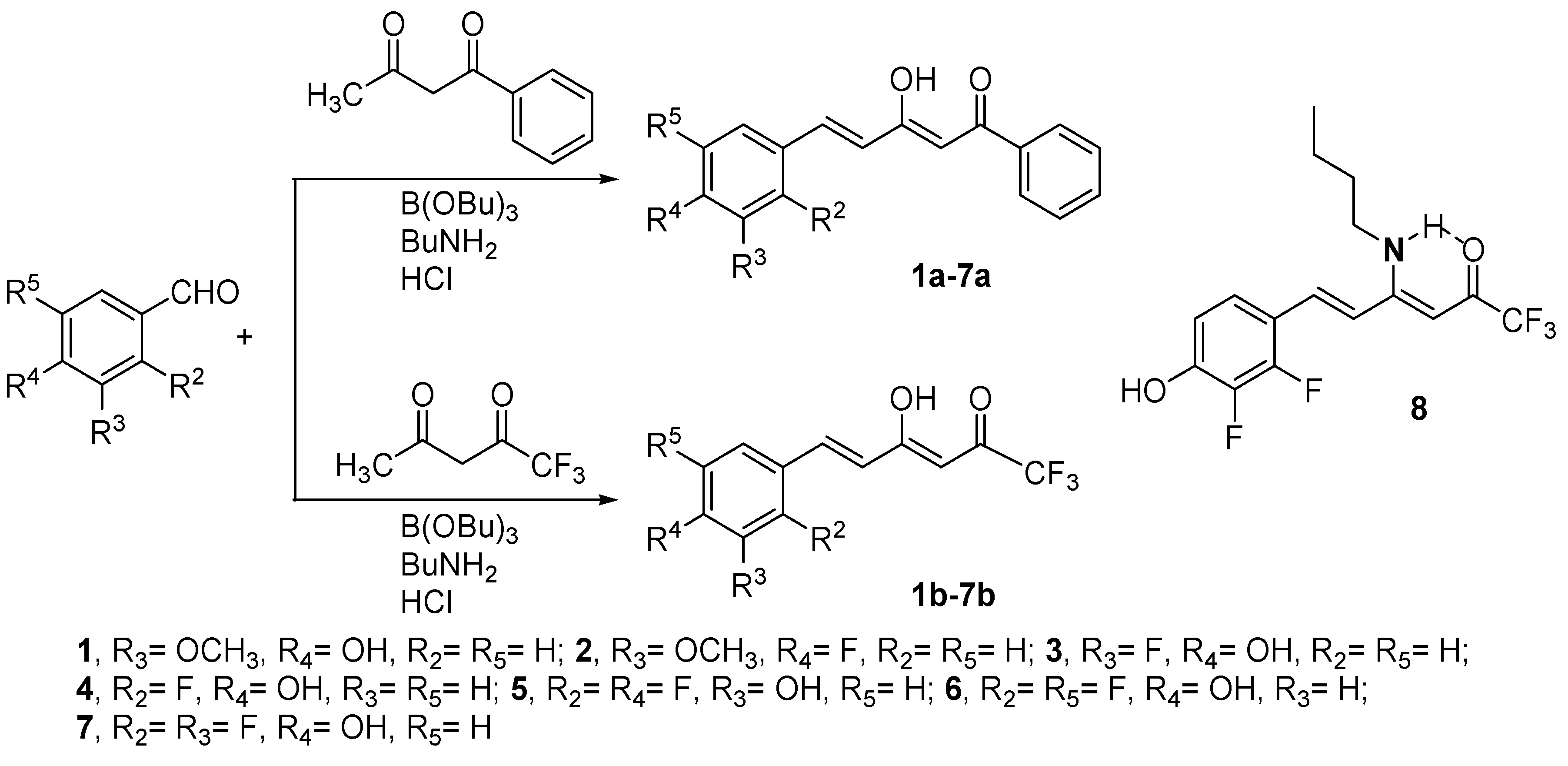
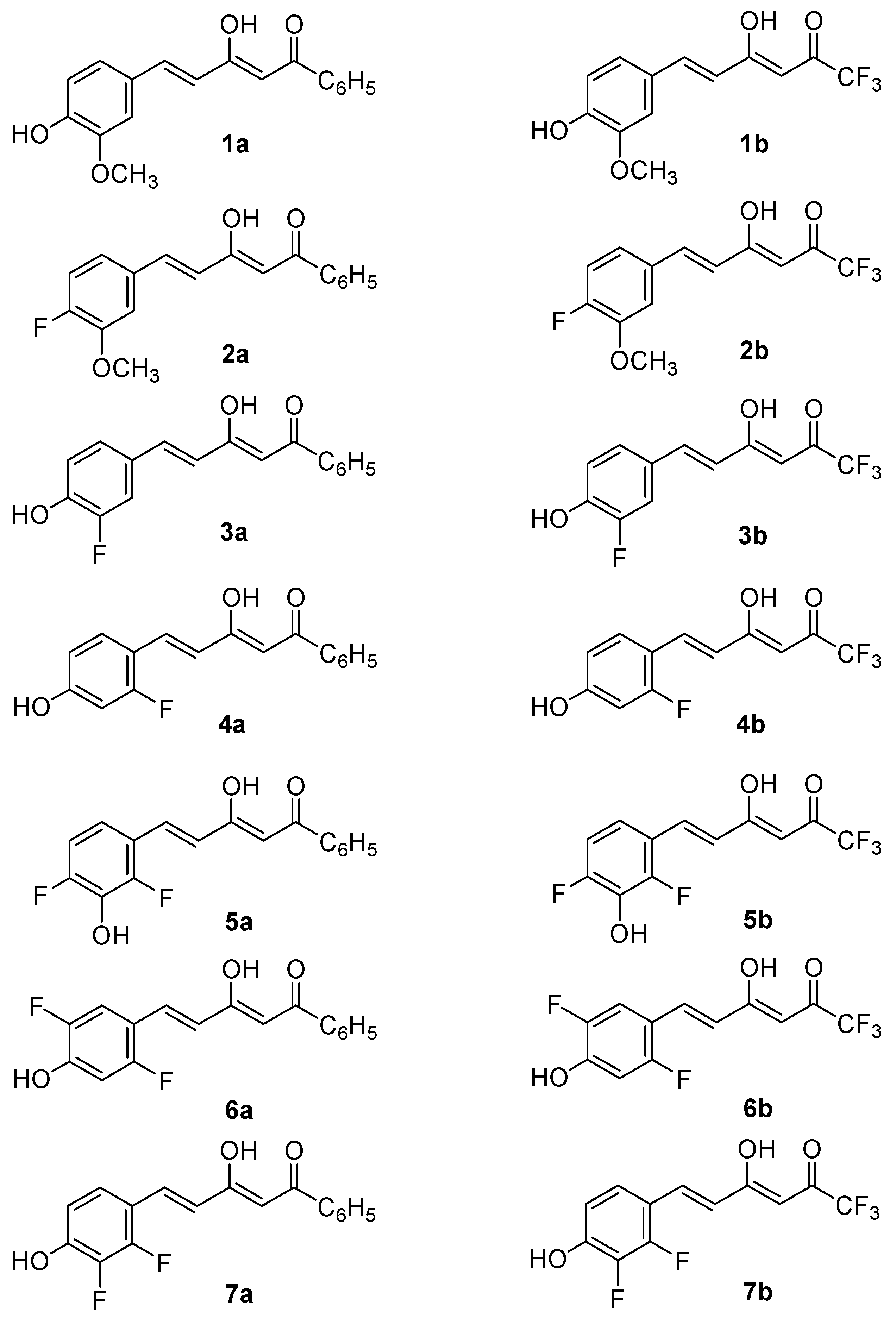
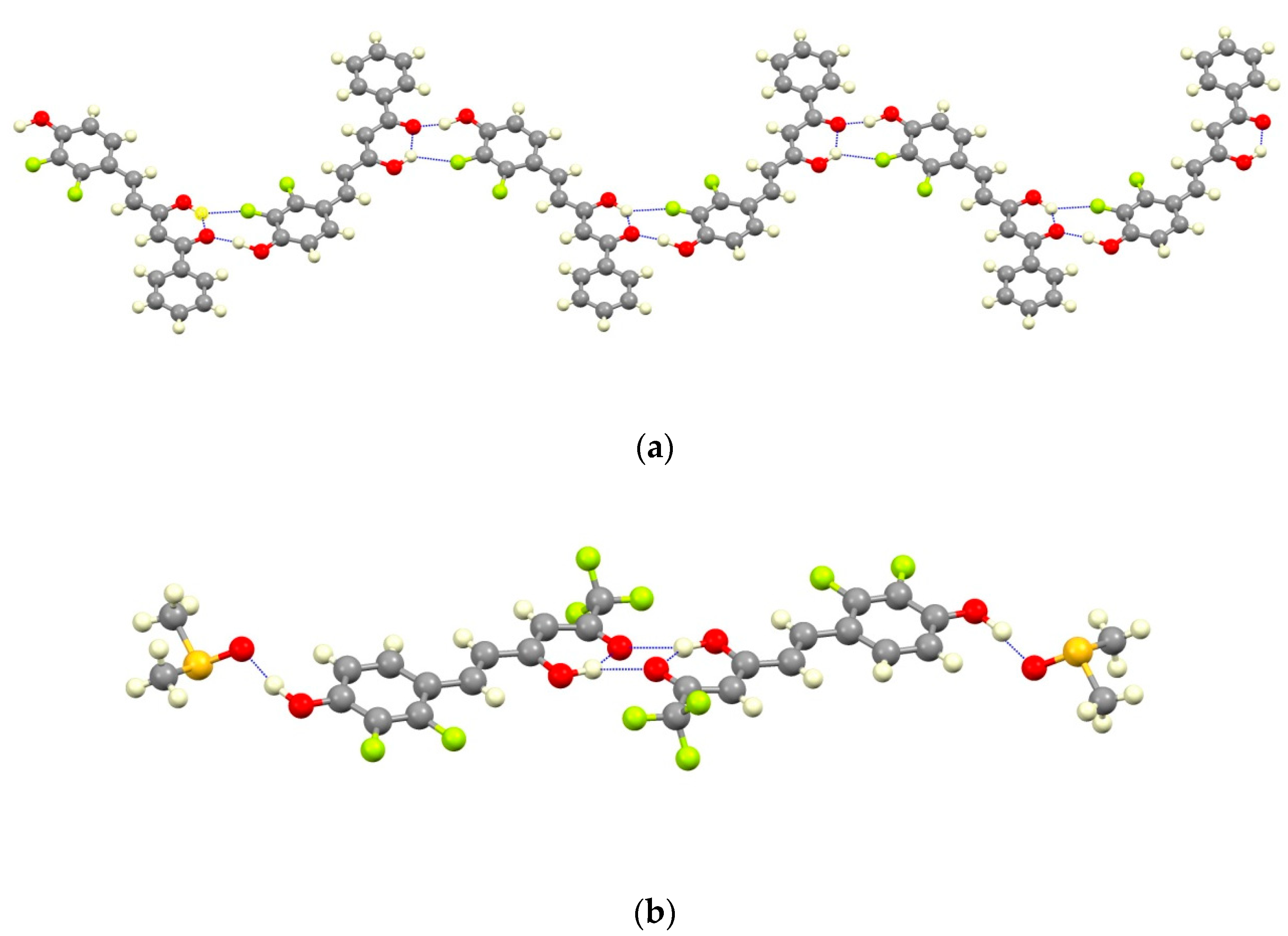
2.1. Synthesis and Structure Characterization
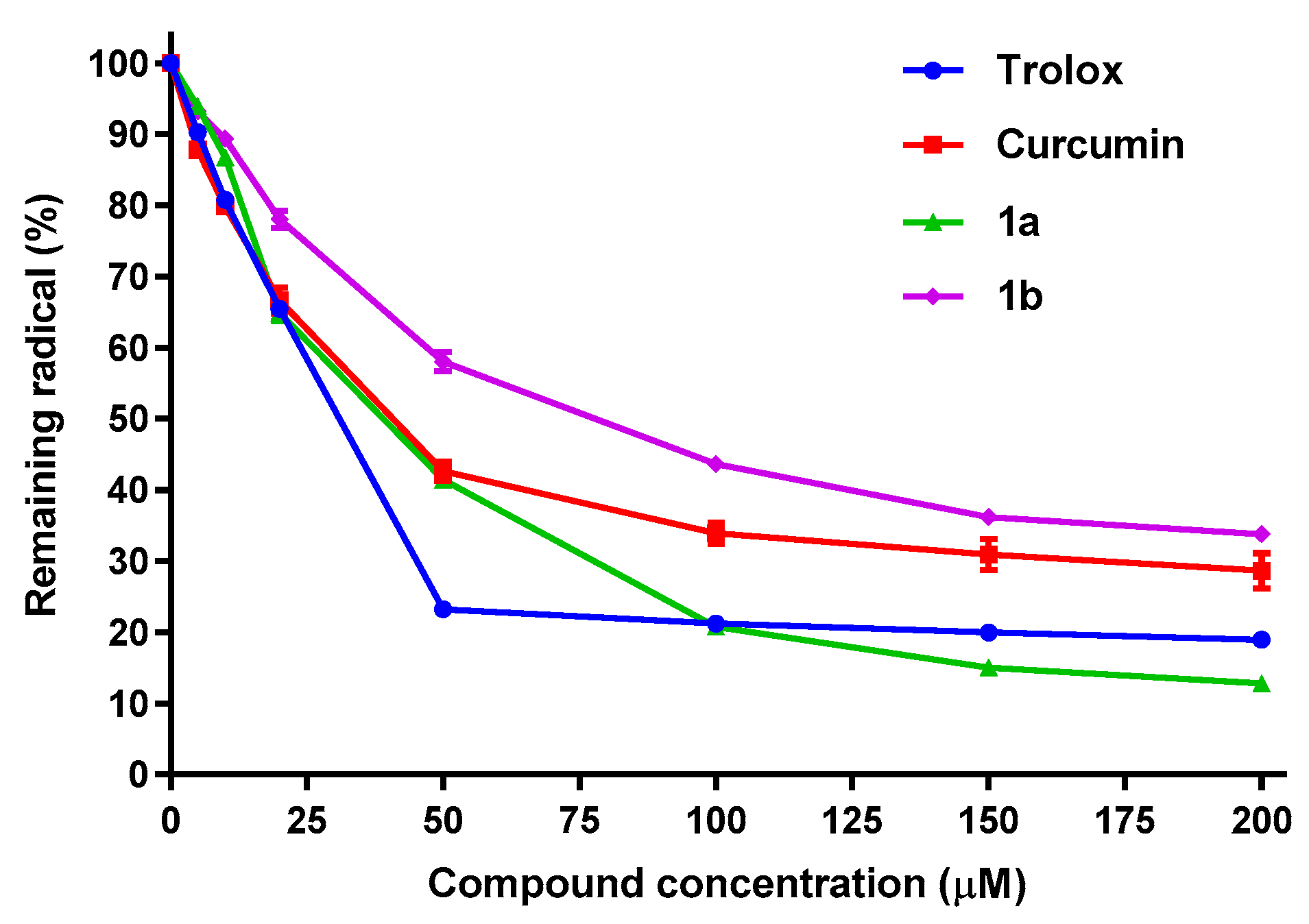
2.2. Antioxidant Properties
2.3. Calculation of Physicochemical Properties
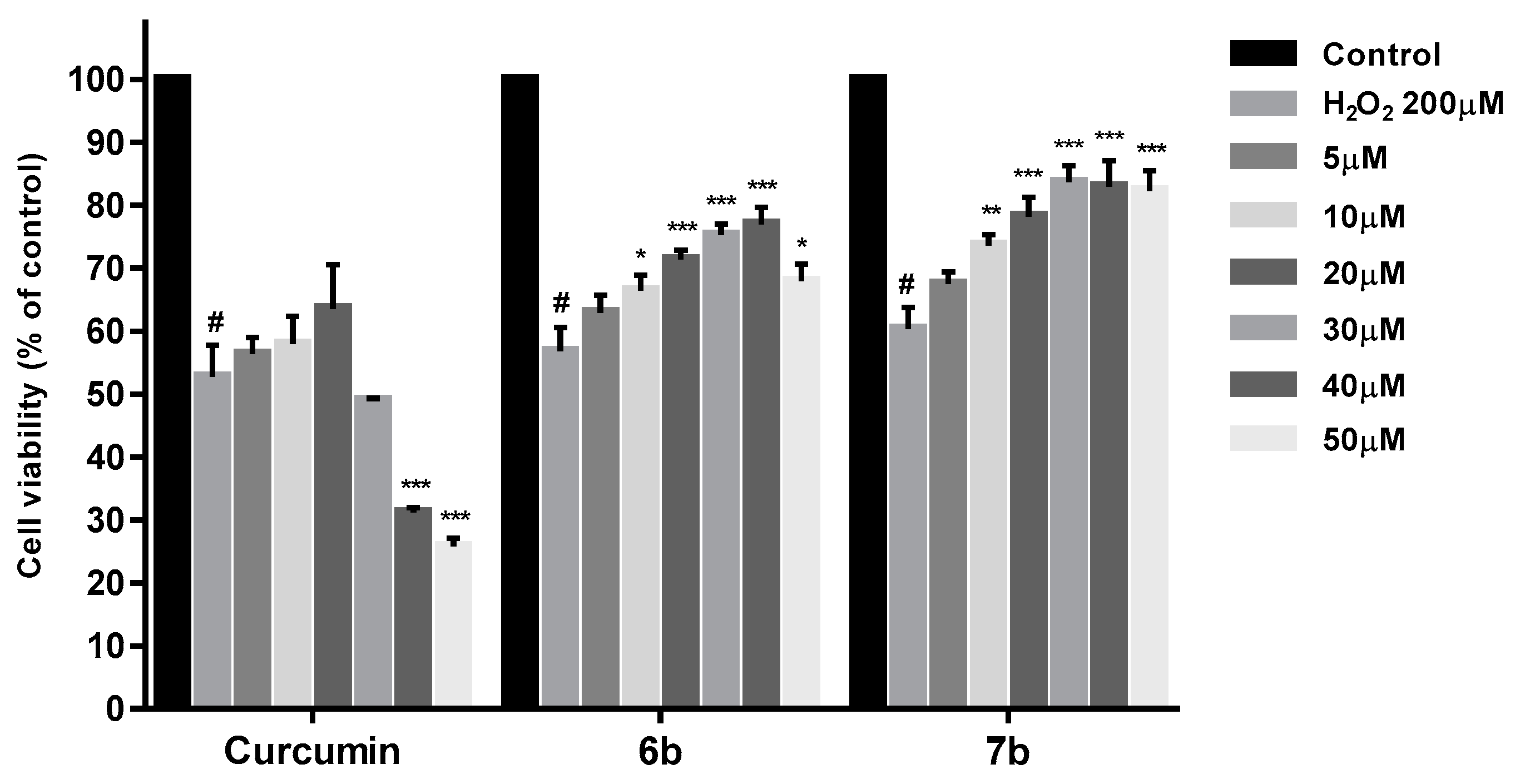
2.4. Cytotoxicity Studies
2.5. Determination of Kinetic Solubility
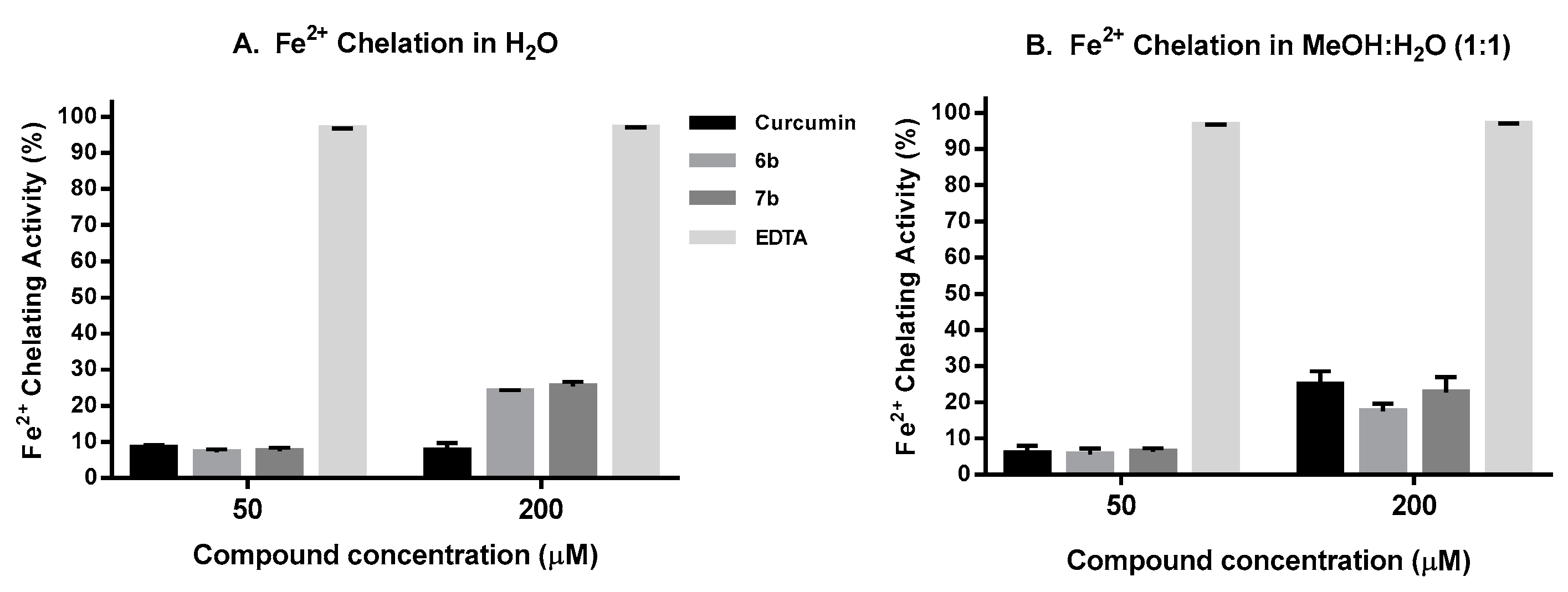
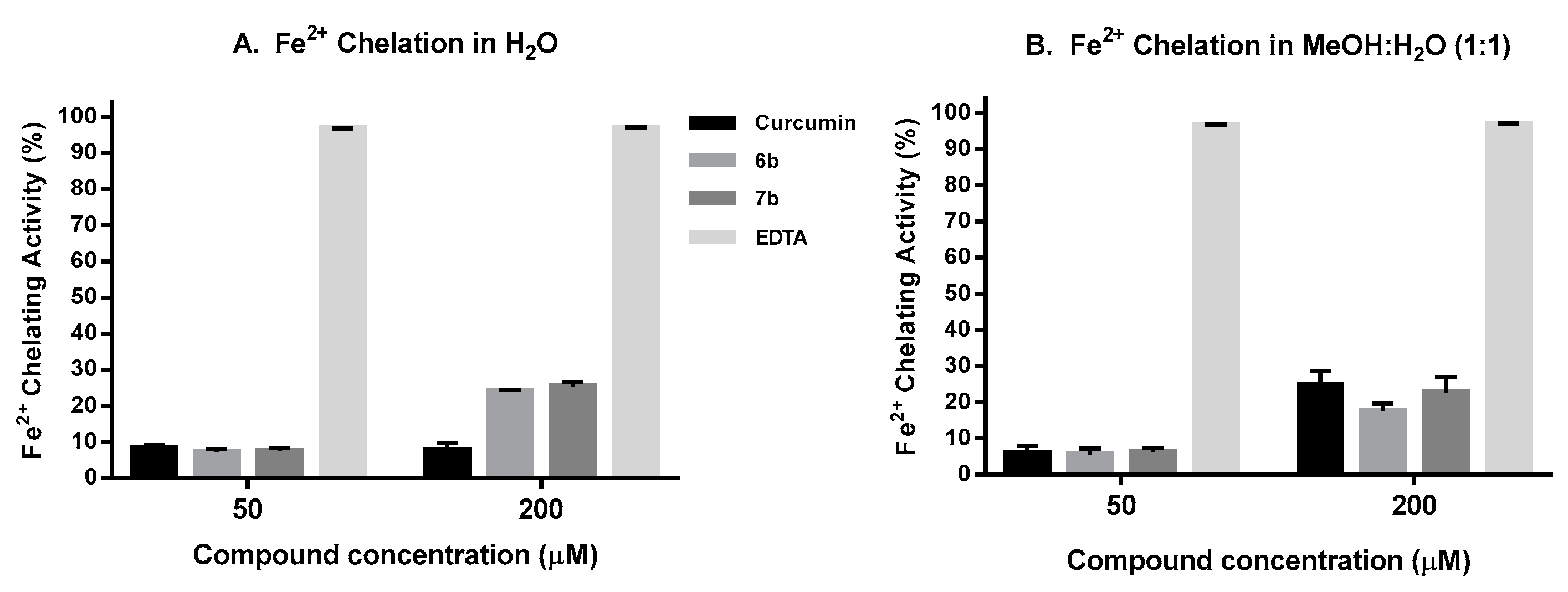
2.6. Iron Chelation Activity
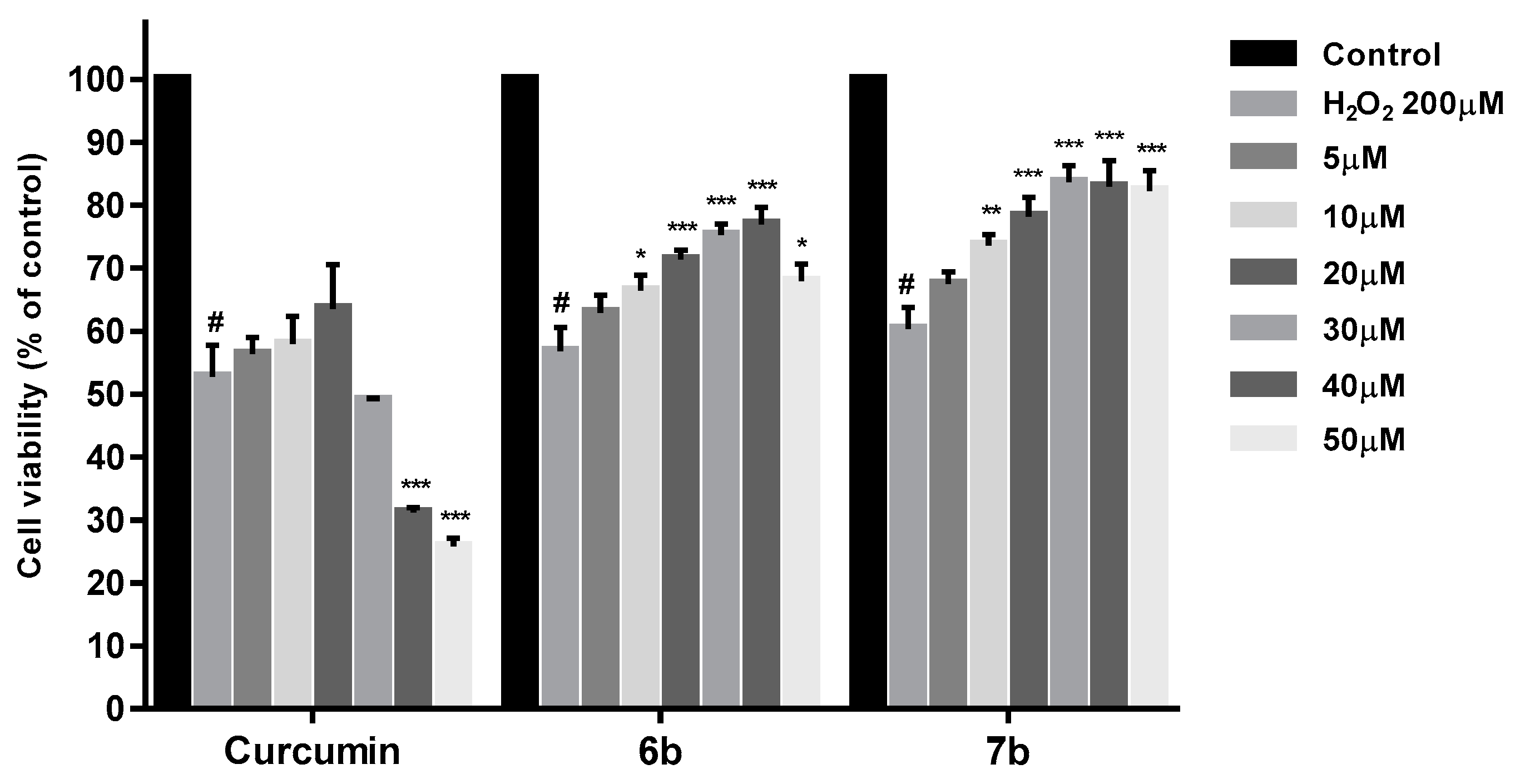
2.7. Neuroprotection Studies
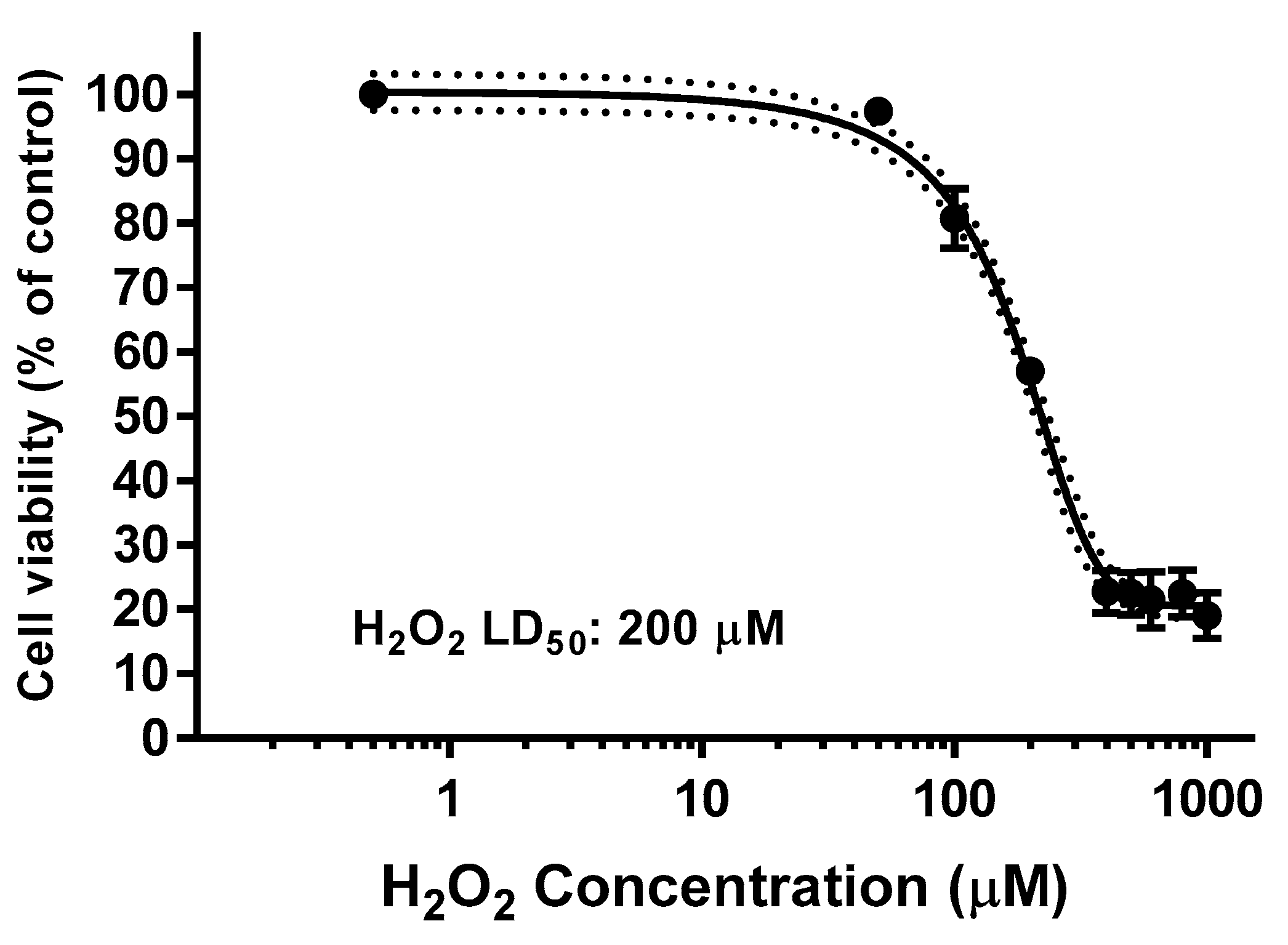
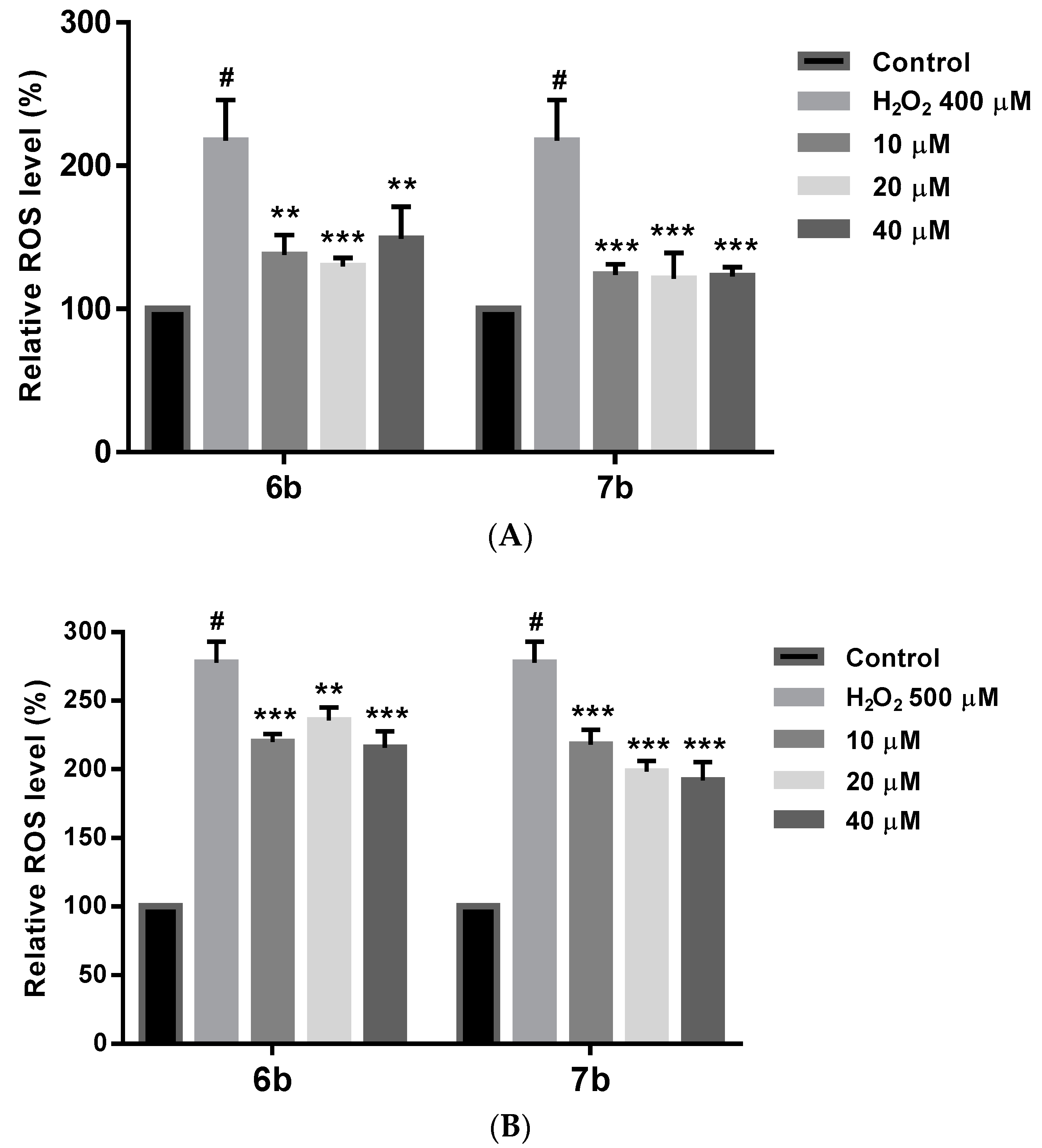
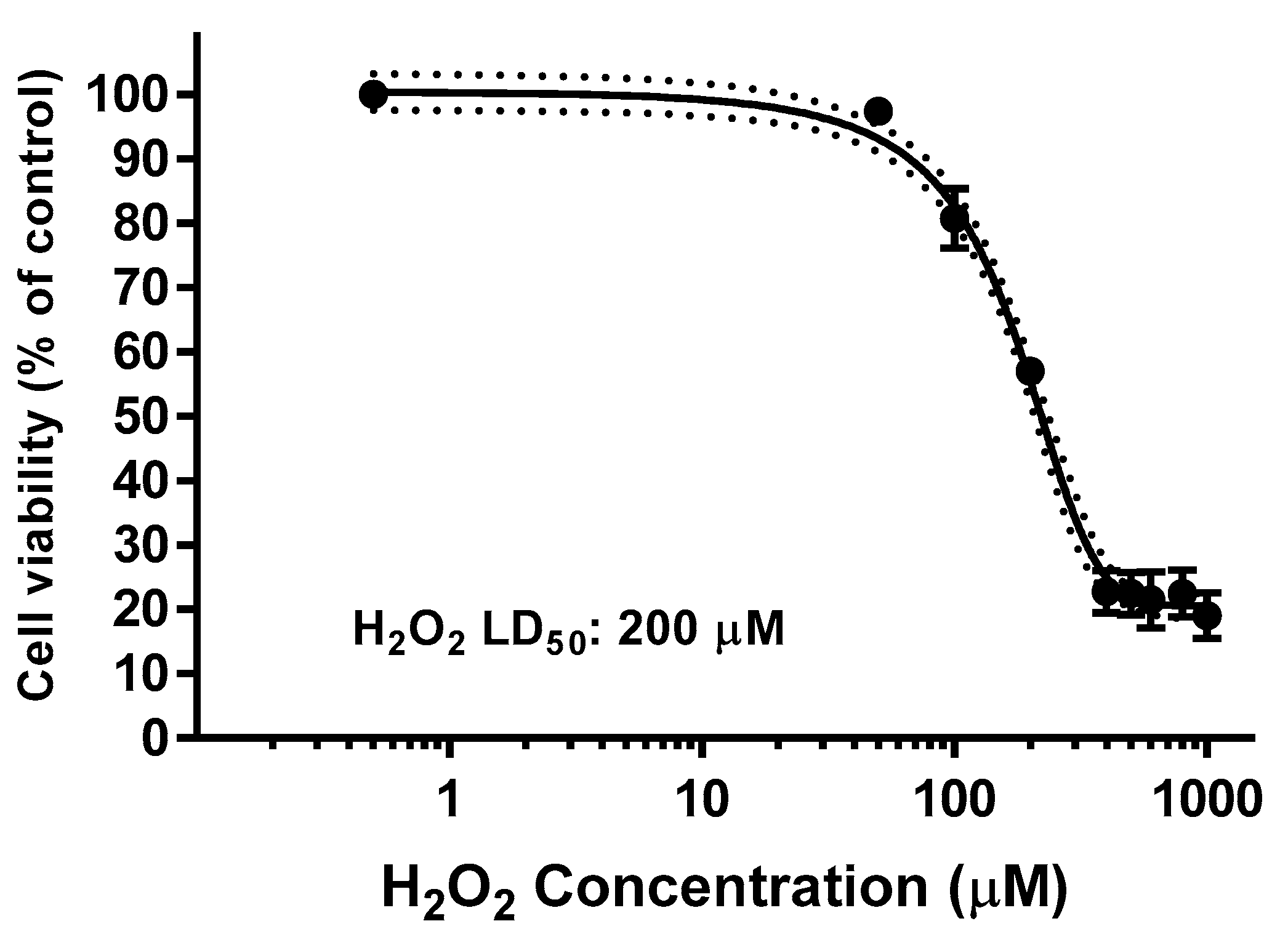
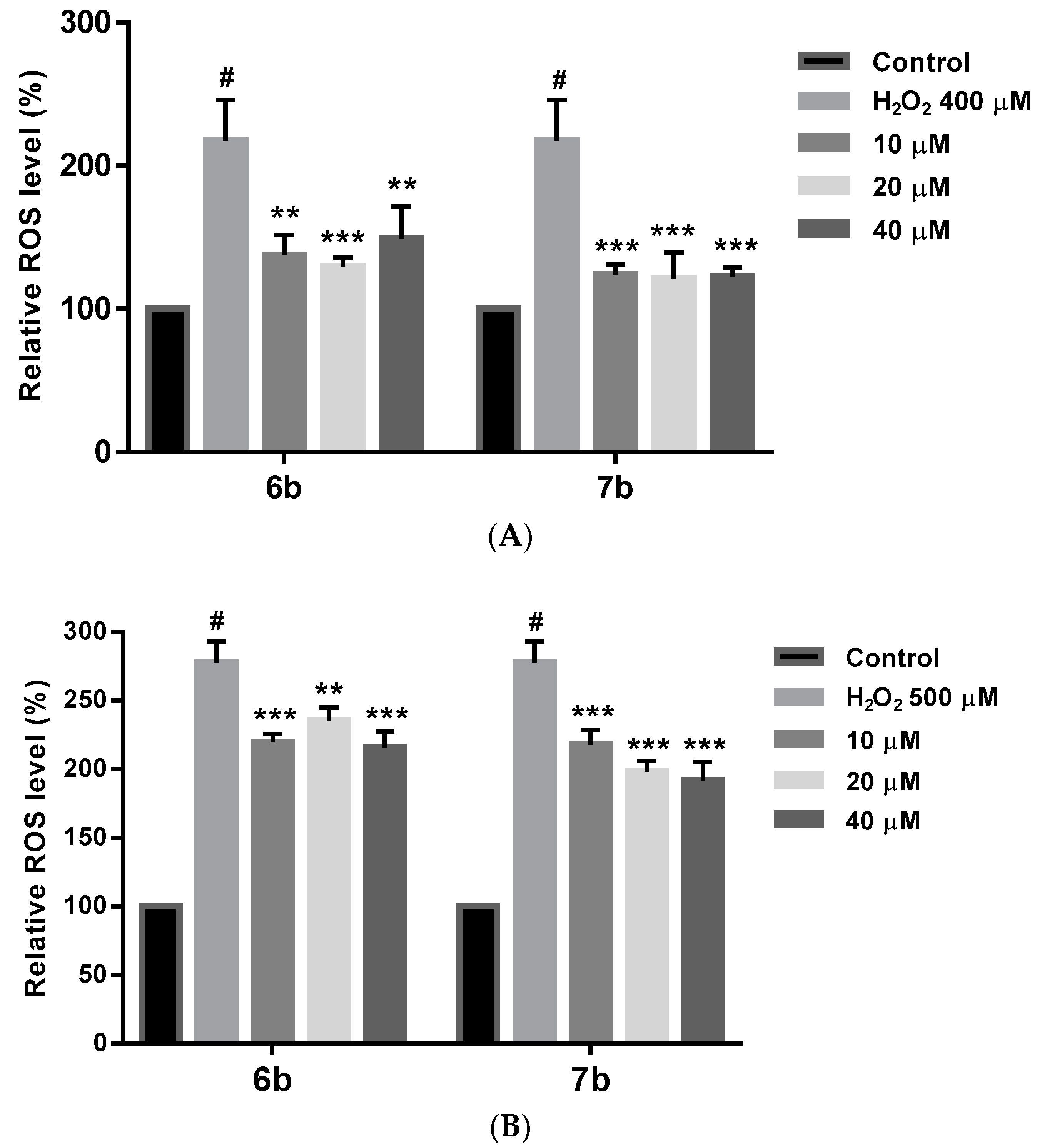
2.8. Effect on ROS Levels in SH-SY5Y Cells under Oxidative Stress
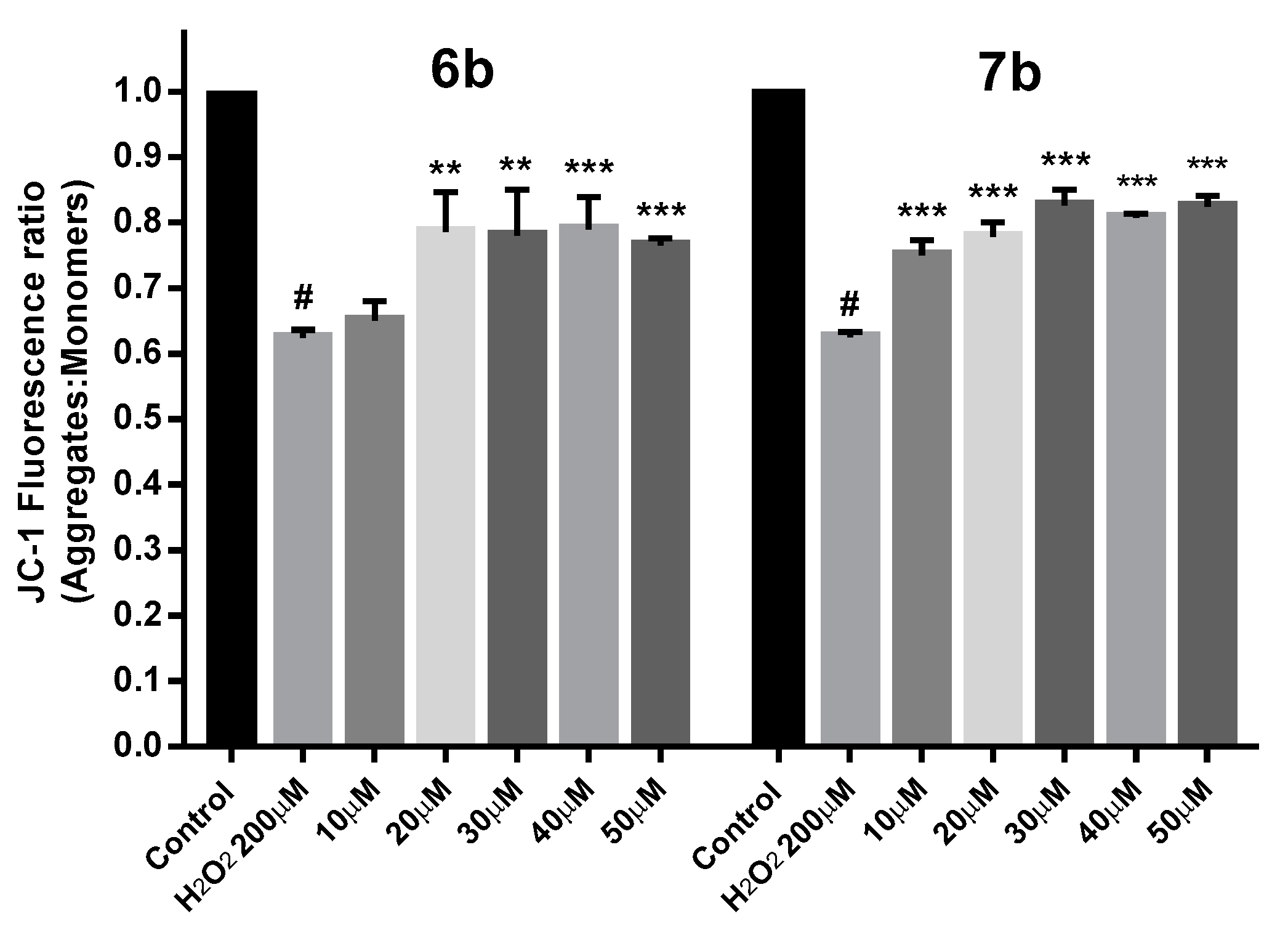
2.9. Protection against Mitochondrial Membrane Depolarization Induced by Oxidative Stress
3. Conclusions
4. Experimental Section
4.1. General
4.2. NMR Parameters
4.3. Preparation of Compounds 1a–7a and 1b–7b
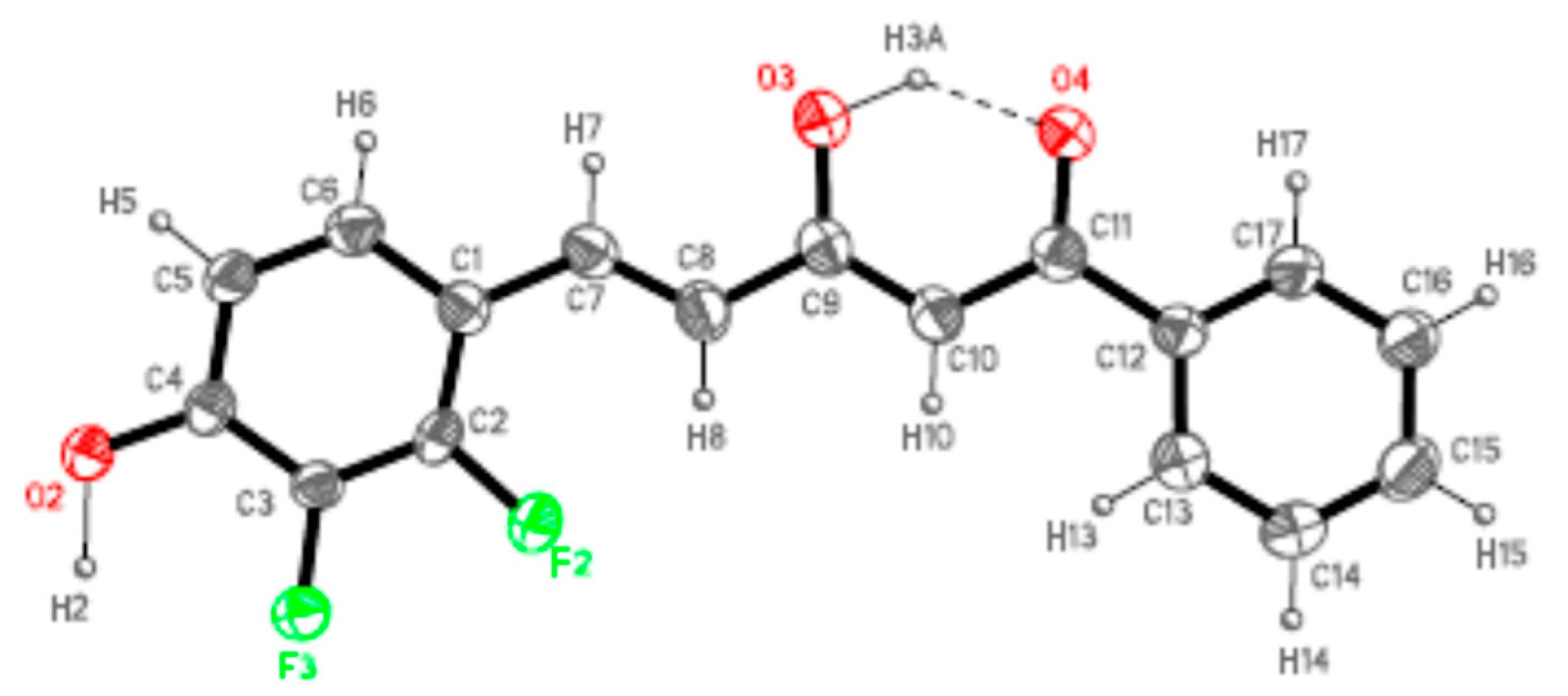
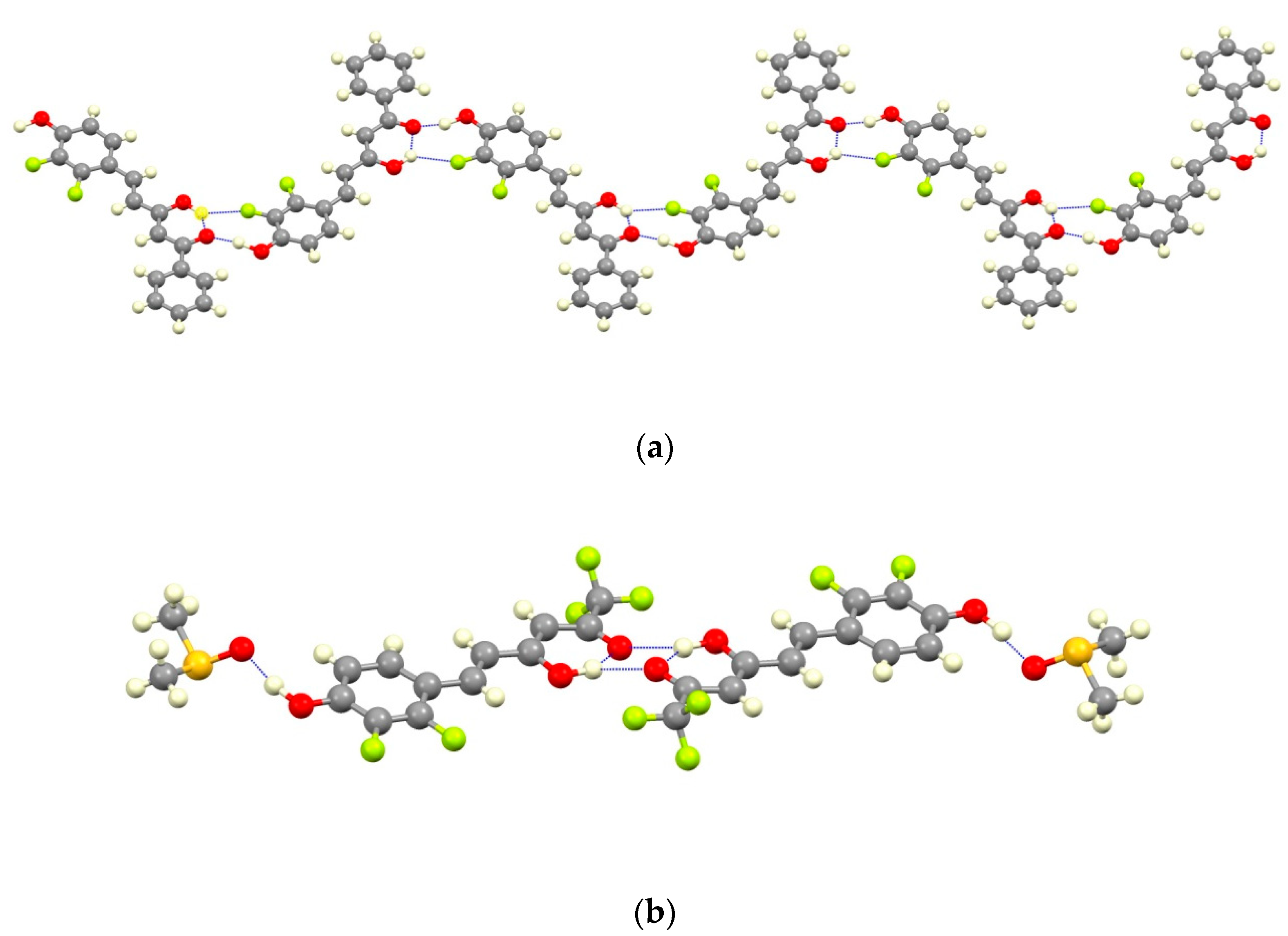
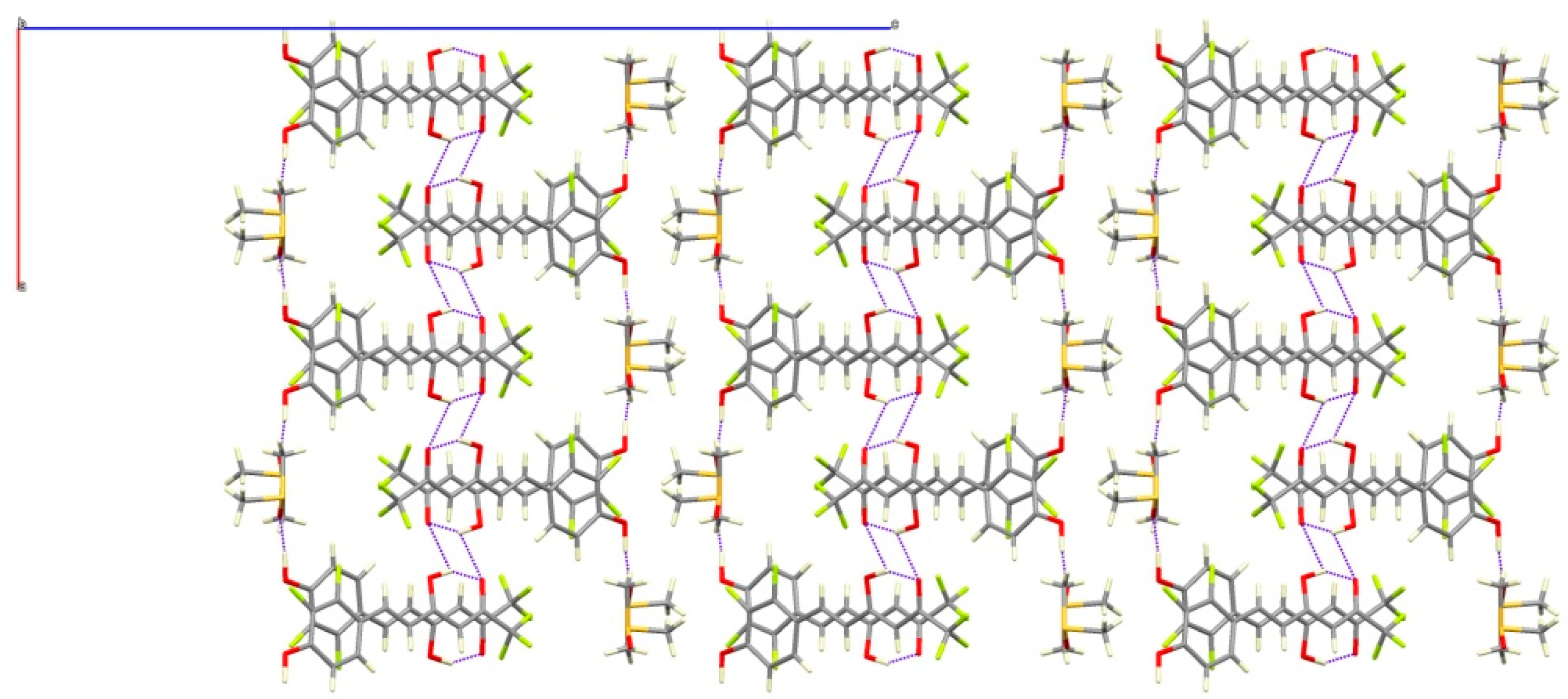
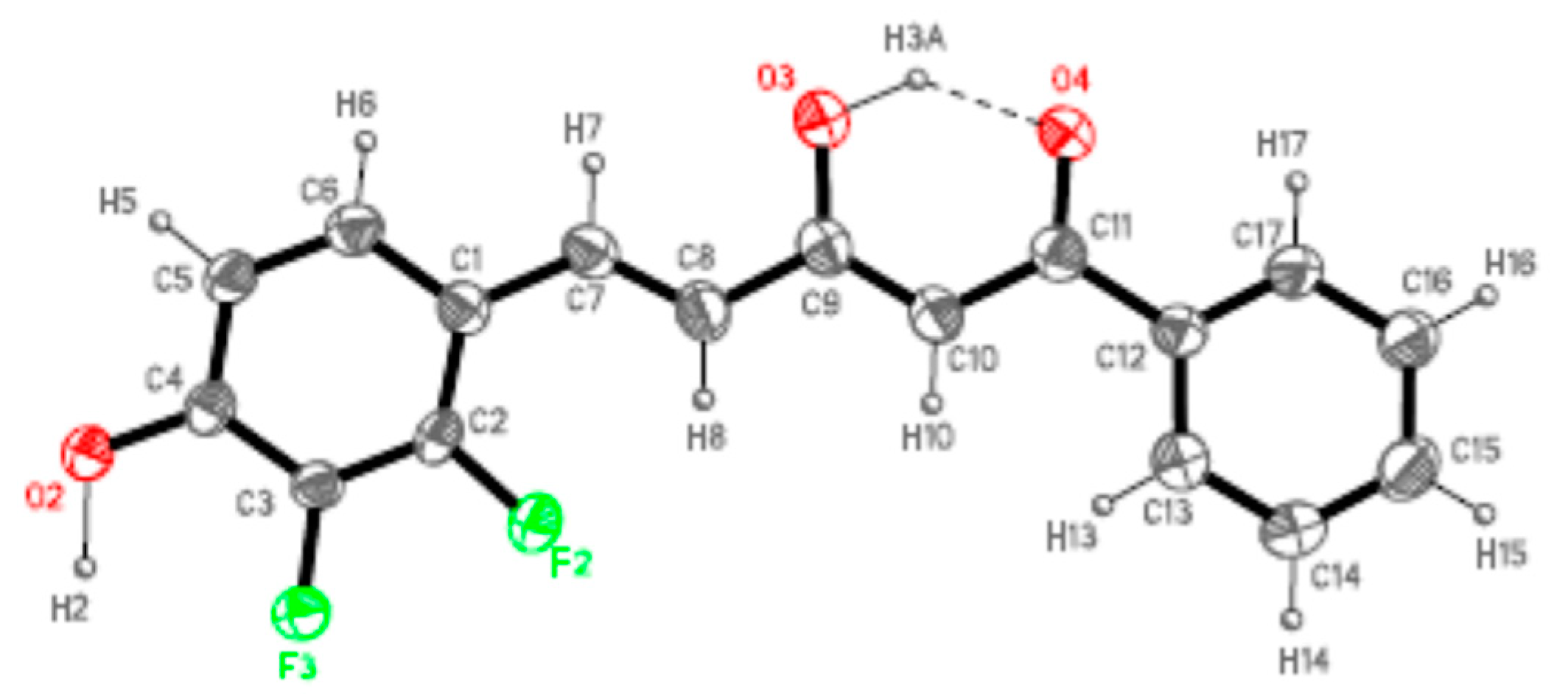
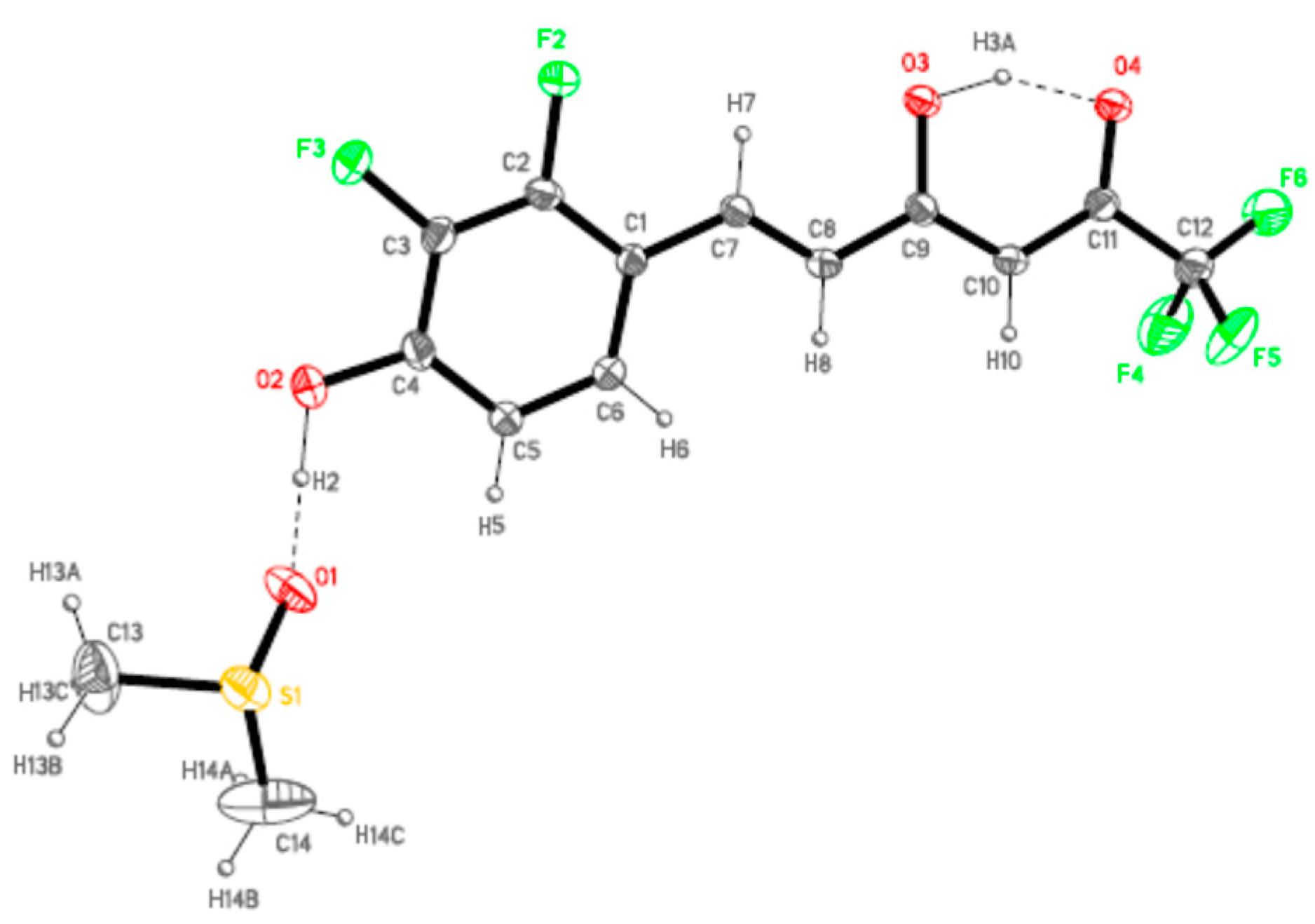
4.4. X-ray Data Collection and Structure Refinement
4.5. Radical Scavenger Capacity Determination
4.5.1. ABTS Assay
4.5.2. FRAP (Ferric Reducing Antioxidant Power) Assay
4.5.3. DPPH Assay
4.6. Cell Culture
4.7. Cytotoxicity Activity In Vitro in SH-SY5Y Cell Line
4.8. SH-SY5Y Cell Neuroprotection Assay
4.9. Lactate Dehydrogenase (LDH) Cell Viability Assay
4.10. Kinetic Solubility by UV-Spectrometry Assay
4.11. Iron Chelation Capacity
4.12. Estimation of Intracellular Reactive Oxygen Species (ROS)
4.13. Determination of Mitochondrial Membrane Potential ΔΨm (MMP)
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- ONU. World Population Ageing; United Nations, Department of Economic Social Affairs Population Division: New York, NY, USA, 2015; 164p. [Google Scholar]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.; Alafuzoff, I. Altered proteins in the aging brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, S.T.; Graham, D.I. Aging-associated changes in human brain. J. Neuropathol. Exp. Neurol. 1997, 56, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Szeto, J.Y.Y.; Lewis, S.J.G. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, A.; Piplani, P. Current pharmacotherapy and putative disease-modifying therapy for Alzheimer’s disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2016, 37, 1403–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Dong, J.; Cheng, C.; Le, W. Therapies for Parkinson’s diseases: Alternatives to current pharmacological interventions. J. Neural Transm. 2016, 123, 1279–1299. [Google Scholar] [CrossRef] [PubMed]
- Huot, P.; Fox, S.H.; Brotchie, J.M. Dopamine reuptake inhibitors in Parkinsons disease: A review of nonhuman primate studies and clinical trials. J. Pharmacol. Exp. Ther. 2016, 357, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Lü, Y.; Zhang, M. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Schapira, A. The genetics of Parkinson’s disease. Br. Med. Bull. 2015, 114, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.; Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 2017, 135, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef] [PubMed]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Coppede, F.; Migliore, L.; Siciliano, G.; Murri, L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimer’s Dis. 2006, 10, 59–73. [Google Scholar] [CrossRef]
- Onyango, I.; Khan, S.; Miller, B.; Swerdlow, R.; Trimmer, P.; Bennett, P.J. Mitochondrial genomic contribution to mitochondrial dysfunction in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 183–193. [Google Scholar] [CrossRef]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Castello, M.A.; Soriano, S. On the origin of Alzheimer’s disease. Trials and tribulations of the amyloid hypothesis. Ageing Res. Rev. 2014, 13, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. Propagation of Tau aggregates Tim Bliss. Mol. Brain 2017, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Cali, T.; Szabo, I.; Brini, M. Alpha-synuclein at the intracellular and the extracellular side: Functional and dysfunctional implications. Biol. Chem. 2017, 398, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. Alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between A, Tau, and -Synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T. Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 155–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, S.; Outeiro, T.F. Epigenetics in Parkinson’s and Alzheimer’s diseases. Subcell. Biochem. 2013, 61, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J.; Sonawane, B.; Butler, R.N.; Trasande, L.; Callan, R.; Droller, D. Early environmental origins of neurodegenerative disease in later life. Environ. Health Perspect. 2005, 113, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.H.; Basha, M.R. Environmental risk factors and the developmental basis for Alzheimer’s disease. Rev. Neurosci. 2005, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W.; Schumacher, A.; Maloney, B.; Lahiri, D.K.; Zawia, N.H. Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): Towards a new paradigm. Curr. Alzheimer Res. 2012, 9, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A mini review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regener. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Fukui, H.; Moraes, C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: Reality or just an attractive hypothesis? Trends Neurosci. 2008, 31, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed]
- Tabner, B.J.; El-Agnaf, O.M.A.; German, M.J.; Fullwood, N.J.; Allsop, D. Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 2005, 33, 1082–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, I.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Neurochem. Res. 2008, 33, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.G.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Ghosh, R.; Mandal, S.C. Antioxidant protection: A promising therapeutic intervention in neurodegenerative disease. Free Radic. Res. 2011, 45, 888–905. [Google Scholar] [CrossRef] [PubMed]
- Gelain, D.P.; Antonio Behr, G.; Birnfeld De Oliveira, R.; Trujillo, M. Antioxidant therapies for neurodegenerative diseases: Mechanisms, current trends, and perspectives. Oxid. Med. Cell. Longev. 2012, 2012, 895153. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Lin, M.T.; Beal, M.F. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S633–S643. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.A.; Ernst, M.E. Antioxidants, supplements, and Parkinson’s disease. Ann. Pharmacother. 2006, 40, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Khandker, S.S.; Alam, F.; Khalil, M.I.; Kamal, M.A.; Gan, S.H. Alzheimer’s disease and natural products: Future regimens emerging from nature. Curr. Top. Med. Chem. 2017, 17, 1408–1428. [Google Scholar] [CrossRef] [PubMed]
- Muller, T. New small molecules for the treatment of Parkinson’s disease. Expert Opin. Investig. Drugs 2010, 19, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Morris, J.K. New therapeutics to modulate mitochondrial function in neurodegenerative disorders. Curr. Pharm. Des. 2017, 23, 731–752. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Moriano, C.; González-Burgos, E.; Gómez-Serranillos, M.P. Mitochondria-targeted protective compounds in Parkinson’s and Alzheimer’s diseases. Oxid. Med. Cell. Longev. 2015, 2015, 408927. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-G.; Sun, M.-X.; Zhang, W.-L.; Wang, W.-W.; Jin, Y.-M.; Xie, C.-L. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: A meta-analysis of randomized controlled trials. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2017, 38, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Molino, S.; Dossena, M.; Buonocore, D.; Ferrari, F.; Venturini, L.; Ricevuti, G.; Verri, M. Polyphenols in dementia: From molecular basis to clinical trials. Life Sci. 2016, 161, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Taghibiglou, C. The mechanisms of action of curcumin in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 58, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-H.; Loo, C.-Y.; Bebawy, M.; Luk, F.; Mason, R.; Rohanizadeh, R. Curcumin and its Derivatives: Their application in neuropharmacology and neuroscience in the 21st century. Curr. Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef] [PubMed]
- Lopresti, A.L. Curcumin for neuropsychiatric disorders: A review of in vitro, animal and human studies. J. Psychopharmacol. 2017, 31, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.A.; Jantan, I. Synthetic curcumin analogs as inhibitors of beta-amyloid peptide aggregation: potential therapeutic and diagnostic agents for Alzheimer’s disease. Mini Rev. Med. Chem. 2015, 15, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Mithu, V.S.; Sarkar, B.; Bhowmik, D.; Das, A.K.; Chandrakesan, M.; Maiti, S.; Madhu, P.K. Curcumin alters the salt bridge-containing turn region in amyloid β(1-42) aggregates. J. Biol. Chem. 2014, 289, 11122–11131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Browne, A.; Child, D.; Tanzi, R.E. Curcumin decreases amyloid-beta peptide levels by attenuating the maturation of amyloid-beta precursor protein. J. Biol. Chem. 2010, 285, 28472–28480. [Google Scholar] [CrossRef] [PubMed]
- Calcul, L.; Zhang, B.; Jinwal, U.K.; Dickey, C.A.; Baker, B.J. Natural products as a rich source of tau-targeting drugs for Alzheimer’s disease. Future Med. Chem. 2012, 4, 1751–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.K.; Kotia, V.; Ghosh, D.; Mohite, G.M.; Kumar, A.; Maji, S.K. Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem. Neurosci. 2013, 4, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in alpha-synuclein by increasing reconfiguration rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Pfeiffer, E.; Schulz, S.I.; Dempe, J.S. Curcumin uptake and metabolism. Biofactors 2013, 39, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, C.; Zhang, X.; Zhang, C.; Zhong, Q.; Chen, G.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. Structure-activity relationship and pharmacokinetic studies of 1,5-Diheteroarylpenta-1,4-dien-3-ones: A class of promising curcumin-based anticancer agents. J. Med. Chem. 2015, 58, 4713–4726. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Gordon, O.N.; Edwards, R.L.; Luis, P.B. Degradation of curcumin: From mechanism to biological implications. J. Agric. Food Chem. 2015, 63, 7606–7614. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-H.; Cheng, A.-L. Clinical studies with curcumin. Adv. Exp. Med. Biol. 2007, 595, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Su, X.; Liu, A.; Zhang, L.; Yu, A.; Xi, Y.; Zhai, G. Advances in clinical study of curcumin. Curr. Pharm. Des. 2013, 19, 1966–1973. [Google Scholar] [PubMed]
- Gonzalez-Albadalejo, J.; Sanz, D.; Claramunt, R.M.; Lavandera, J.L.; Alkorta, I.; Elguero, J. Curcumin and curcuminoids: Chemistry, structural studies and biological properties. An. Real Acad. Nac. Farm. 2015, 81, 278–310. [Google Scholar]
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112. [Google Scholar] [CrossRef] [PubMed]
- Pabon, H.J.J. A synthesis of curcumin and related compounds. Recl. Trav. Chim. Pays-Bas 1964, 83, 379–386. [Google Scholar] [CrossRef]
- Claramunt, R.M.; Bouissane, L.; Cabildo, M.P.; Cornago, M.P.; Elguero, J.; Radziwon, A.; Medina, C. Synthesis and biological evaluation of curcuminoid pyrazoles as new therapeutic agents in inflammatory bowel disease: Effect on matrix metalloproteinases. Bioorg. Med. Chem. 2009, 17, 1290–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornago, P.; Cabildo, P.; Sanz, D.; Claramunt, R.M.; Torralba, M.C.; Torres, M.R.; Elguero, J. Structures of hemi-curcuminoids in the solid state and in solution. Eur. J. Org. Chem. 2013, 2013, 6043–6054. [Google Scholar] [CrossRef]
- Nieto, C.I.; Cabildo, P.; Claramunt, R.M.; Cornago, P.; Sanz, D.; Torralba, M.C.; Torres, M.R.; Ferraro, M.B.; Alkorta, I.; Marín-Luna, M.; et al. The structure of β-diketones related to curcumin determined by X-ray crystallography, NMR (solution and solid state) and theoretical calculations. Struct. Chem. 2016, 27, 705–730. [Google Scholar] [CrossRef]
- Berger, S.; Braun, S.; Kalinowski, H.O. NMR Spectroscopy of the Non-Metallic Elements; Wiley: Chichester, UK, 1997; ISBN 0-471-96763-7. [Google Scholar]
- Sheldrick, G.M. SHELX-97: Programs for Crystal Structure Analysis; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Huang, D.; Ou, B.; Prior, R.L. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J. Agric. Food Chem. 2005, 53, 4290–4302. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin. Sci. 1993, 84, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Bravo, L.; Saura-Calixto, F. Antioxidant activity of dietary polyphenols as determined by a modified ferric reducing/antioxidant power assay. J. Agric. Food Chem. 2000, 48, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Sreejayan, N.; Rao, M.N. Free radical scavenging activity of curcuminoids. Arzneimittel-forschung 1996, 46, 169–171. [Google Scholar] [PubMed]
- Venkatesan, P.; Rao, M.N. Structure-activity relationships for the inhibition of lipid peroxidation and the scavenging of free radicals by synthetic symmetrical curcumin analogues. J. Pharm. Pharmacol. 2000, 52, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Bondet, V.; Brand-Williams, W.; Berset, C. Kinetics and mechanisms of antioxidant activity using the DPPH. Free radical method. LWT Food Sci. Technol. 1997, 30, 609–615. [Google Scholar] [CrossRef]
- Ligeret, H.; Barthelemy, S.; Bouchard Doulakas, G.; Carrupt, P.-A.; Tillement, J.-P.; Labidalle, S.; Morin, D. Fluoride curcumin derivatives: New mitochondrial uncoupling agents. FEBS Lett. 2004, 569, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Improving drug design: An update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol. 2016, 29, 564–616. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. Abstr. Pap. Am. Chem. Soc. 1999, 217, U696. [Google Scholar]
- MedChem Designer(TM), version 3.1.0.30; MedChem-Designer-Simulations-Plus-Inc.: Lancaster, CA, USA, 2014.
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Ghribi, O. Cellular model of Alzheimer’s disease—Relevance to therapeutic testing. Exp. Neurol. 2012, 233, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Hoelke, B.; Gieringer, S.; Arlt, M.; Saal, C. Comparison of nephelometric, UV-spectroscopic, and HPLC methods for High-throughput determination of aqueous drug solubility in microtiter plates. Anal. Chem. 2009, 81, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H. Brain iron deficiency and excess; cognitive impairment and neurodegeneration with involvement of striatum and hippocampus. Neurotox. Res. 2008, 14, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. The relevance of metals in the pathophysiology of neurodegeneration, pathological considerations. Int. Rev. Neurobiol. 2013, 110, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.L.; Williamson, S.J.M.; Paleologou, K.E.; Hewitt, R.; El-Agnaf, O.M.A.; Allsop, D. Fe(II)-induced DNA damage in α-synuclein-transfected human dopaminergic BE(2)-M17 neuroblastoma cells: Detection by the Comet assay. J. Neurochem. 2003, 87, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, H.; Song, N.; Xie, J. Oxidative stress partially contributes to iron-induced alpha-synuclein aggregation in SK-N-SH cells. Neurotox. Res. 2011, 19, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Telling, N.D.; Everett, J.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Gallagher, J.J.; Wang, J.; Hitchcock, A.P. Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer’s disease. Cell Chem. Biol. 2017, 24, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Mandel, S.; Youdim, M.B.H.; Amit, T. Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic. Biol. Med. 2013, 62, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Dinis, T.C.; Maderia, V.M.; Almeida, L.M. Action of phenolic derivatives (acetaminophen, salicylate, and 5-aminosalicylate) as inhibitors of membrane lipid peroxidation and as peroxyl radical scavengers. Arch. Biochem. Biophys. 1994, 315, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Dairam, A.; Fogel, R.; Daya, S.; Limson, J.L. Antioxidant and iron-binding properties of curcumin, capsaicin, and S-allylcysteine reduce oxidative stress in rat brain homogenate. J. Agric. Food Chem. 2008, 56, 3350–3356. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Salvioli, S.; Ardizzoni, A.; Franceschi, C.; Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411, 77–82. [Google Scholar] [CrossRef]
- Cossarizza, A.; Salvioli, S. Flow cytometric analysis of mitochondrial membrane potential using JC-1. Curr. Protoc. Cytom. 2001, 13, 9–14. [Google Scholar] [CrossRef]
- GraphPadPrism-v7; GraphPad-Software Inc.: La Jolla, CA, USA, 2016.
Sample Availability: Samples of the compounds are not available from the authors. |




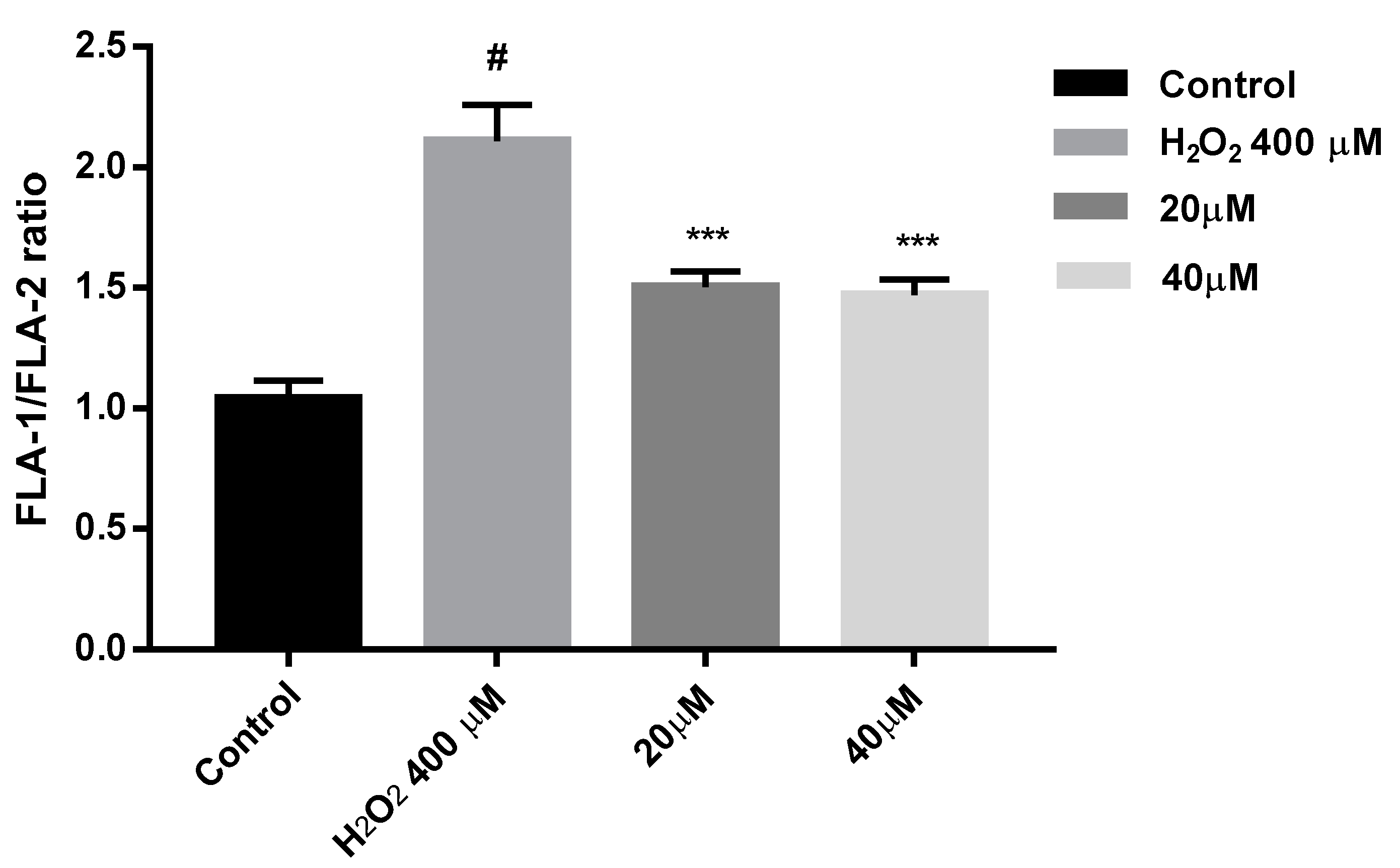
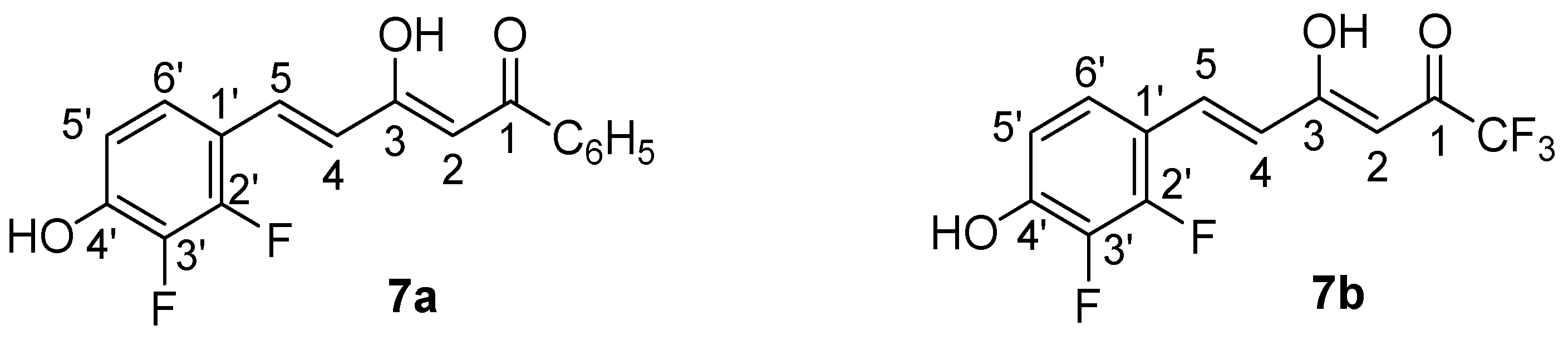

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
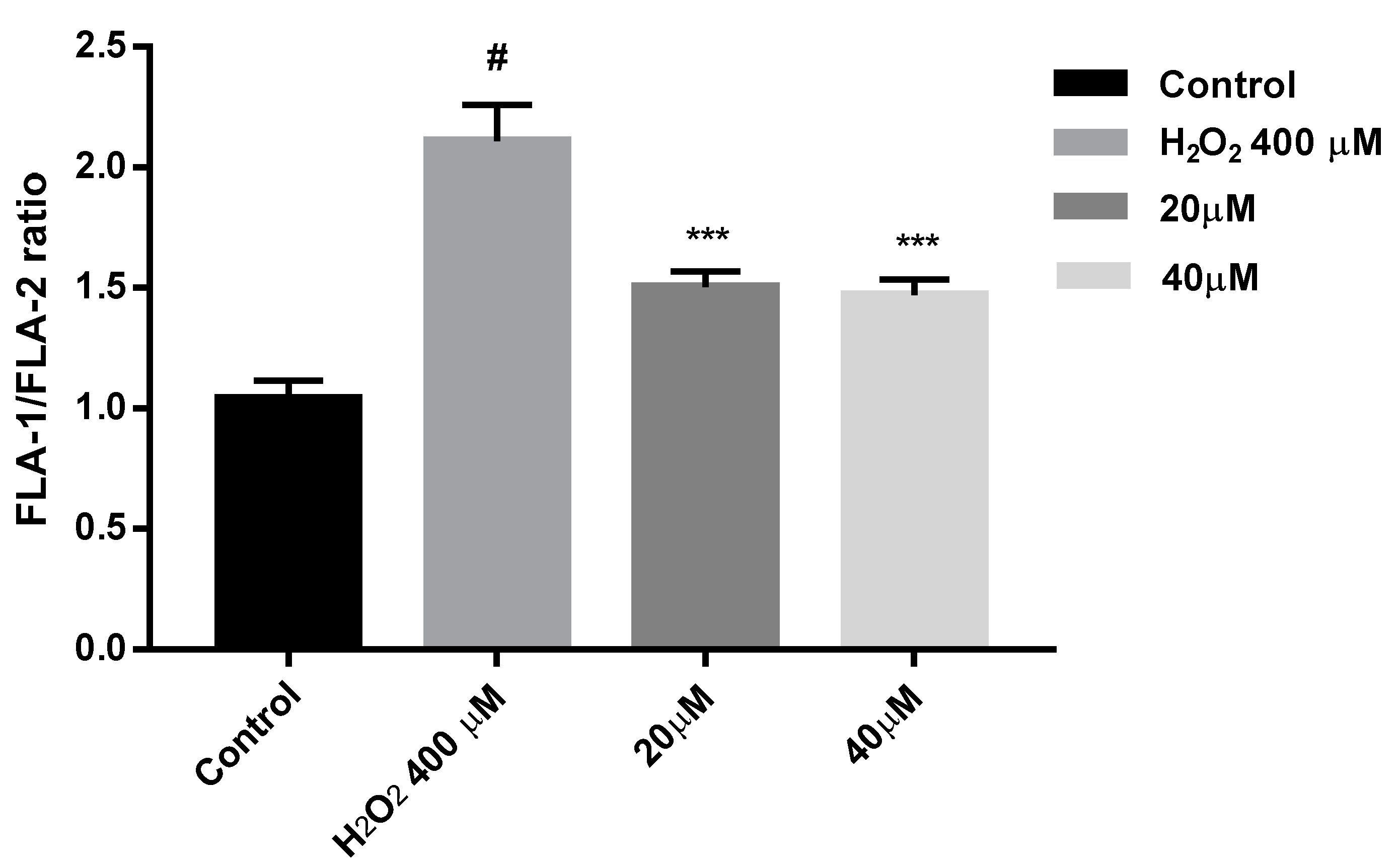{kind=link}
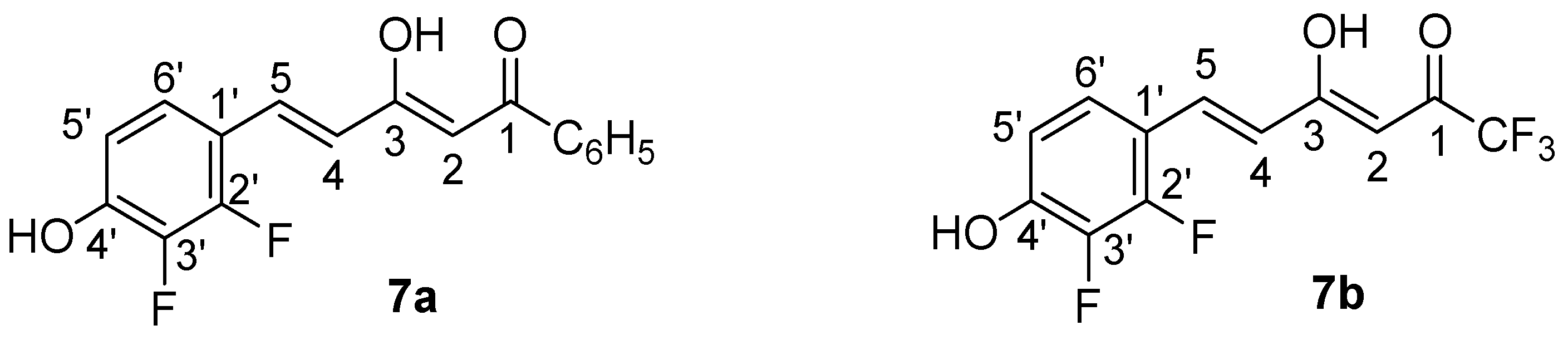{kind=link}
{kind=link}
| Compound | D-H···A | Symmetry Operations | d(D-H) | d(H···A) | d(D···A) | <DHA |
|---|---|---|---|---|---|---|
| 7a | O3H3A···O4 | #1 − x + 1/2, y − 1/2, z + 3/2 | 1.09 | 1.66 | 2.551(2) | 135.3 |
| O2-H2···O4#1 | 1.19 | 1.56 | 2.720(2) | 164.5 | ||
| 7b | O2-H2···O1 | 1.10 | 1.50 | 2.583(6) | 166.7 | |
| O3-H3A···O4 | 1.10 | 1.54 | 2.557(5) | 150.7 |
| ABTS Assay | FRAP Assay (%) | ||||
|---|---|---|---|---|---|
| Compound | IC50 (µM) | ABTS-TEAC100 * | at 100 µM | at 200 µM | FRAP-TEAC100 ** |
| Trolox | 34.0 ± 2.3 | 1 | 53.0 | 100.0 | 1 |
| Curcumin | 22.4 ± 0.9 | 1.05 | 47.0 | 74.4 | 0.86 |
| 1a | 50.5 ± 2.0 | 0.88 | 32.2 | 55.7 | 0.59 |
| 2a | >600 | 0.21 | 0.1 | 0.7 | 0.00 |
| 3a | 60.2 ± 2.2 | 0.51 | 20.5 | 31.0 | 0.33 |
| 4a | 107.8 ± 7.6 | 0.40 | 9.8 | 14.3 | 0.15 |
| 5a | 274.7 ± 35.3 | 0.23 | 3.4 | 5.3 | 0.05 |
| 6a | 67.2 ± 2.7 | 0.51 | 15.5 | 25.2 | 0.25 |
| 7a | 79.6 ± 4.3 | 0.69 | 14.0 | 20.7 | 0.22 |
| 1b | 34.6 ± 2.7 | 0.73 | 36.6 | 61.4 | 0.64 |
| 2b | >600 | 0.23 | 1.9 | 3.1 | 0.03 |
| 3b | 113.3 ± 12.4 | 0.67 | 19.7 | 32.6 | 0.34 |
| 4b | 216.1 ± 39.5 | 0.52 | 4.8 | 8.5 | 0.09 |
| 5b | 518.1 ± 94.6 | 0.32 | 3.0 | 4.7 | 0.05 |
| 6b | 115.5 ± 11 | 0.67 | 6.7 | 11.3 | 0.15 |
| 7b | 70.9 ± 5.8 | 0.62 | 8.8 | 15.3 | 0.16 |
| Compound | IC50 (µM) * |
|---|---|
| Trolox | 19.0 ± 1.8 |
| Curcumin | 26.5 ± 1.5 |
| 1a | 42.3 ± 3.4 |
| 1b | 53.8 ± 2.6 |
| Compound | MWt | MlogP | logD | NO | TPSA | HBD |
|---|---|---|---|---|---|---|
| Curcumin | 368.39 | 2.256 | 2.778 | 6 | 96.2 | 3 |
| 1a | 296.33 | 2.938 | 2.467 | 4 | 66.8 | 2 |
| 2a | 298.32 | 3.863 | 3.021 | 3 | 46.5 | 1 |
| 3a | 284.29 | 3.628 | 2.813 | 3 | 57.5 | 2 |
| 4a | 284.29 | 3.628 | 3.023 | 3 | 57.5 | 2 |
| 5a | 302.28 | 4.014 | 2.170 | 3 | 57.5 | 2 |
| 6a | 302.28 | 4.014 | 2.885 | 3 | 57.5 | 2 |
| 7a | 302.28 | 4.014 | 2.858 | 3 | 57.5 | 2 |
| 1b | 288.23 | 2.046 | 0.563 | 4 | 66.8 | 2 |
| 2b | 290.22 | 2.981 | 0.945 | 3 | 46.5 | 1 |
| 3b | 276.19 | 2.725 | 0.972 | 3 | 57.5 | 2 |
| 4b | 276.19 | 2.725 | 1.074 | 3 | 57.5 | 2 |
| 5b | 294.18 | 3.122 | 0.974 | 3 | 57.5 | 2 |
| 6b | 294.18 | 3.122 | 1.158 | 3 | 57.5 | 2 |
| 7b | 294.18 | 3.122 | 1.163 | 3 | 57.5 | 2 |
| Compound | IC50 (µM) * |
|---|---|
| Curcumin | 48.8 ± 5.1 |
| 1a | 103.8 ± 6.7 |
| 2a | 75.7 ± 5.4 |
| 3a | 98.1 ± 8.3 |
| 4a | 103.5 ± 7.1 |
| 5a | 118.7 ± 8.5 |
| 6a | 134.2 ± 6.6 |
| 7a | 125.6 ± 8.0 |
| 1b | 101.8 ± 5.6 |
| 2b | 88.0 ± 4.5 |
| 3b | 109.0 ± 7.6 |
| 4b | 138.0 ± 6.5 |
| 5b | 153.0 ± 21.0 |
| 6b | 186.8 ± 13.2 |
| 7b | 200.0 ± 17.1 |
| Compound | Solubility (µg/mL) * |
|---|---|
| 6b | 281.0 ± 9.0 |
| 7b | 298.2 ± 7.0 |
| Curcumin | 24.0 ± 3.0 |
| Compound | Remaining Compound (%) * | |||
|---|---|---|---|---|
| 0 h | 6 h | 24 h | 48 h | |
| 6b | 100 | 96.1 ± 0.5 | 92.2 ± 1.6 | 90.4 ± 3.0 |
| 7b | 100 | 95.7 ± 1.0 | 90.9 ± 1.2 | 87.8 ± 2.3 |
| Curcumin | 100 | 55.9 ± 10.6 | 42.9 ± 9.0 | 38.1 ± 7.4 |
| Crystal Data | 7a | 7b |
|---|---|---|
| CCDC code | 1561929 | 1561930 |
| Empirical formula | C17H12F2O3 | C14H13F5O4S |
| Formula wt | 302.27 | 372.30 |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Pna21 | Pbca |
| a/Å | 21.601(2) | 10.4091(9) |
| b/Å | 12.181(1) | 9.1657(8) |
| c/Å | 5.418(6) | 34.793(3) |
| a α/° | 90 | 90 |
| b β/° | 90 | 90 |
| γ/° | 90 | 90 |
| V/Å3 | 1406.6(7) | 3319.5(5) |
| Z | 4 | 8 |
| Dc/g/cm3 | 1.408 | 1.490 |
| μ(Mo-Kα)/mm−1 | 0.113 | 0.262 |
| F(000) | 624 | 1520 |
| θ range/° | 1.89 to 27.00 | 1.17 to 25.00 |
| Index ranges | −27, −1, −6 to 27, 12, 6 | −10, −10, −41 to 12, 10, 40 |
| Reflections collected | 12,193 | 23,645 |
| Unique reflections | 3098 | 2932 |
| [Rint] | [0.0523] | [0.0985] |
| Completeness to theta (%) | 100.0 | 100.0 |
| Data/restraints/params | 3098/1/199 | 2932/0/217 |
| Goodness-of-fit on F2 | 0.994 | 0.994 |
| R1 (reflns obsd) [I > 2σ(I)] a | 0.0373 (1517) | 0.0670 (1158) |
| ωR2 (all data) b | 0.0740 | 0.2398 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieto, C.I.; Cornago, M.P.; Cabildo, M.P.; Sanz, D.; Claramunt, R.M.; Torralba, M.C.; Torres, M.R.; Martínez Casanova, D.; Sánchez-Alegre, Y.R.; Escudero, E.; et al. Evaluation of the Antioxidant and Neuroprotectant Activities of New Asymmetrical 1,3-Diketones. Molecules 2018, 23, 1837. https://doi.org/10.3390/molecules23081837
Nieto CI, Cornago MP, Cabildo MP, Sanz D, Claramunt RM, Torralba MC, Torres MR, Martínez Casanova D, Sánchez-Alegre YR, Escudero E, et al. Evaluation of the Antioxidant and Neuroprotectant Activities of New Asymmetrical 1,3-Diketones. Molecules. 2018; 23(8):1837. https://doi.org/10.3390/molecules23081837
Chicago/Turabian StyleNieto, Carla I., María Pilar Cornago, María Pilar Cabildo, Dionisia Sanz, Rosa M. Claramunt, María Carmen Torralba, María Rosario Torres, Diana Martínez Casanova, Yaiza Rebeca Sánchez-Alegre, Esther Escudero, and et al. 2018. "Evaluation of the Antioxidant and Neuroprotectant Activities of New Asymmetrical 1,3-Diketones" Molecules 23, no. 8: 1837. https://doi.org/10.3390/molecules23081837