Effect of Dibutyltin Dilaurate on Triglyceride Metabolism through the Inhibition of the mTOR Pathway in Human HL7702 Liver Cells

Abstract
:1. Introduction
2. Results
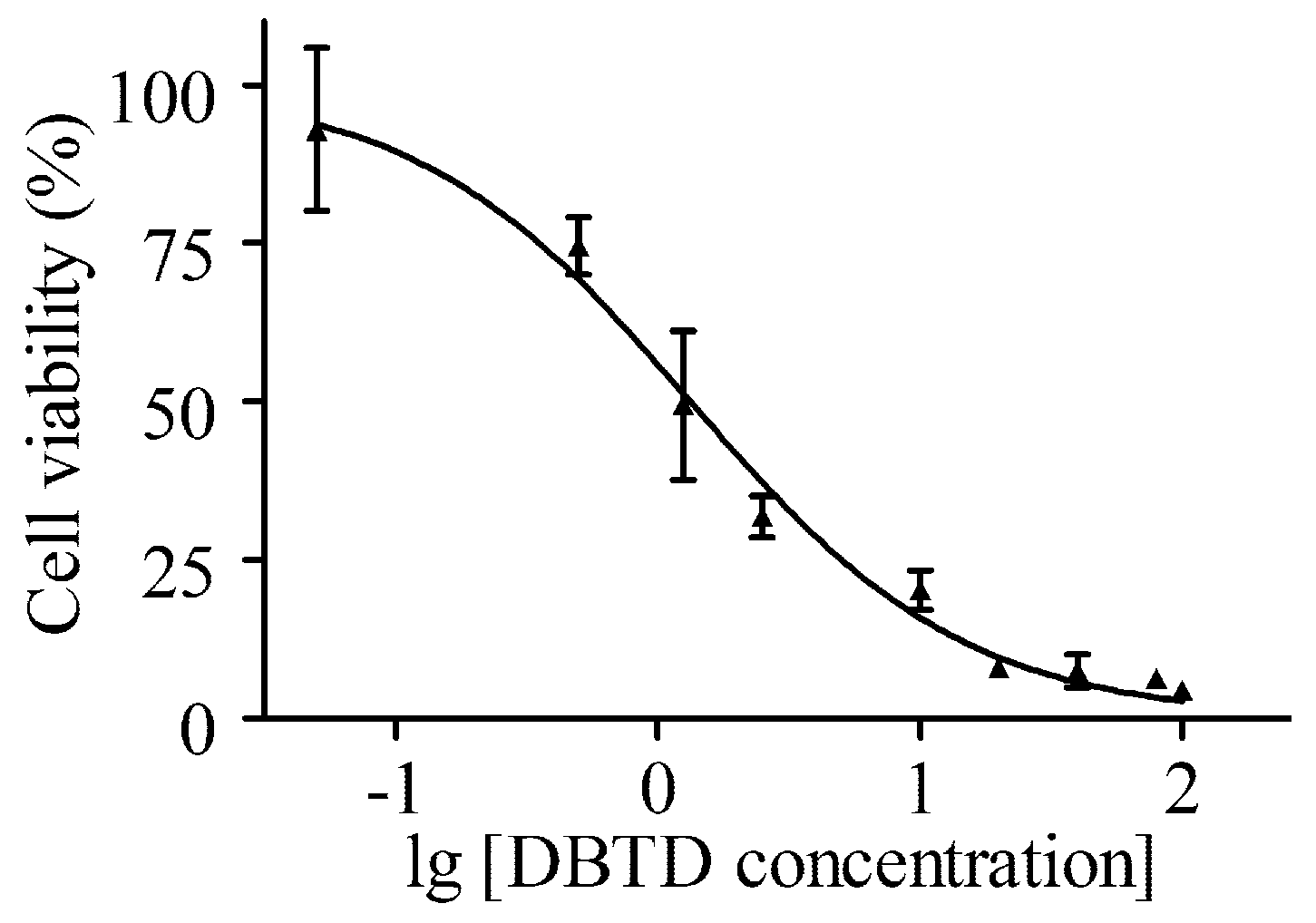
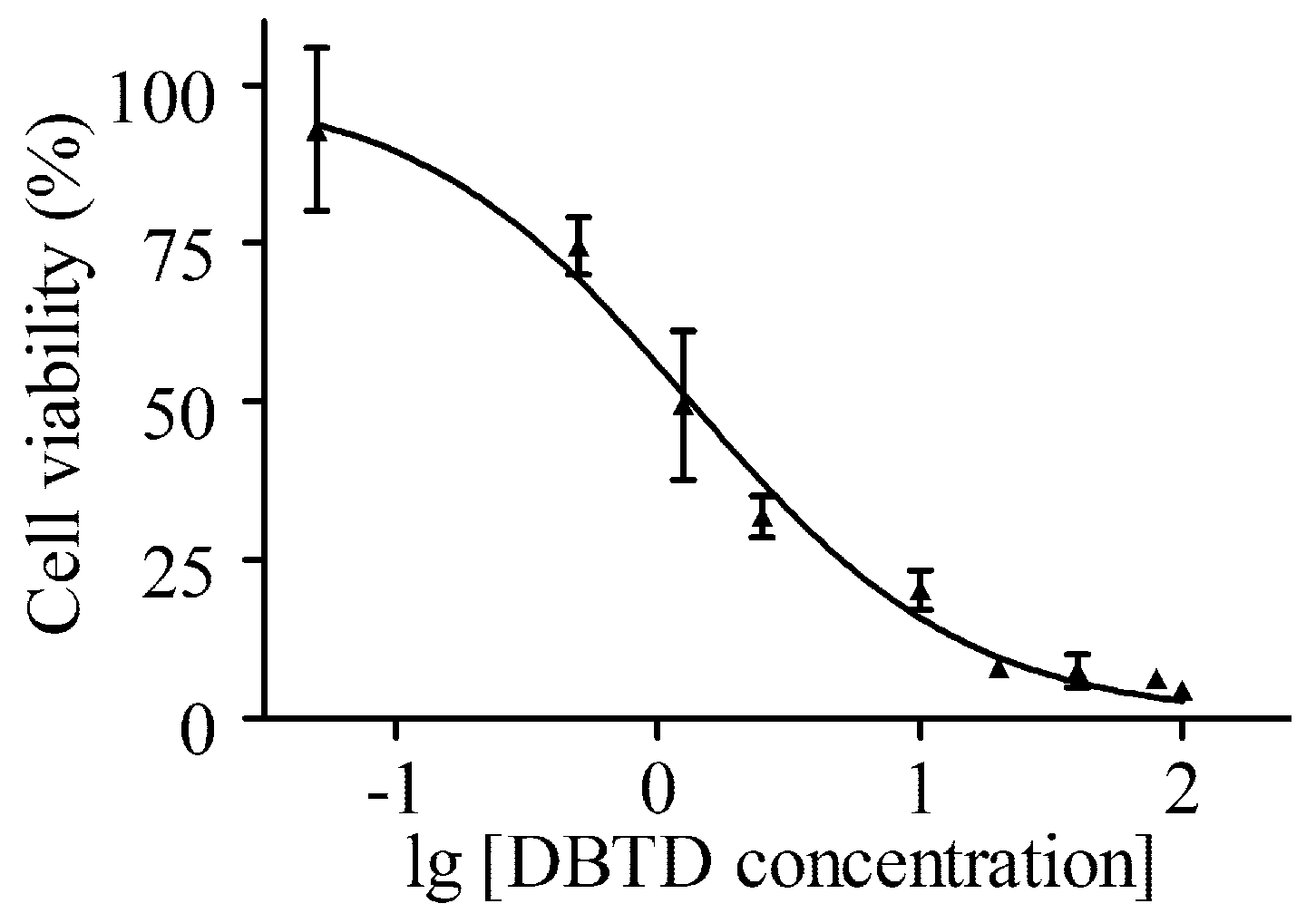
2.1. DBTD Decreases the Viability of Liver Cells
2.2. Activities of Alanine Aminotransferase (ALT) and Aspartate Aminotransferase (AST) Are Elevated in the Cultural HL7702 Cell Media by DBTD
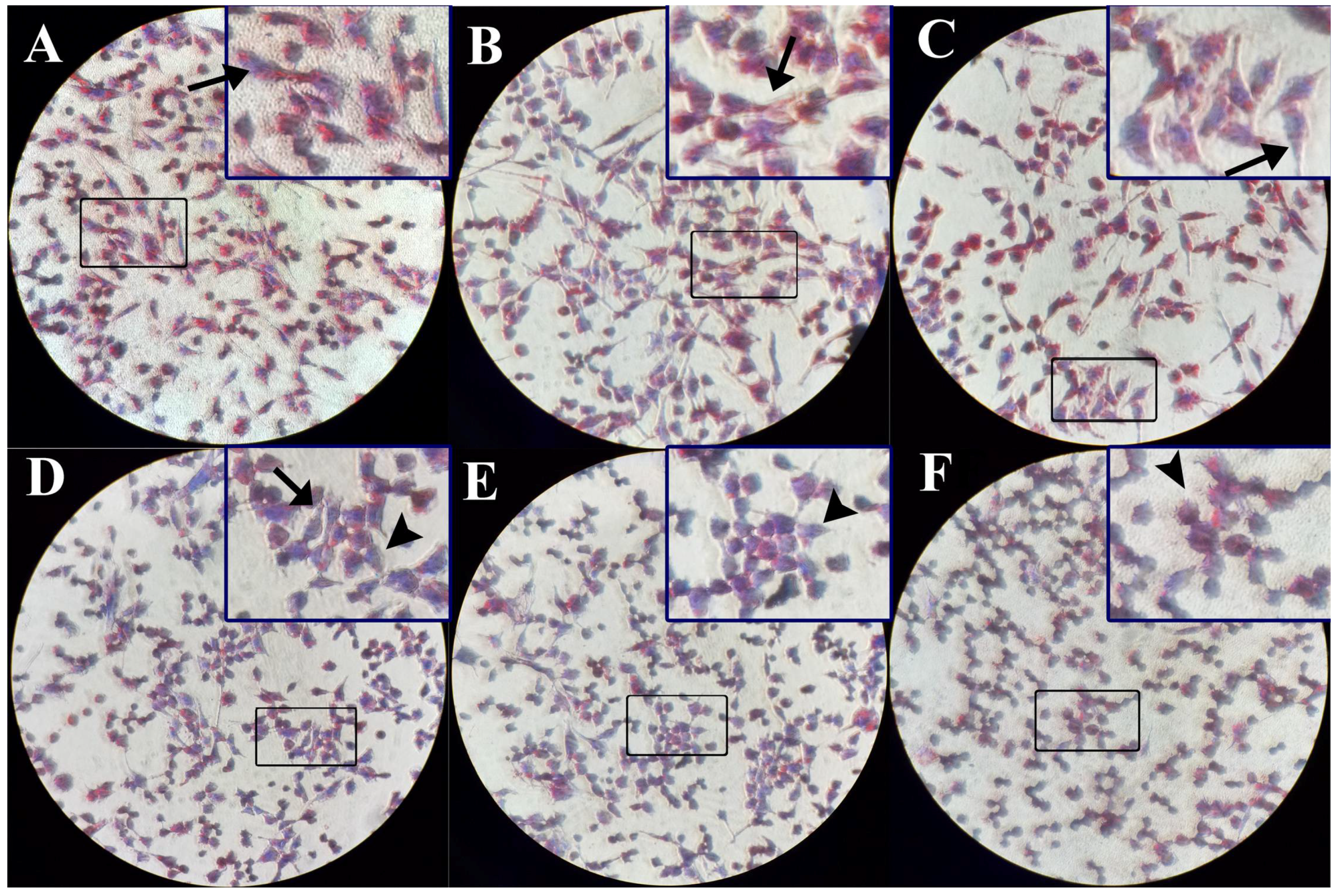
2.3. The Intracellular TG Contents Visualized by Oil-Red O Staining Were Reduced
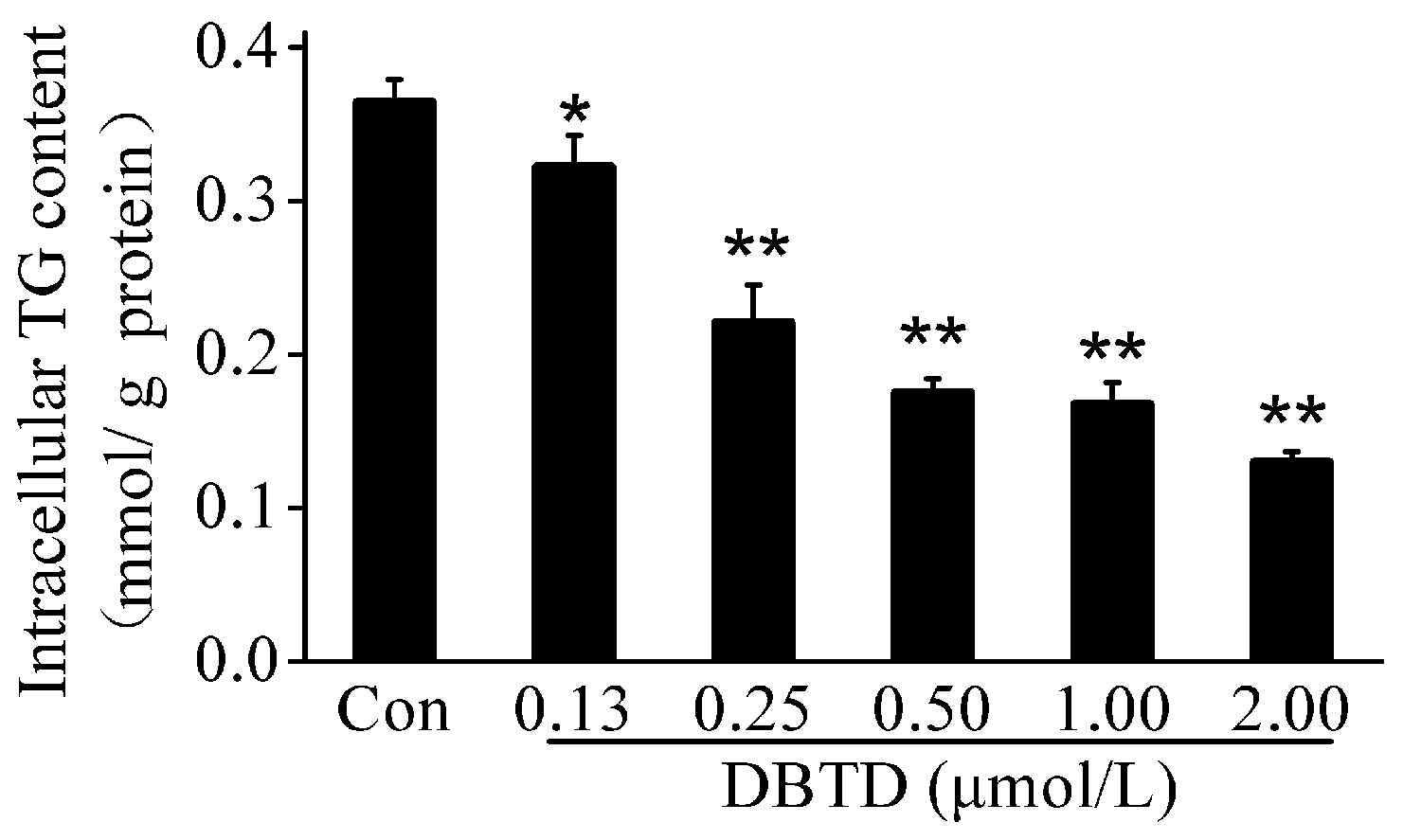
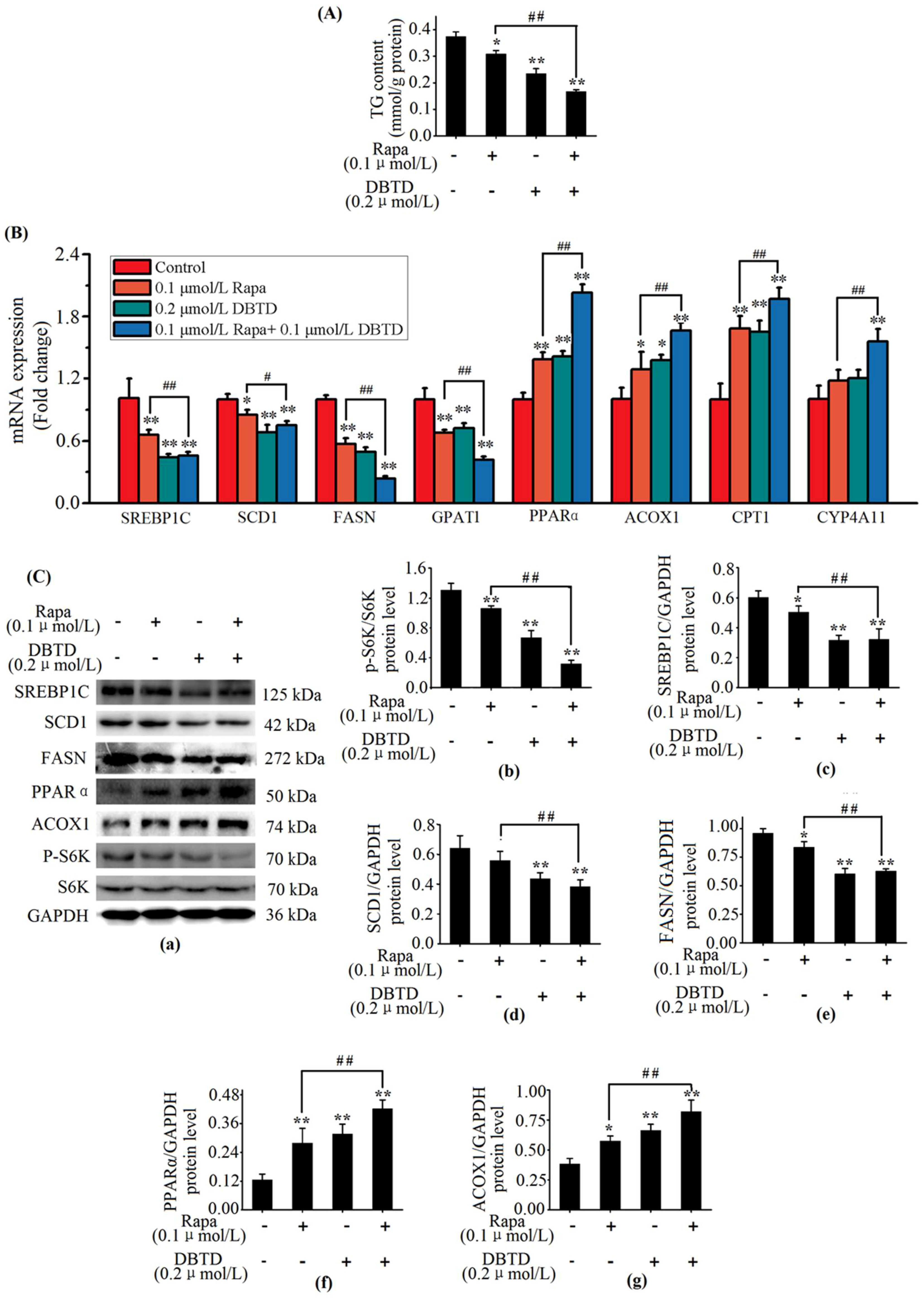
2.4. The TG Contents Were Dose-Dependently Decreased by DBTD in Liver Cells
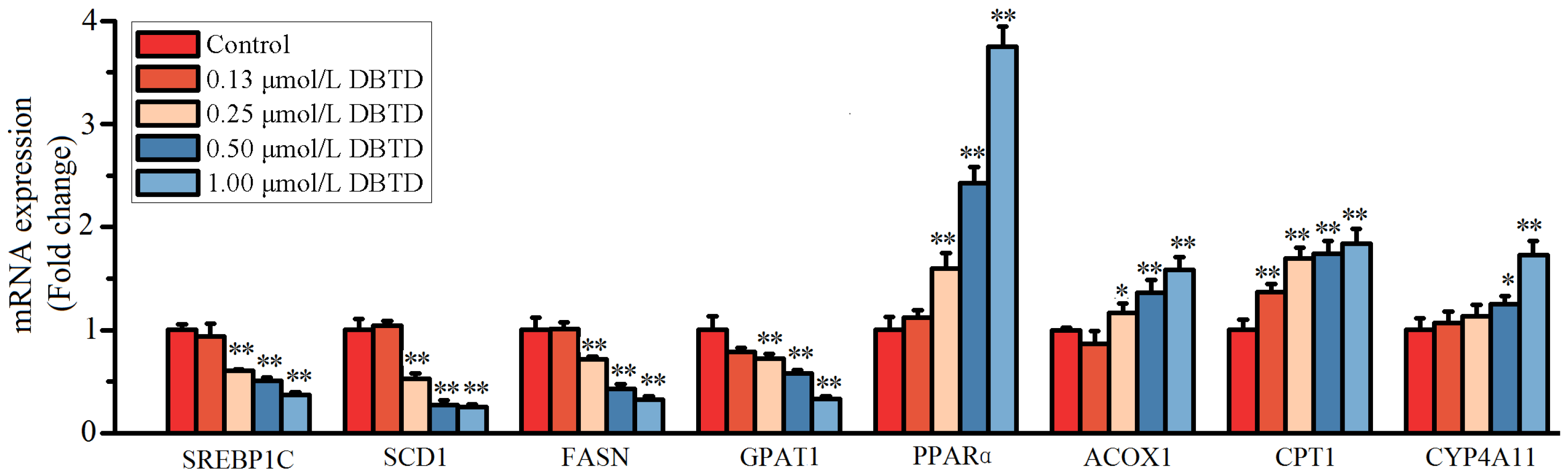
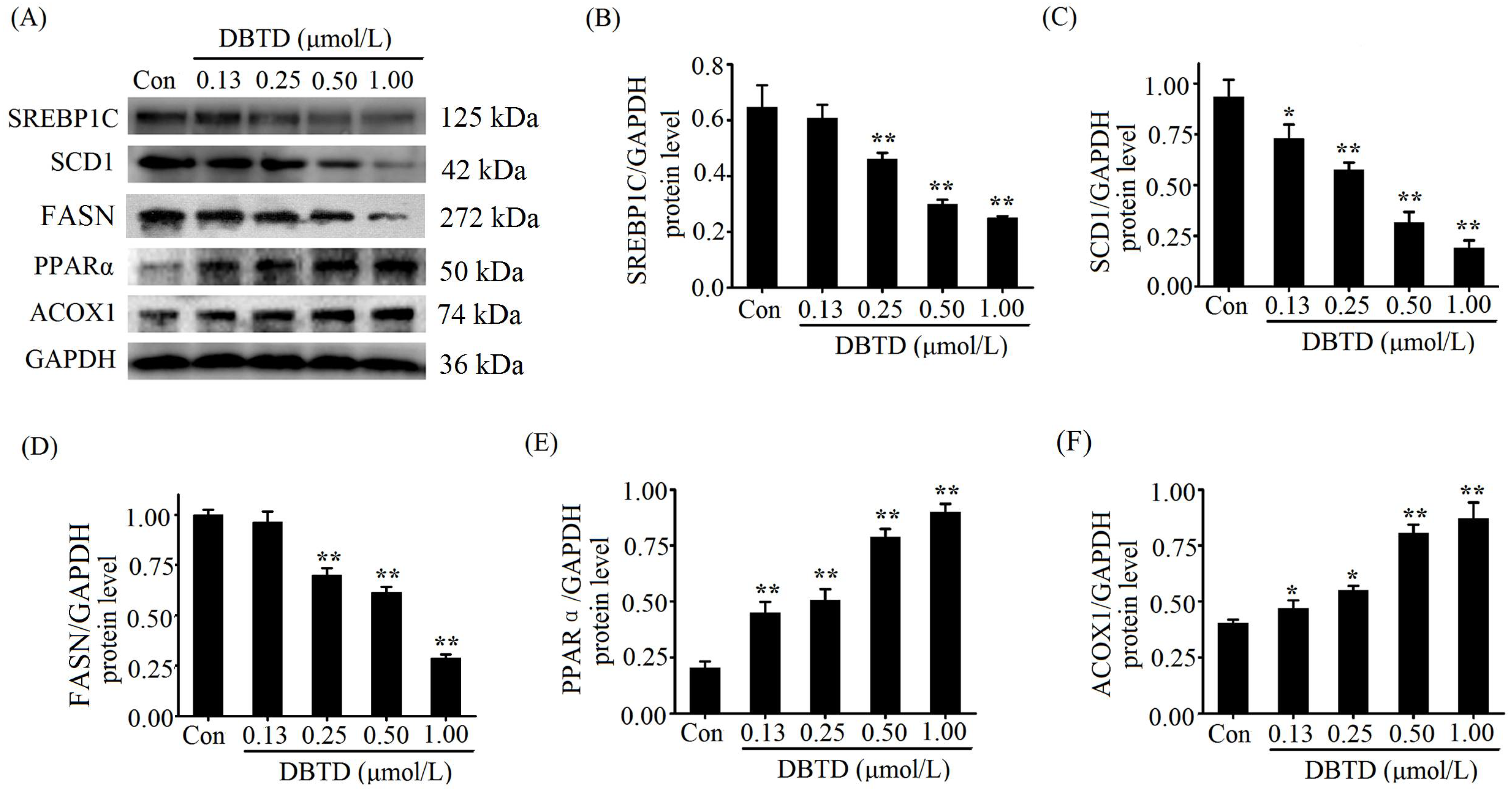
2.5. Exposure to DBTD Leads to Low Expression of Lipogenic Genes and High Expression of Lipolytic Genes in Liver Cells
2.6. Lipogenic Proteins Are Decreased and Lipolytic Proteins Are Increased in DBTD-Treated Liver Cells
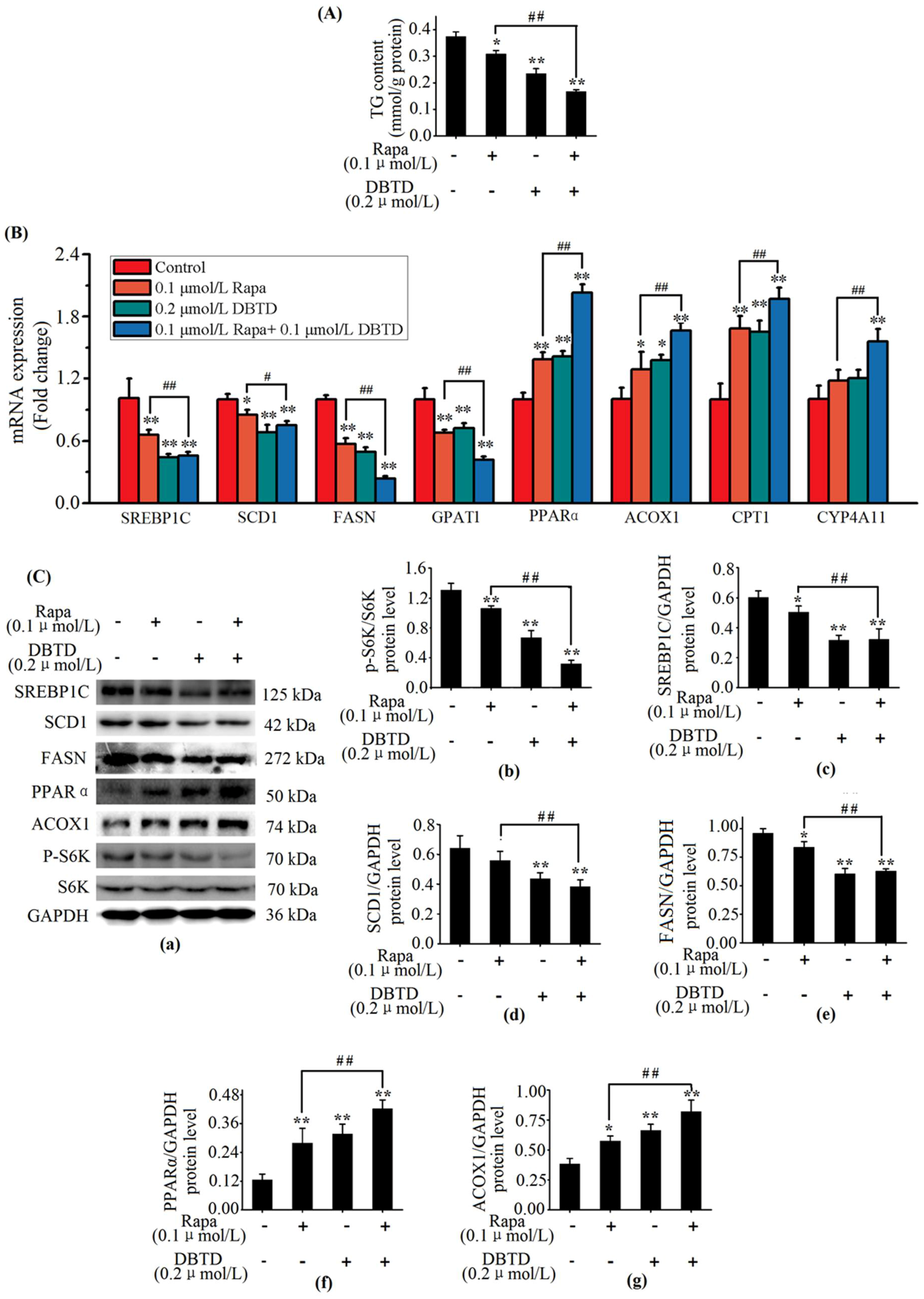
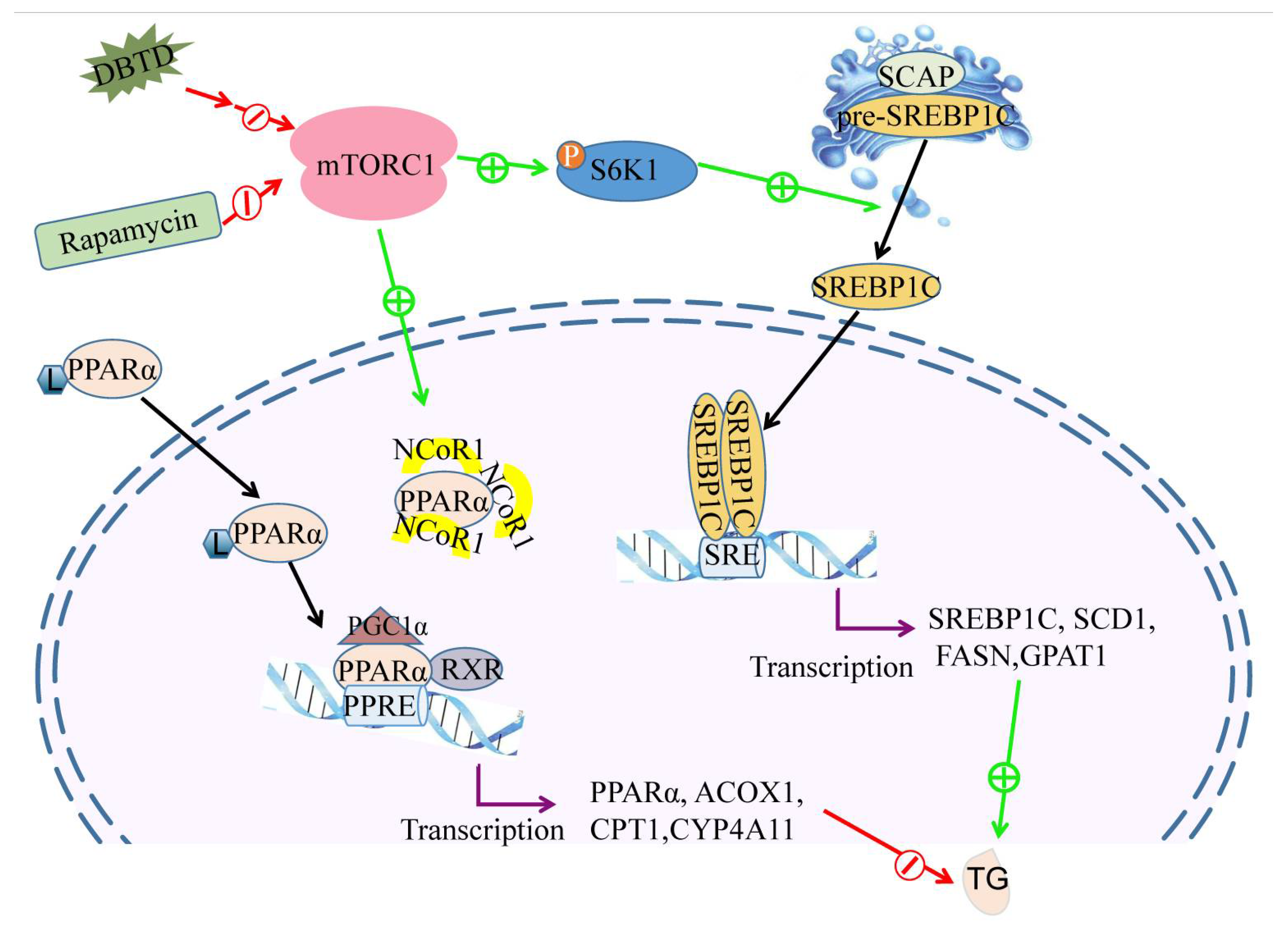
2.7. DBTD Affects TG Metabolism through Inhibiting the mTOR Pathway
3. Discussion
4. Materials and Methods
4.1. Reagent
4.2. Cell Culture and Treatment
4.3. Viability of Cells
4.4. Activities of ALT and AST and Intracellular TG Contents Measurement
4.5. Oil Red O Staining
4.6. Quantitative Real-Time PCR
4.7. Western Blot Analyses
4.8. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xu, R.; Zhou, D.; Zhao, D. Effect of organotin stabiliser on the thermal stabilisation of PVC: Part I—The influence of dibutyltin dilaurate on polyene sequences. Polym. Degrad. Stab. 1990, 27, 203–210. [Google Scholar] [CrossRef]
- Harford, A.J.; O’Halloran, K.; Wright, P.F. The effects of in vitro pesticide exposures on the phagocytic function of four native Australian freshwater fish. Aquat. Toxicol. 2005, 75, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Brito, Y.C.; Ferreira, D.A.C.; Fragoso, D.M.D.A.; Mendes, P.R.; de Oliveira, C.M.J.; Meneghetti, M.R.; Meneghetti, S.M.P. Simultaneous conversion of triacylglycerides and fatty acids into fatty acid methyl esters using organometallic tin (IV) compounds as catalysts. Appl. Catal. A Gen. 2012, 443–444, 202–206. [Google Scholar] [CrossRef]
- Lin, S.C. Grafting of sulfobetaine onto a polyurethane surface to improve blood compatibility. Chin. J. Polym. Sci. 2003, 21, 419–426. [Google Scholar] [CrossRef]
- Mrówka, P.; Kozakiewicz, J.; Jurkowska, A.; Sienkiewicz, E.; Przybylski, J.; Lewandowski, Z.; Przybylski, J.; Lewandowska-Szumiel, M. Moisture-cured silicone-urethanes-candidate materials for tissue engineering: A biocompatibility study in vitro. J. Biomed. Mater. Res. Part A 2010, 94A, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Moon, H.B.; An, Y.R.; Choi, S.G.; Choi, H.G. Accumulation of butyltin compounds in cetaceans from Korean coastal waters. Mar. Pollut. Bull. 2011, 62, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, D.M.; Turra, A.; de Mary, R.R.M.; Rosalinda, C.M. Distribution of butyltin compounds in Brazil's southern and southeastern estuarine ecosystems: Assessment of spatial scale and compartments. Environ. Sci. Pollut. Res. Int. 2016, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Strand, S. Butyltin compounds in human liver. Environ. Res. 2002, 8, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Merkord, J.; Weber, H.; Hennighausen, G. Ethanol increases the toxic effects of dibutyltin dichloride (DBTC) on pancreas and liver of rats. Toxicol. Lett. 1996, 88, 56. [Google Scholar] [CrossRef]
- Ueno, S.; Suzuki, T.; Susa, N.; Furukawa, Y.; Sugiyama, M. Effect of SKF-525A on liver metabolism and hepatotoxicity of tri- and dibutyltin compounds in mice. Arch. Toxicol. 1997, 71, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.S. Multiorgan chronic inflammatory hepatobiliary pancreatic murine model deficient in tumor necrosis factor receptors 1 and 2. World J. Gastroenterol. 2016, 22, 4988. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, X.; Niu, L.; Li, Q. Proteomics analysis reveals an important role for the PPAR signaling pathway in DBDCT-induced hepatotoxicity mechanisms. Molecules 2017, 22, 1113. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3281–3282. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-H.; Savas, U.; Griffin, K.J.; Eric Johnson, F. Identification of Peroxisome Proliferator-responsive Human Genes by Elevated Expression of the Peroxisome Proliferator-activated Receptor in HepG2 Cells. J. Biol. Chem. 2001, 276, 27950–27958. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Naqanuma, T.; Kusunose, E.; Kono, T.; Yasumoto, R.; Suqimura, K.; Kishimoto, T. Human Fatty Acid Hydroxylase, CYP4A11 Determination of Complete Genomic Sequence and Characterization of Purified Recombinant Protein. Arch. Biochem. Biophys. 2000, 378, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Barrer, G.; Toaldo, C.; Pizzimenti, S.; Cerbone, A.; Pettazzoni, P.; Dianzani, M.U.; Ferretti, C. The Role of PPAR Ligands in Controlling Growth-Related Gene Expression and their Interaction with Lipoperoxidation Products. PPAR Res. 2008, 2008, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Näär, A.M. SREBPs: Regulators of cholesterol/lipids as therapeutic targets in metabolic disorders, cancers and viral diseases. Clin. Lipidol. 2012, 7, 27–36. [Google Scholar] [CrossRef]
- Koo, S.-H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhu, Q.; Chen, M.; Huang, Q.; Wang, W.; Li, Q.; Huang, Y.; Di, W. The changing 50% inhibitory concentration (IC50) of cisplatin: A pilot study on the artifacts of the MTT assay and the precise measurement of density-dependent chemoresistance in ovarian cancer. Oncotarget 2016, 7, 70803–70821. [Google Scholar] [CrossRef] [PubMed]
- Ozer, J.; Ratner, M.; Shaw, M.; Bailey, W.; Schomaker, S. The current state of serum biomarkers of hepatotoxicity. Toxicology 2008, 245, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Kashimoto, T.; Susa, N.; Shiota, Y.; Okuda, M.; Mutoh, K.-I.; Hoshi, F.; Watanabe, K.; Tsuda, S.; Kawazoe, S.; et al. Effects of butyltin compounds on mitochondrial respiration and its relation to hepatotoxicity in mice and Guinea pigs. Toxicol. Sci. 2003, 75, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.D.; Han, C.C.; Wan, H.F.; He, F.; Xu, H.Y.; Wei, S.H.; Du, X.H.; Xu, F. Effects of inhibiting PI3K-Akt-mTOR pathway on lipid metabolism homeostasis in goose primary hepatocytes. Animal 2016, 10, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, R.; Fok, W.C.; Coles, A.; Salmon, A.B.; Pérez, V.I. Rapamycin and dietary restriction induce metabolically distinctive changes in mouse liver. J. Gerontol. 2015, 70, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of TOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Lemke, M.; Görl, N.; Berg, A.; Weber, H.; Hennighausen, G.; Merkord, J. Influence of selenium treatment on the acute toxicity of dibutyltin dichloride in rats. Pancreatology 2006, 6, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Boyer, I.J. Toxicity of dibutyltin, tributyltin and other organotin compounds to humans and to experimental animals. Toxicology 1989, 55, 253–298. [Google Scholar] [CrossRef]
- Milton, F.A.; Lacerda, M.G.; Sinoti, S.B.P.; Mesquita, P.G.; Prakasan, D.; Coelho, M.S.; de Lima, C.L.; Martini, A.G.; Pazzine, G.T.; Borin, M.F.; et al. Dibutyltin Compounds Effects on PPARγ/RXRα Activity, Adipogenesis, and Inflammation in Mammalians Cells. Front. Pharmacol. 2017, 8, 507. [Google Scholar] [CrossRef] [PubMed]
- Hiromori, Y.; Nishikawa, J.-I.; Yoshida, I.; Nagase, H.; Nakanishi, T. Structure-dependent activation of peroxisome proliferator-activated receptor gamma by organotin compounds. Chem.-Biol. Interact. 2009, 180, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, J.; Obradovic, M.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Pitt, S.J.; Stewart, A.J.; Isenovic, E.R. A high fat diet induces sex-specific differences in hepatic lipid metabolism and nitrite/nitrate in rats. Nitric Oxide 2016, 54, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Sharma, S. Mammalian target of rapamycin (m TOR) as a potential therapeutic target in various diseases. Inflammopharmacology 2017, 25, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Yoshino, K.-I.; Hidayat, S.; Tokunaga, C.; Hara, K.; Eguchi, S.; Avruch, J.; Yonezawa, K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 2004, 9, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Zacarias, J.L.; Castro-Munozledo, F.; Kuri-Harcuch, W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil Red O. Histochemistry 1992, 97, 493–497. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sample of dibutyltin dilaurate (DBTD) is available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DBTD (μmol/L) | ||||||
|---|---|---|---|---|---|---|
| 0 | 0.13 | 0.25 | 0.50 | 1.00 | 2.00 | |
| ALT (IU/L) | 0.93 ± 0.69 | 0.86 ± 0.51 | 1.52 ± 1.00 | 2.41 ± 1.58 * | 3.99 ± 0.32 ** | 5.45 ± 0.44 ** |
| AST (IU/L) | 2.94 ± 1.89 | 2.26 ± 1.45 | 4.41 ± 0.65 | 5.77 ± 1.13 * | 9.72 ± 0.84 ** | 10.51 ± 1.37 ** |
| DBTD (μmol/L) | ||||||
|---|---|---|---|---|---|---|
| 0 | 0.13 | 0.25 | 0.5 | 1.0 | 2.0 | |
| Absorbance | 0.194 ± 0.008 | 0.193 ± 0.004 | 0.188 ± 0.006 | 0.156 ± 0.007 ** | 0.136 ± 0.006 ** | 0.102 ± 0.006 ** |
| Group | Blank Group | Rapa (0.1 μmol/L) | DBTD (0.2 μmol/L) | Rapa (0.1 μmol/L) + DBTD (0.2 μmol/L) |
|---|---|---|---|---|
| Viability (%) | 100 | 102 ± 11 | 93 ± 8 | 98 ± 5 |
| Group | ALT (IU/L) | AST (IU/L) |
|---|---|---|
| Blank group | 0.91 ± 0.61 | 2.83 ± 0.58 |
| Rapa (0.1 μmol/L) | 1.05 ± 0.39 | 2.77 ± 0.2 |
| DBTD (0.2 μmol/L) | 1.01 ± 0.59 | 2.79 ± 0.52 |
| Rapa (0.1 μmol/L) + DBTD (0.2 μmol/L) | 1.06 ± 0.47 | 2.83 ± 0.52 |
| Genes | Primer Sequence (5′-3′) | Length (bp) | |
|---|---|---|---|
| SREBP1C | Forward | CGGAACCATCTTGGCAACAGT | 141 |
| Reverse | CGCTTCTCAATGGCGTTGT | ||
| SCD1 | Forward | TTCCCGACGTGGCTTTTTCT | 149 |
| Reverse | AGCCAGGTTTGTAGTACCTCC | ||
| FASN | Forward | TTCCGAGATTCCATCCTACG | 122 |
| Reverse | AGGCTCACAAACGAATGGAC | ||
| GPAT1 | Forward | ATGGCATTCTTACAGTGGCAGAGC | 183 |
| Reverse | GCTCCTTGGATTGGCTCACCTTC | ||
| PPARα | Forward | ACGATTCGACTCAAGCTGGT | 123 |
| Reverse | GTTGTGTGACATCCCGACAG | ||
| ACOX1 | Forward | GCCTCTGGATCTTCACTTGG | 123 |
| Reverse | GTCTGGGCATAAGTGCCAAT | ||
| CPT1 | Forward | CAGACACCATCCAGCACATGAGAG | 157 |
| Reverse | TGAGGCTCCGAGGTATTGTCCAG | ||
| CYP4A11 | Forward | TACATACAGGCCATTAGTGACC | 83 |
| Reverse | CTGTAGATGGTGTCATTCTGGT | ||
| GAPDH | Forward | TGGTGAAGACGCCAGTGGA | 138 |
| Reverse | GCACCGTCAAGGCTGAGAAC | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, X.; Li, Y.; Mai, J.; Ji, X.; Li, Q. Effect of Dibutyltin Dilaurate on Triglyceride Metabolism through the Inhibition of the mTOR Pathway in Human HL7702 Liver Cells. Molecules 2018, 23, 1654. https://doi.org/10.3390/molecules23071654
Qiao X, Li Y, Mai J, Ji X, Li Q. Effect of Dibutyltin Dilaurate on Triglyceride Metabolism through the Inhibition of the mTOR Pathway in Human HL7702 Liver Cells. Molecules. 2018; 23(7):1654. https://doi.org/10.3390/molecules23071654
Chicago/Turabian StyleQiao, Xiaozhi, Yunlan Li, Jiaqi Mai, Xiaoqing Ji, and Qingshan Li. 2018. "Effect of Dibutyltin Dilaurate on Triglyceride Metabolism through the Inhibition of the mTOR Pathway in Human HL7702 Liver Cells" Molecules 23, no. 7: 1654. https://doi.org/10.3390/molecules23071654