Liquid Chromatography Coupled with Linear Ion Trap Hybrid OrbitrapMass Spectrometry for Determination of Alkaloids in Sinomeniumacutum


Abstract
1. Introduction
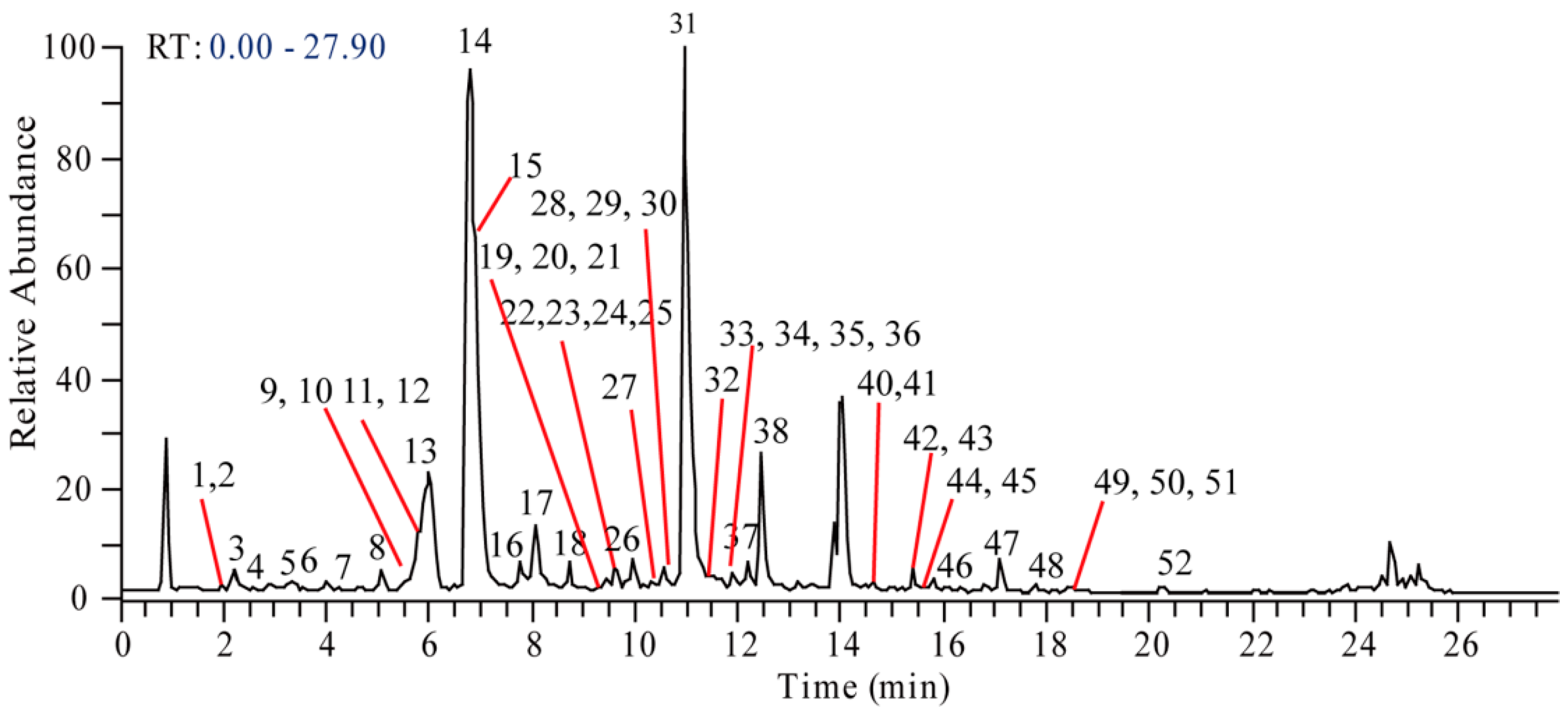
2. Results and Discussion
2.1. Detection of Morphinan Alkaloids
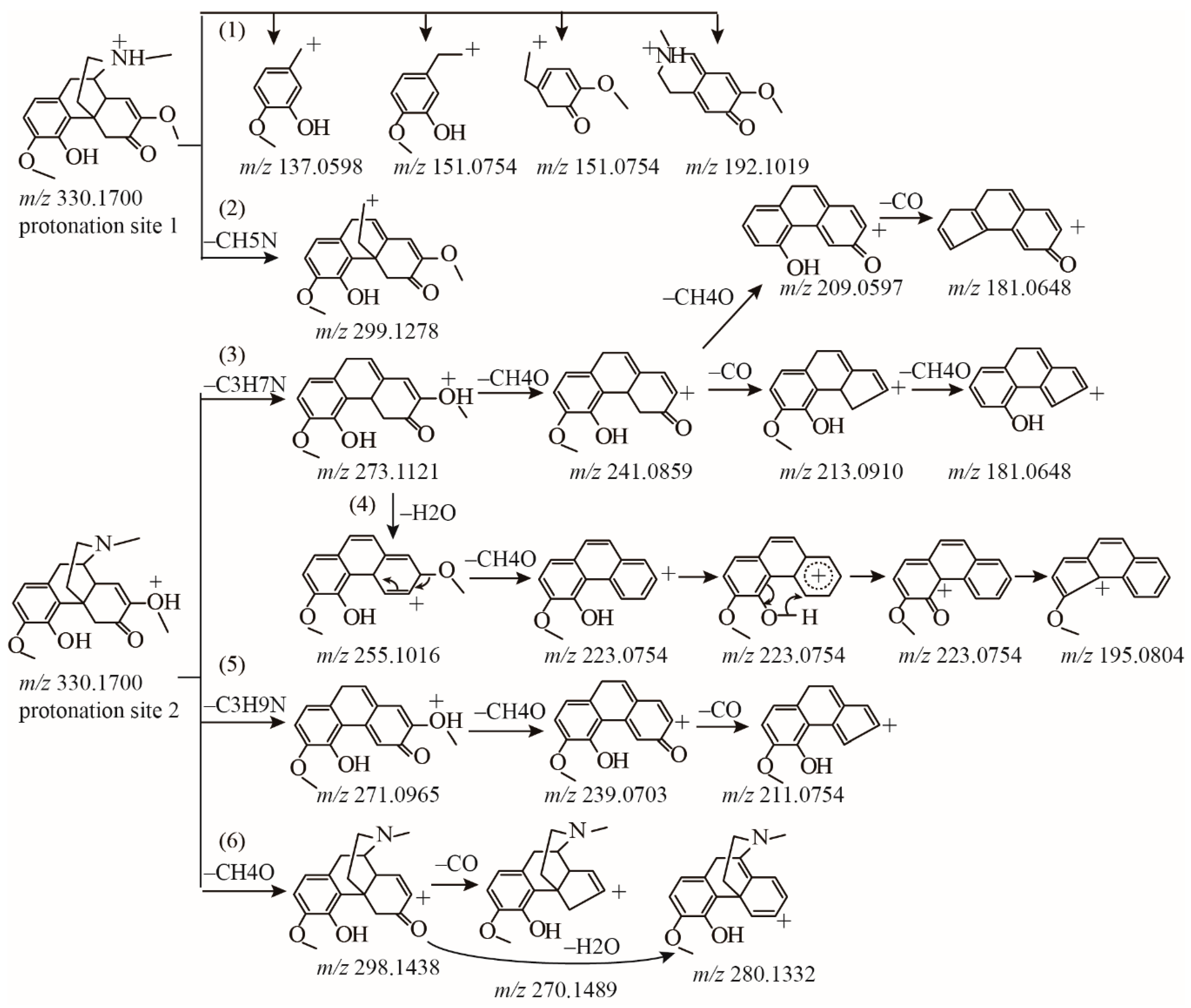
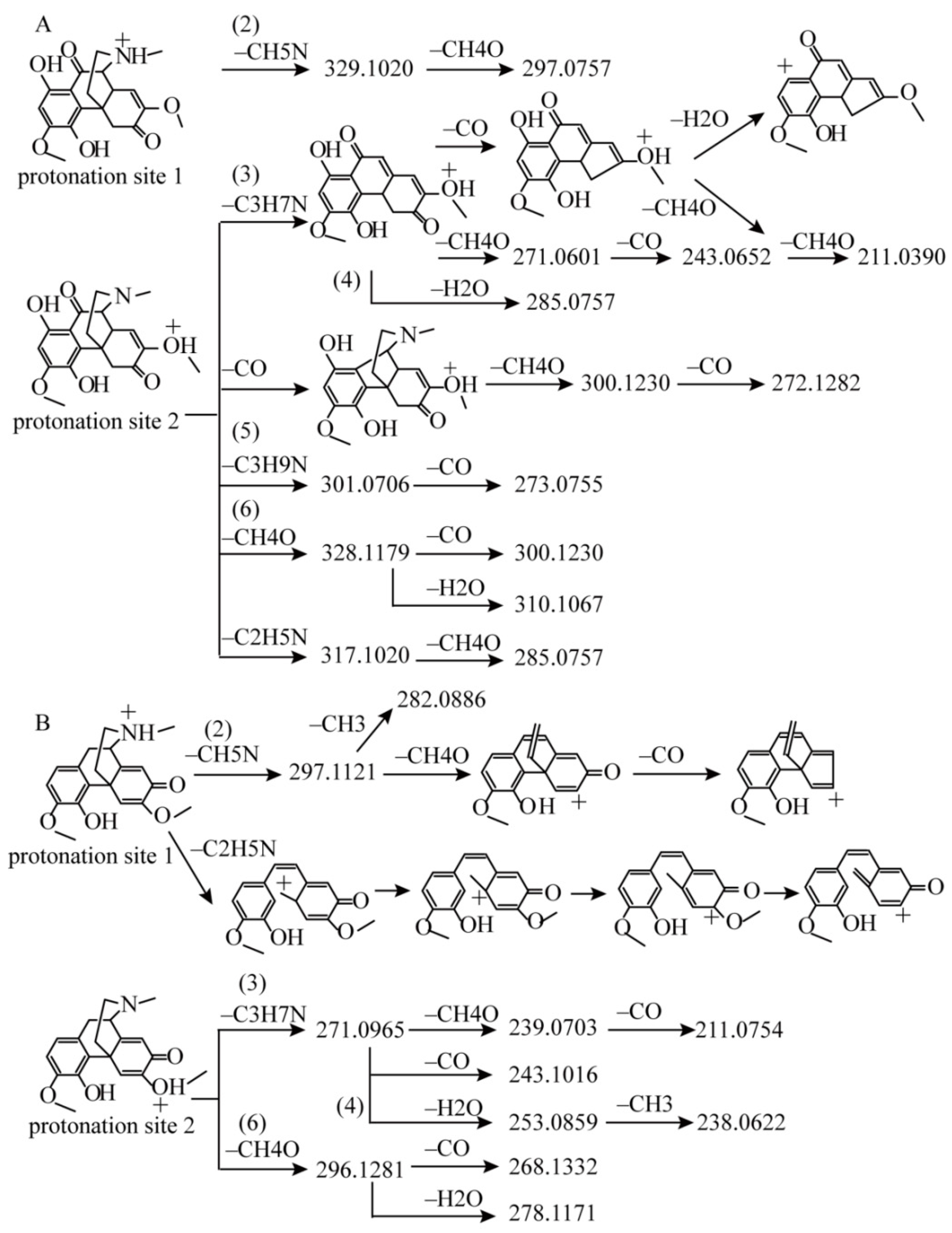
2.1.1. Fragmentation Analysis of Standard Sinomenine
2.1.2. Detection of Sinomenine Analogues
2.2. Detection of Aporphines and Benzylisoquinolines
2.3. Detection of Protoberberines
2.4. Other Alkaloids
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Sample Preparation
3.3. Chromatographic Separation Conditions
3.4. MS Conditions
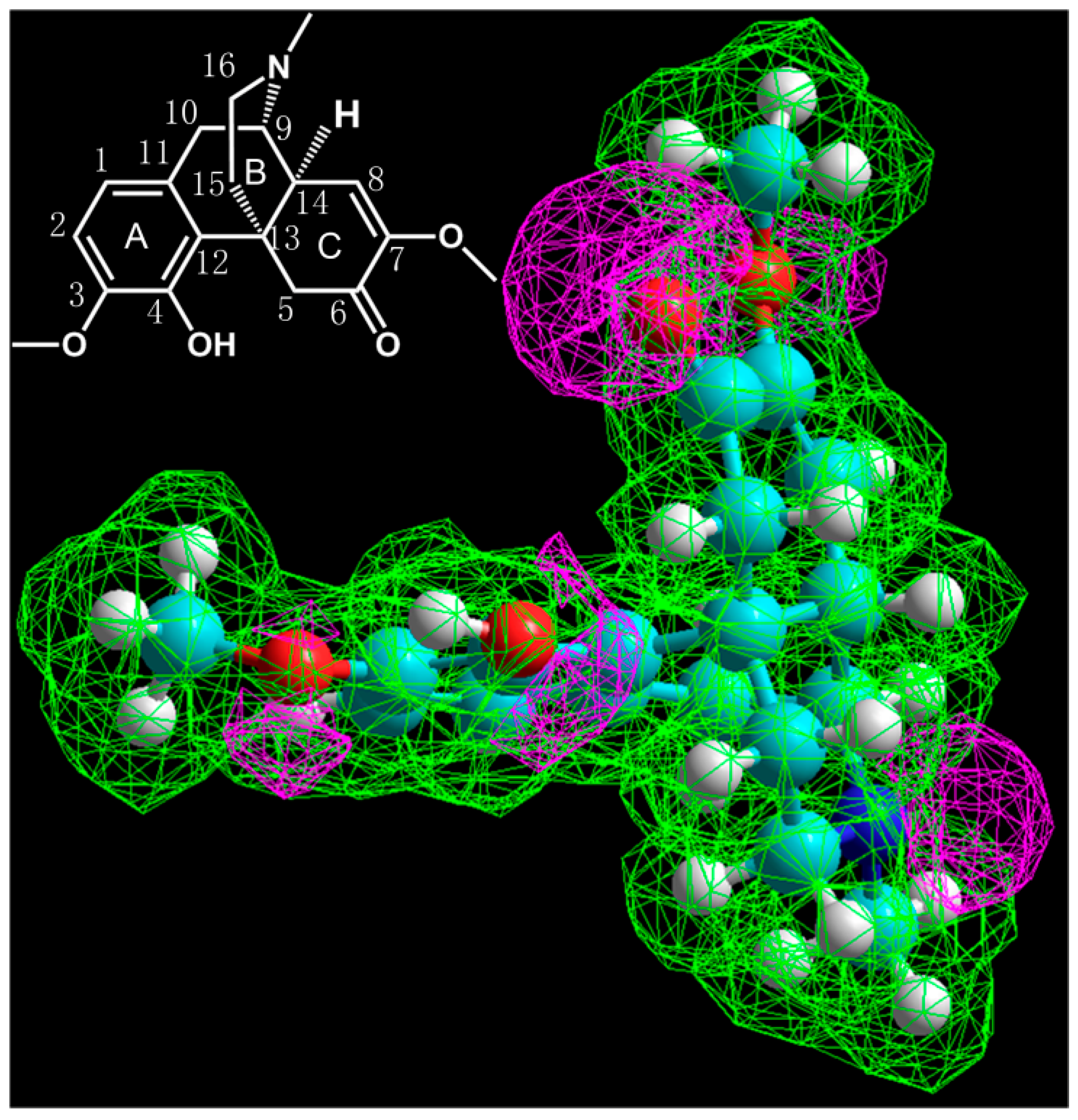
3.5. Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ganzera, M.; Sturm, S. Recent advances on HPLC/MS in medicinal plant analysis-An update covering 2011–2016. J. Pharm. Biomed. Anal. 2017, 147, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural alkaloids as P-gp inhibitors for multidrug resistance reversal in cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Guo, Z.Q.; Xu, X.Y.; Wu, Z.J. Recent (2000–2015) developments in the analysis of minor unknown natural products based on characteristic fragment information using LC-MS. Mass Spectrom. Rev. 2016, 37, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Qing, Z.X.; Cheng, P.; Liu, X.B.; Liu, Y.S.; Zeng, J.G. Systematic identification of alkaloids in macleaya microcarpa fruits by liquid chromatography tandem mass spectrometry combined with the isoquinoline alkaloids biosynthetic pathway. J. Pharm. Biomed. Anal. 2015, 103, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.B.; Li, L.M.; Fang, D.M.; Li, G.Y.; Zhang, G.L.; Wu, Z.J. Electrospray ionization tandem mass spectrometry of chaetoglobosins. Rapid Commun. Mass Spectrom. 2012, 26, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Deevanhxay, P.; Suzuki, M.; Maeshibu, N.; Li, H.; Tanaka, K.; Hirose, S. Simultaneous characterization of quaternary alkaloids, 8-oxoprotoberberine alkaloids, and a steroid compound in coscinium fenestratum by liquid chromatography hybrid ion trap time-of-flight mass spectrometry. J. Pharm. Biomed. Anal. 2009, 50, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Smyth, W.F. Recent studies on the electrospray ionisation mass spectrometric behaviour of selected nitrogen-containing drug molecules and its application to drug analysis using liquid chromatography-electrospray ionisation mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 824, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Liu, H.L.; Zhang, L.; Feng, L.L.; Huai, Y.; Deng, Z.S.; Sun, Y.; Xu, Q.; Li, J.X. Synthesis and biological evaluation of novel sinomenine derivatives as anti-inflammatory agents. Eur. J. Med. Chem. 2012, 50, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Liang, Z.T.; Peng, Y.; Yao, X.; Chen, H.B.; Zhao, Z.Z. Tissue-specific metabolite profiling of alkaloids in Sinomenii Caulis using laser microdissection and liquid chromatography-quadrupole/time of flight-mass spectrometry. J. Chromatogr. A 2012, 1248, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, B.; Liu, K.; Bo, T.; Liao, Y.; Liu, H. Fragmentation pathways of heroin-related alkaloids revealed by ion trap and quadrupole time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 2851–2862. [Google Scholar] [CrossRef] [PubMed]
- Raith, K.; Neubert, R.; Poeaknapo, C.; Boettcher, C.; Zenk, M.H.; Schmidt, J. Electrospray tandem mass spectrometric investigations of morphinans. J. Am. Soc. Mass Spectrom. 2003, 14, 1262–1269. [Google Scholar] [CrossRef]
- Hu, N.; Tu, Y.P.; Liu, Y.; Jiang, K.; Pan, Y. Dissociative protonation and proton transfers: Fragmentation of alpha, beta-unsaturated aromatic ketones in mass spectrometry. J. Org. Chem. 2008, 73, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.P. Dissociative protonation sites: Reactive centers in protonated molecules leading to fragmentation in mass spectrometry. J. Org. Chem. 2006, 71, 5482–5488. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.Z.; Wang, X.L.; Wang, H.B.; Wang, Y.B.; Lin, L.P.; Ding, J.; Qin, G.W. Morphinane alkaloid dimers from Sinomenium acutum. J. Nat. Prod. 2008, 71, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Liu, B.R.; Wang, J.R.; Chen, C.K.; Qin, G.W.; Lee, S.S. Two new morphinane alkaloids from Sinomenium acutum. J. Asian Nat. Prod. Res. 2011, 13, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.J.; Tsai, T.H.; Lin, L.C. New alkaloids and cytotoxic principles from Sinomenium acutum. Planta Med. 2012, 78, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Li, M.; Liu, J.J.; Yan, Q.; Zhong, M.; Liu, J.X.; Di, D.L.; Liu, J.X. Simultaneous determination of the content of isoquinoline alkaloids in Dicranostigma leptopodum (Maxim) Fedde and the effective fractionation of the alkaloids by high-performance liquid chromatography with diode array detection. J. Sep. Sci. 2015, 38, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, I.K.; Park, J.E.; Oh, J.Y.; Lee, K.R. Two new alkaloids from the rhizomes of Sinomenium acutum. Bull. Korean Chem. Soc. 2012, 33, 3455–3457. [Google Scholar] [CrossRef]
- Chen, Y.; Le Droumaguet, C.; Li, K.; Cotham, W.E.; Lee, N.; Walla, M.; Wang, Q. A novel rearrangement of fluorescent human thymidylate synthase inhibitor analogues in ESI tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yue, L.; Guo, M.; Jiang, K.; Pan, Y. Elimination of benzene from protonated N-benzylindoline: Benzyl cation/proton transfer or direct proton transfer? J. Am. Soc. Mass Spectrom. 2013, 24, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhu, Y.; Shao, Q.; Wang, Y.; Fan, X.; Cheng, Y. Identification of chemical constituents in two traditional Chinese medicine formulae by liquid chromatography-mass spectrometry and off-line nuclear magnetic resonance. J. Pharm. Biomed. Anal. 2016, 117, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Le, P.M.; McCooeye, M.; Windust, A. Characterization of the alkaloids in goldenseal (Hydrastis canadensis) root by high resolution Orbitrap LC-MSn. Anal. Bioanal. Chem. 2013, 405, 4487–4498. [Google Scholar] [CrossRef] [PubMed]
- Li, X.N.; Zhang, A.; Sun, H.; Song, Y.; Zou, D.; Wang, X. Rapid discovery of absorbed constituents and metabolites in rat plasma after the oral administration of Zi Shen Wan using high-throughput UHPLC-MS with a multivariate analysis approach. J. Sep. Sci. 2016, 39, 4700–4711. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Liang, J.; Yang, W.Z.; Hou, J.J.; Yang, M.; Da, J.; Wang, Y.; Jiang, B.H.; Liu, X.; Wu, W.Y.; et al. HPLC/qTOF-MS-oriented characteristic components data set and chemometric analysis for the holistic quality control of complex TCM preparations: Niuhuang Shangqing pill as an example. J. Pharm. Biomed. Anal. 2014, 89, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.X.; Fang, L.L.; Liang, X.L.; Su, D.; Guo, X.J. A sensitive and selective UPLC-MS/MSmethod for simultaneous determination of 10 alkaloids from Rhizoma Menispermi in rat plasma and its application to a pharmacokinetic study. Talanta 2015, 144, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Hu, R.; Sun, C.; Pan, Y. Comprehensive separation and analysis of alkaloids from Stephania yunnanensis by counter-current chromatography coupled with liquid chromatography tandem mass spectrometry analysis. J. Chromatogr. A 2012, 1226, 18–23. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | tR (min) | m/z | Formula ([M + H]+) | ppm | Identification |
|---|---|---|---|---|---|
| 1 | 1.97 | 492.2225 | C25H34O9N | −0.308 | Sinomenine glucoside |
| 2 | 2.00 | 346.1650 | C19H24O5N | 0.111 | Sinomenine N-oxide |
| 3 | 2.05 | 434.1818 | C22H28O8N | 0.817 | Higenamine glucoside |
| 4 | 2.74 | 522.2331 | C26H36O10N | −0.283 | Cephatonine glucoside |
| 5 | 3.21 | 346.1643 | C19H24O5N | −1.789 | Unknown |
| 6 | 3.76 | 318.1699 | C18H24O4N | −0.065 | N-demethyl dihydrosinomenine |
| 7 1 | 4.48 | 272.1285 | C16H18O3N | 0.330 | Higenamine |
| 8 | 4.88 | 384.1209 | C18H23O6NCl | 0.256 | Acutumidine |
| 9 | 4.93 | 344.1854 | C20H26O4N | −0.215 | N-methyl sinomenine |
| 10 | 4.99 | 316.1545 | C18H22O4N | 0.145 | N-demethyl sinomenine |
| 11 | 5.59 | 332.1854 | C19H26O4N | −0.827 | Dihydrosinomenine |
| 12 | 5.69 | 448.1973 | C23H30O8N | 0.707 | Coclaurine glucoside |
| 13 | 5.78 | 478.2079 | C24H32O9N | 1.656 | 3′-hydroxy-N-methyl coclaurine glucoside |
| 14 1 | 6.61 | 330.1700 | C19H24O4N | 0.167 | Sinomenine |
| 15 | 6.74 | 398.1362 | C19H25O6NCl | −0.657 | Acutumine |
| 16 | 7.64 | 316.1543 | C18H22O4N | −0.110 | 3′-hydroxy-N-methylcoclaurine |
| 17 | 7.94 | 314.1394 | C18H20O4N | 2.182 | Feruloyltyramine |
| 18 | 8.62 | 476.2283 | C25H34O8N | 0.407 | Magnocurarine4′-O-glucopyranoside |
| 19 | 8.92 | 358.1646 | C20H24O5N | −0.319 | Hydroxyl magnoflorine |
| 20 | 9.23 | 360.1438 | C19H22O6N | −0.334 | 1-hydroxy-10-oxo-sinomenine |
| 21 | 9.25 | 358.1646 | C20H24O5N | −0.319 | Hydroxyl magnoflorine |
| 22 | 9.56 | 286.1442 | C17H20O3N | 0.400 | Coclaurine |
| 23 | 9.57 | 657.3165 | C38H45O8N2 | −0.780 | Disinomenine |
| 24 | 9.69 | 360.1802 | C20H26O5N | −0.309 | Cephatonine |
| 25 | 9.79 | 300.1595 | C18H22O3N | 0.030 | N-methylcoclaurine |
| 26 | 9.86 | 298.1442 | C18H20O3N | 1.442 | Stepharine |
| 27 | 9.98 | 358.1647 | C20H24O5N | −0.229 | Hydroxyl magnoflorine |
| 28 | 10.47 | 344.1852 | C20H26O4N | −1.147 | Tembetarine |
| 29 | 10.56 | 328.1547 | C19H22O4N | 0.325 | Unknown |
| 30 | 10.70 | 370.1646 | C21H24O5N | −0.863 | Unknown |
| 31 1 | 10.91 | 342.1698 | C20H24O4N | −0.185 | Magnoflorine |
| 32 | 11.16 | 328.1545 | C19H22O4N | 0.145 | Unknown |
| 33 | 11.52 | 330.1699 | C19H24O4N | −0.196 | Isosinomenine |
| 34 | 11.59 | 328.1547 | C19H22O4N | 0.991 | Sinoacutine |
| 35 | 11.85 | 360.1437 | C19H22O6N | −1.177 | Unknown |
| 36 | 11.96 | 340.1543 | C20H22O4N | −0.102 | N-Methyl bulbocapnine |
| 37 | 12.19 | 314.1753 | C19H24O3N | 0.795 | Magnocurarine |
| 38 | 12.44 | 342.1698 | C20H24O4N | −0.54 | Laurifoline |
| 39 | 13.98 | 356.1856 | C21H26O4N | −0.238 | N-methyl isocorydine |
| 40 | 14.62 | 390.1548 | C20H24O7N | −0.406 | Unknown |
| 41 | 14.67 | 368.1492 | C21H22O5N | −0.215 | Oxypalmatine |
| 42 | 15.34 | 322.1078 | C19H16O4N | 1.166 | Menisporphine |
| 43 | 15.43 | 324.1231 | C19H18O4N | 0.048 | Stepharanine |
| 44 | 15.74 | 322.1076 | C19H16O4N | 0.793 | Menisporphine |
| 45 | 15.77 | 356.1854 | C21H26O4N | −0.603 | N-methyl corydine |
| 46 | 16.15 | 374.1596 | C20H24O6N | −0.625 | Unknown |
| 47 | 17.77 | 338.1386 | C20H20O4N | −0.221 | Dehydrocorydalmine or its isomer |
| 48 | 18.15 | 338.1387 | C20H20O4N | −0.043 | Dehydrocorydalmine or its isomer |
| 49 1 | 18.42 | 338.1384 | C20H20O4N | −0.842 | Jatrorrhizine |
| 50 | 18.42 | 336.1231 | C20H18O4N | 0.314 | Pseudoberberine |
| 51 1 | 18.74 | 338.1383 | C20H20O4N | −1.108 | Columbamin |
| 52 1 | 21.38 | 352.1543 | C21H22O4N | −0.098 | Palmatine |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, J.; Zhao, X.; Shen, C.; Ji, J.; Xu, J.; Wang, S.; Xie, T.; Tong, W. Liquid Chromatography Coupled with Linear Ion Trap Hybrid OrbitrapMass Spectrometry for Determination of Alkaloids in Sinomeniumacutum. Molecules 2018, 23, 1634. https://doi.org/10.3390/molecules23071634
Shan J, Zhao X, Shen C, Ji J, Xu J, Wang S, Xie T, Tong W. Liquid Chromatography Coupled with Linear Ion Trap Hybrid OrbitrapMass Spectrometry for Determination of Alkaloids in Sinomeniumacutum. Molecules. 2018; 23(7):1634. https://doi.org/10.3390/molecules23071634
Chicago/Turabian StyleShan, Jinjun, Xia Zhao, Cunsi Shen, Jianjian Ji, Jianya Xu, Shouchuan Wang, Tong Xie, and Wenjun Tong. 2018. "Liquid Chromatography Coupled with Linear Ion Trap Hybrid OrbitrapMass Spectrometry for Determination of Alkaloids in Sinomeniumacutum" Molecules 23, no. 7: 1634. https://doi.org/10.3390/molecules23071634
APA StyleShan, J., Zhao, X., Shen, C., Ji, J., Xu, J., Wang, S., Xie, T., & Tong, W. (2018). Liquid Chromatography Coupled with Linear Ion Trap Hybrid OrbitrapMass Spectrometry for Determination of Alkaloids in Sinomeniumacutum. Molecules, 23(7), 1634. https://doi.org/10.3390/molecules23071634