(-)-Shikimic Acid as a Chiral Building Block for the Synthesis of New Cytotoxic 6-Aza-Analogues of Angucyclinones

 , ,
, ,
Abstract
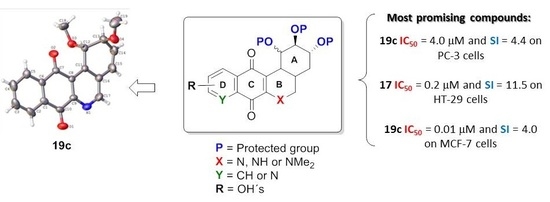
:
1. Introduction
2. Results and Discussion
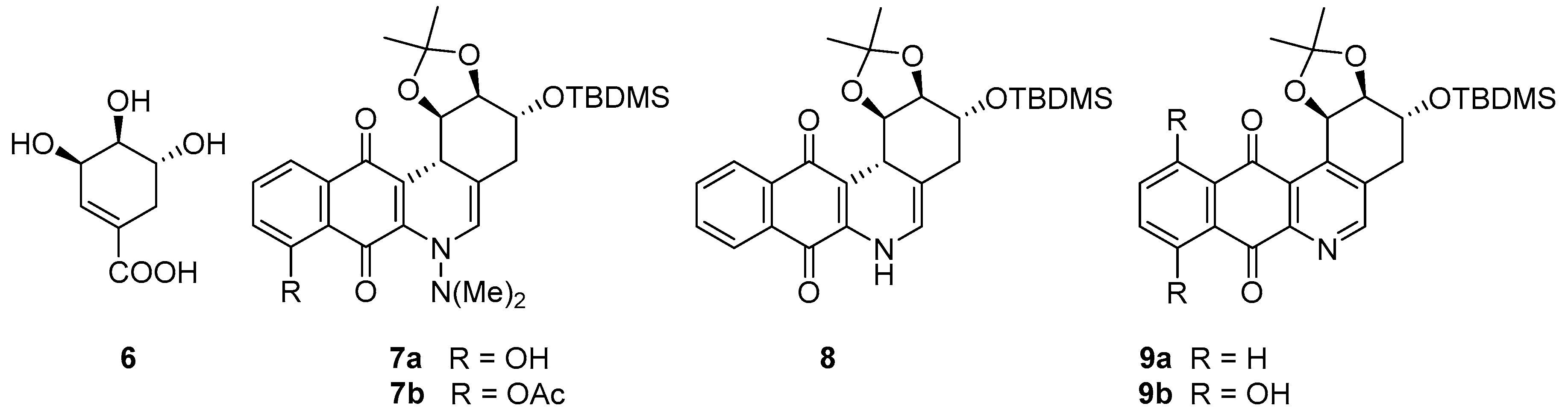
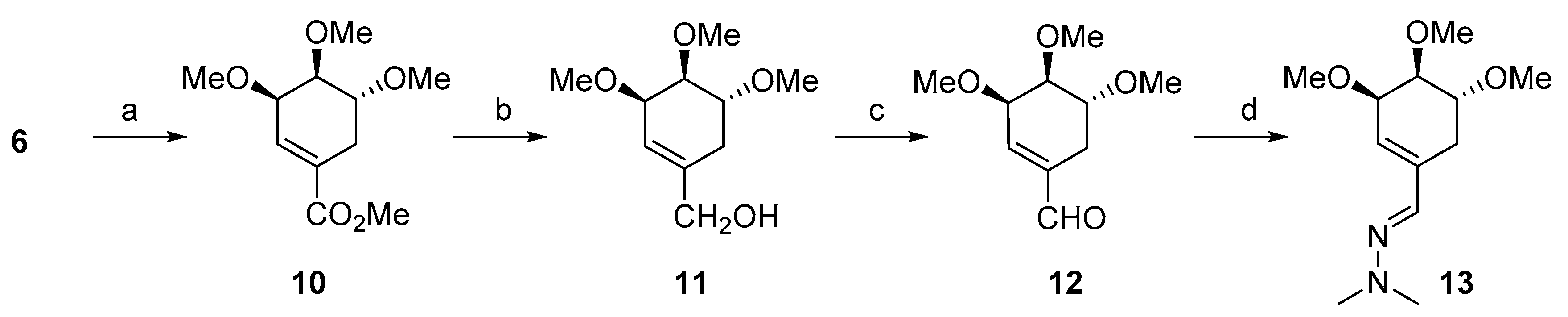
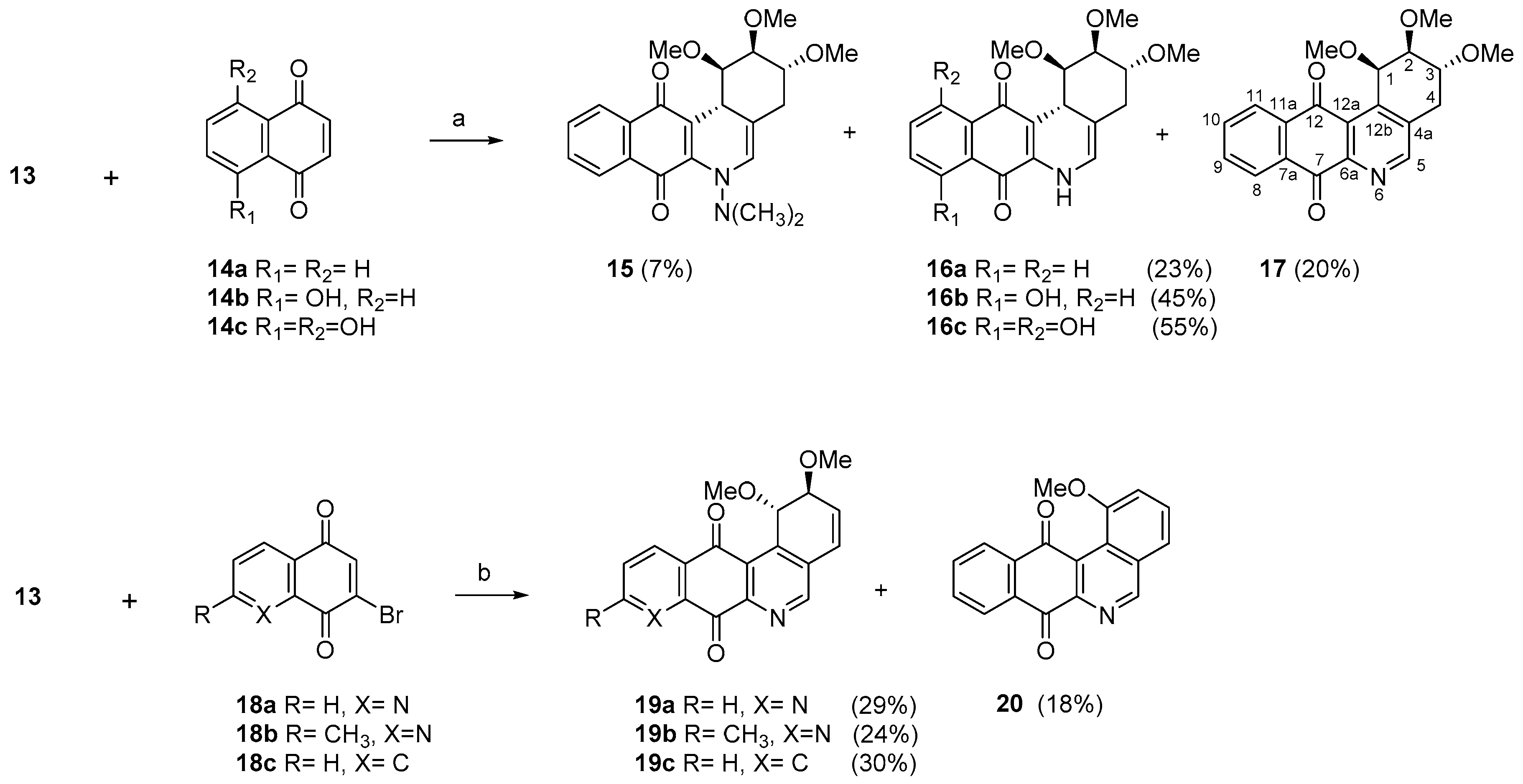
2.1. Synthesis
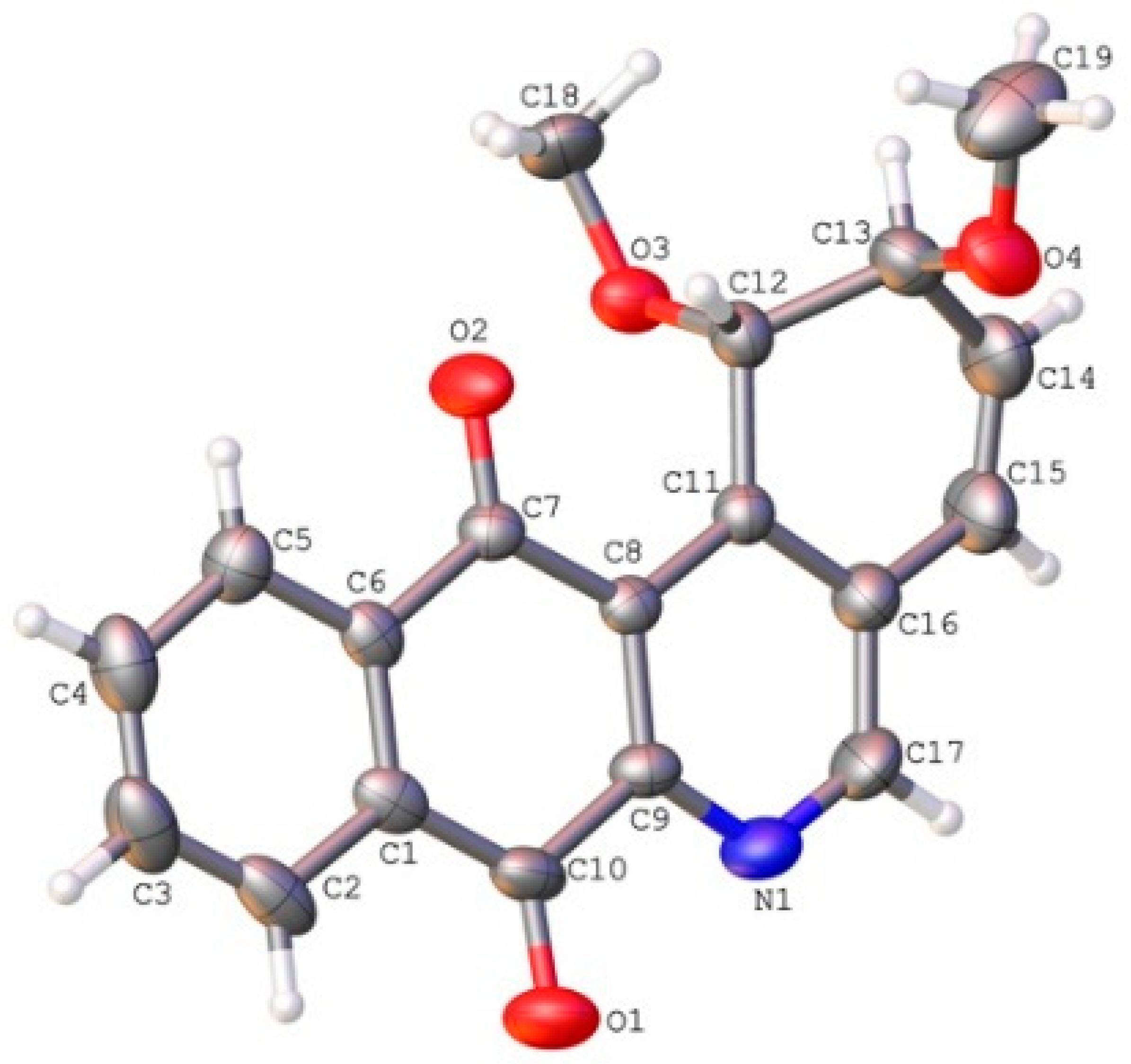
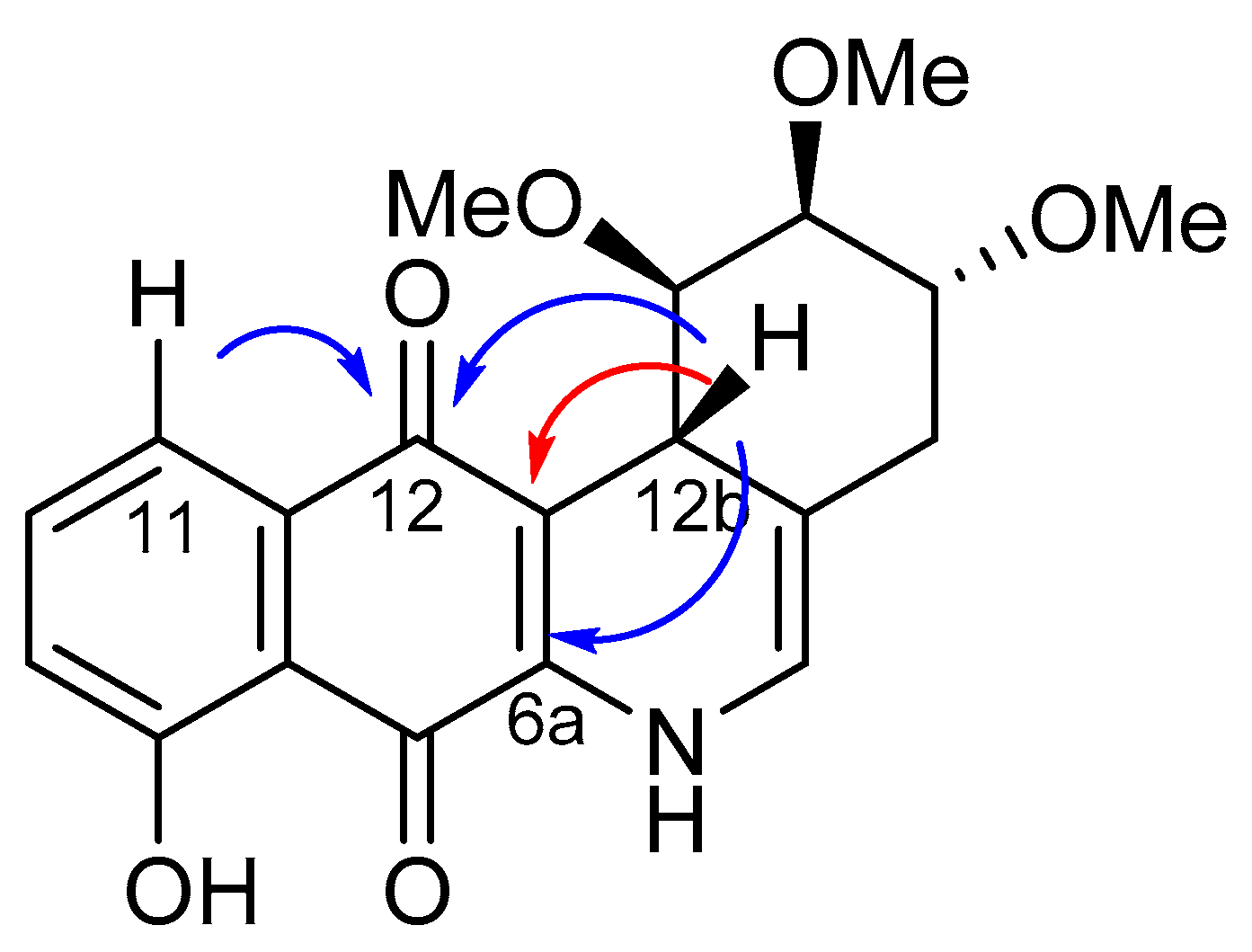
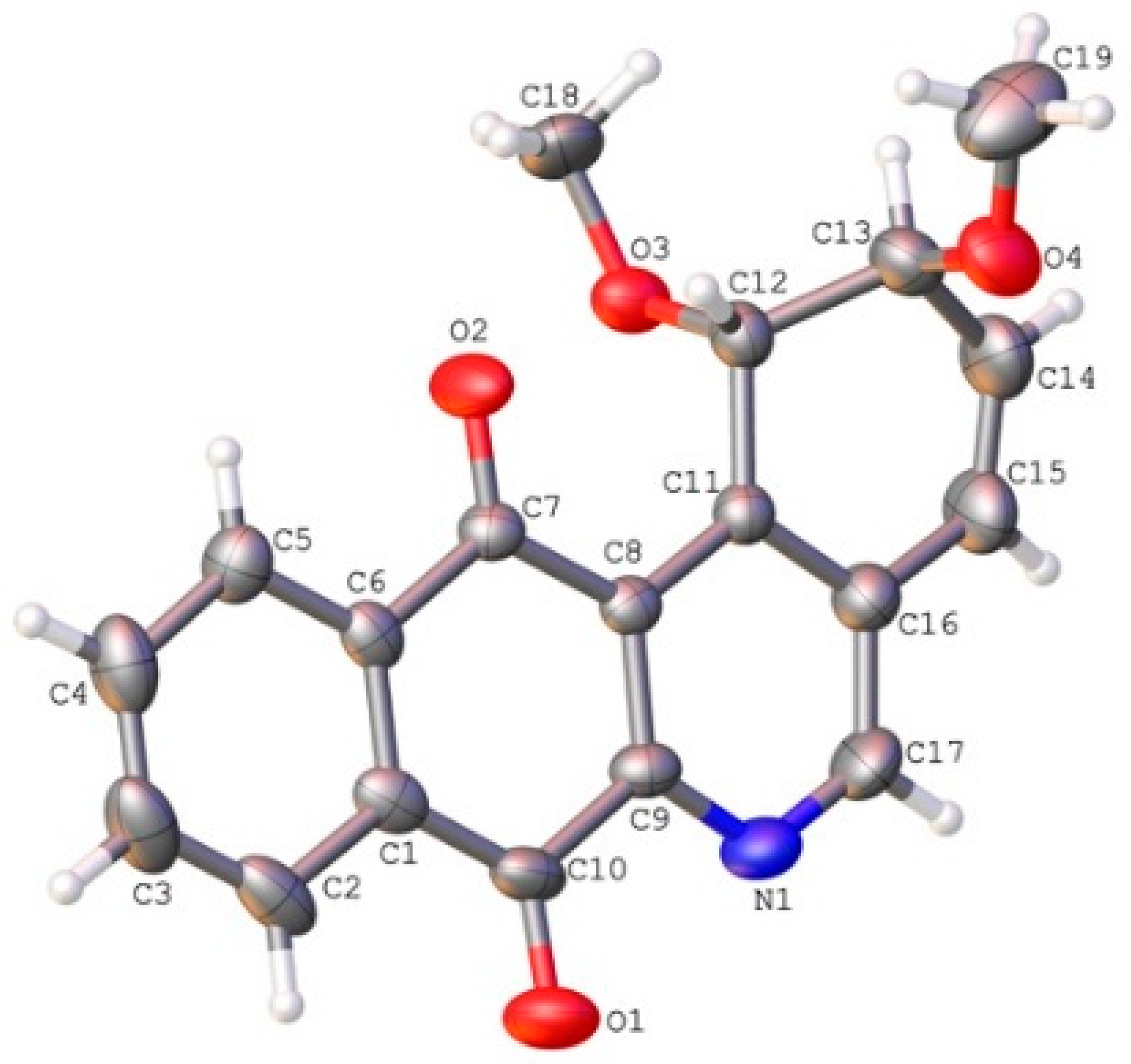
2.2. Crystallographic Studies
2.3. Biology
- (i)
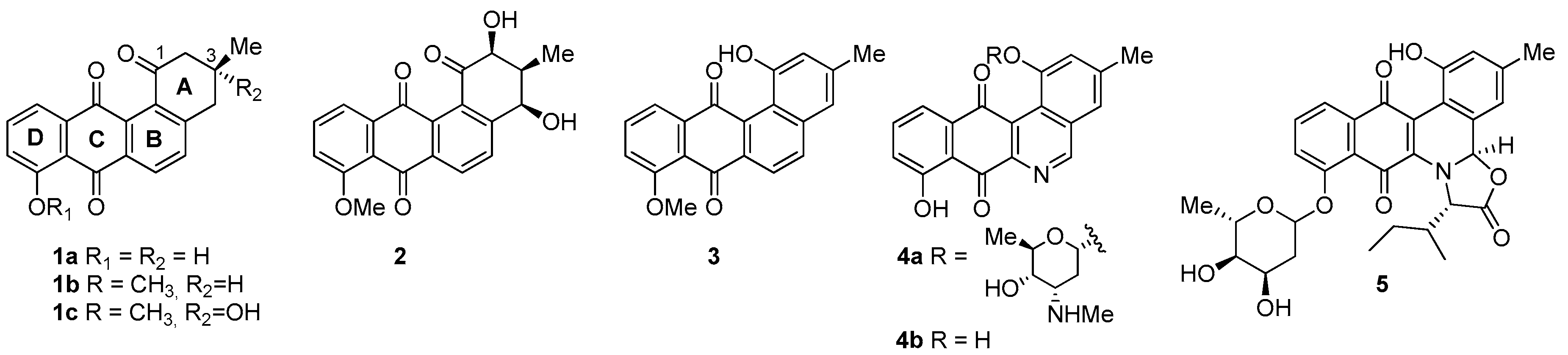
- For PC-3 cells, compounds 7a, 19a, and 19b were the most potent compounds of all assayed, with IC50 values less than 1.0 μM. Nevertheless, among these three, 7a was more selective (SI value = 3.3). Although 16a, 17, 19c, and 20 were less active than the last-mentioned compounds, they showed the highest selectivity (SI values = 4.9, 3.0, 4.4, and 3.0, respectively).
- (ii)
- For HT-29 cells, once more the compounds 7a, 19a, and 19b were the most potent derivatives (IC50 values < 0.4 μM). It should be noted that 7a elicited the best result in selectivity in respect to all assayed compounds in this study. Nevertheless, 19a and 19b showed SI values less than 1.0. In addition, 17 and 20 were also interesting compounds due to their moderate cytotoxicity (IC50 values ~ 3.0 μM), as well as their good selectivity (SI values = 3.6 and 6.4, respectively).
- (iii)
- For MCF-7, we observed that 19a was the best compound in potency (IC50 value ~ 10 nM) and selectivity (SI value = 4.0). This is an interesting result because this cell line was less sensitive than the other cell lines assayed.
- (i)
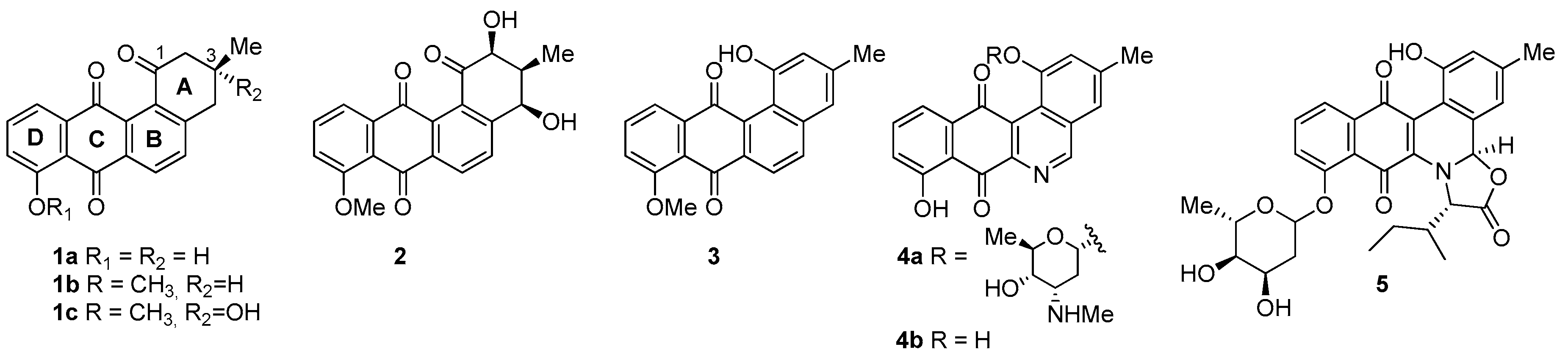
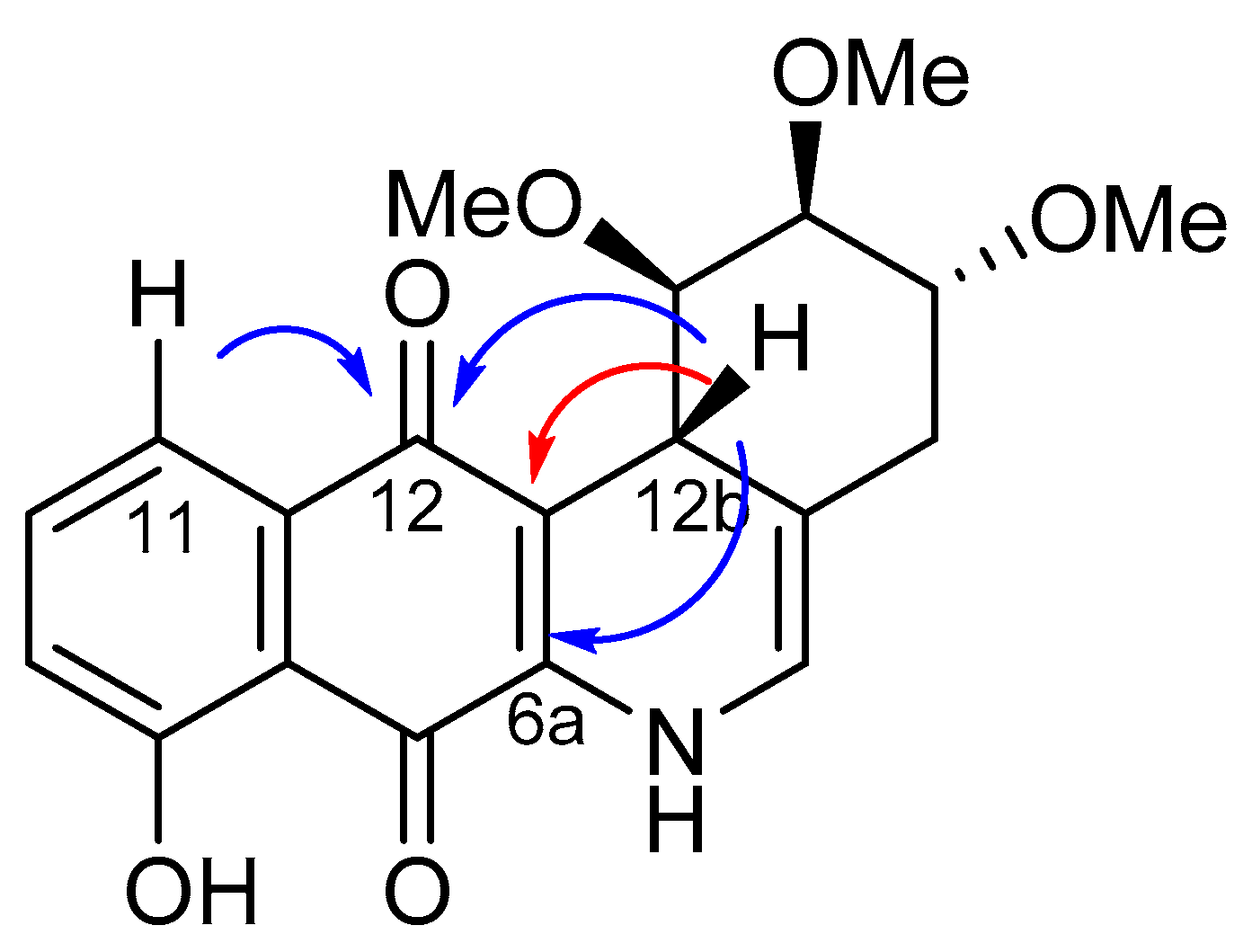
- Considering the size of the protected groups on ring A, the evidence could indicate that groups that are more voluminous generate an increase in cytotoxic activity. This behaviour is clearly observed in all cancer cell lines, when comparing 8 versus 16a and 7b with 15. The same slight tendency is demonstrated when comparing 7a with 16b.
- (ii)
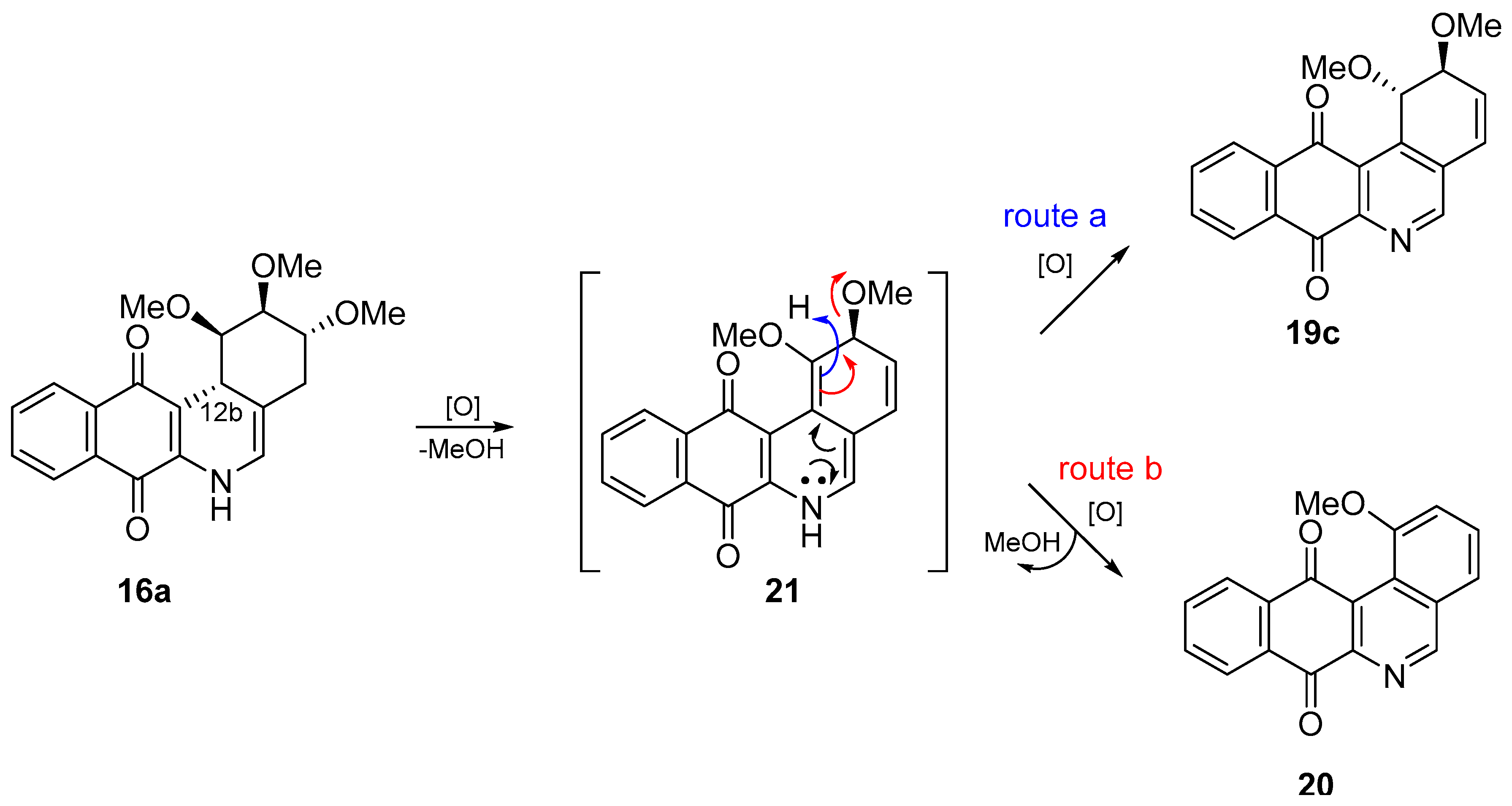
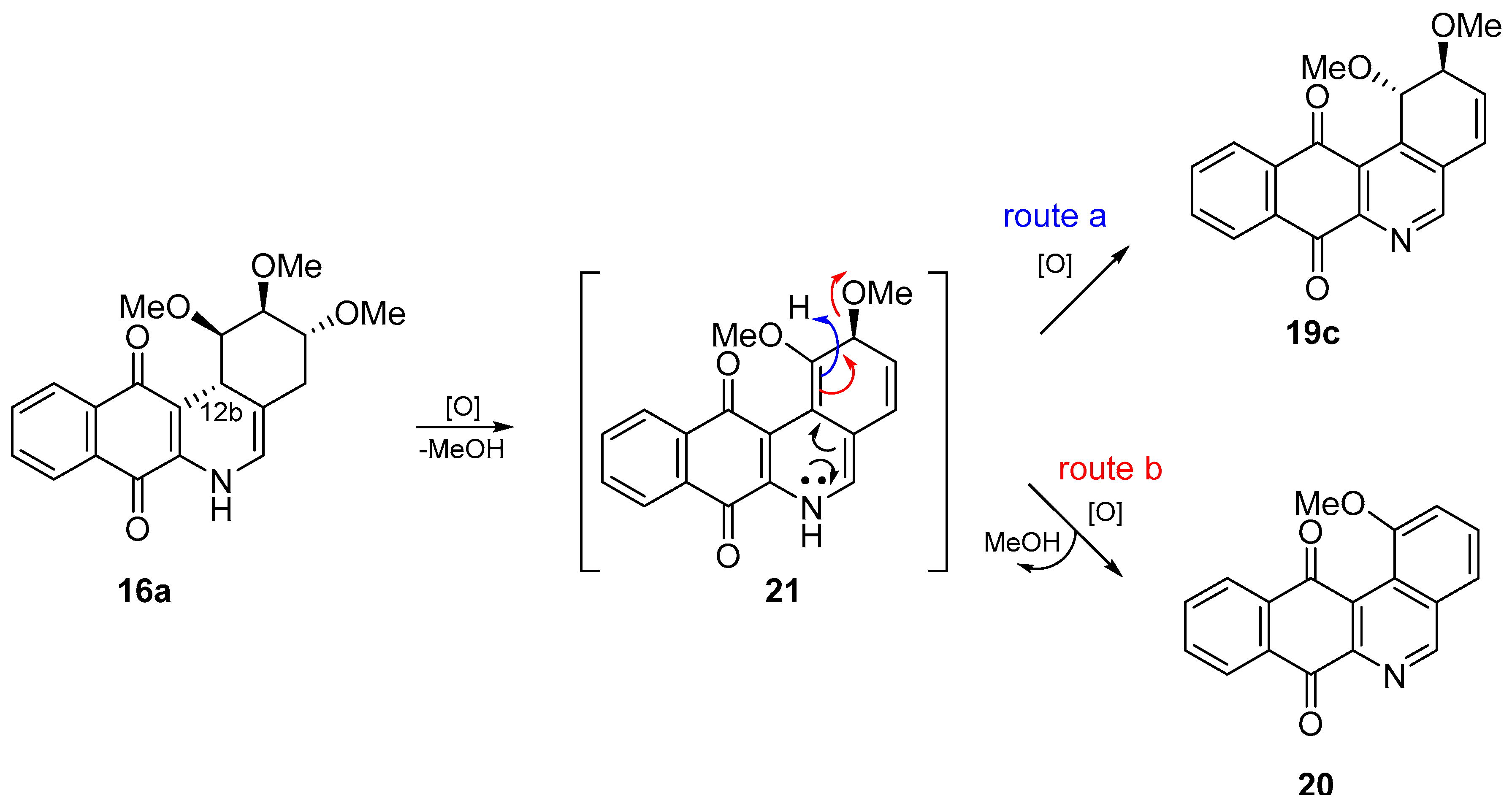
- With respect to ring B, it was not possible to indicate that the cytotoxicity of these angucyclines could be related only to the aromatic features of this ring due to the influence of the protected groups. In fact, for voluminous groups on ring A, the cytotoxicity was reduced in the aromatic system (8 versus 9). However, when the protected groups were methoxy, a significant increase of activity (almost tenfold) was elicited when this ring was aromatic (16a versus 17). This effect was observed on the three cancer cell lines.
- (iii)
- Finally, when the benzene ring (ring D) was substituted in C-8 for an electron-donating group (hydroxyl group), this modification generated an increase in the cytotoxic activity. This effect was observed for 7a when comparing it with its analogous 8 on two cancer cell lines (PC-3 and HT-29 cells). This behaviour was similar for 16a and its related compound 16b, but not in all three cancer cell lines. On the other hand, the isosteric replacement of the benzene ring by a pyridine ring led to an increase in the potency of the respective derivatives, which is shown when comparing the IC50 values between 19c and 19a–b. These results are in agreement with the data reported by several other authors [23,34,40,41]. Our results indicate that, on three cell lines, the addition of an extra nitrogen atom in the aromatic ring increases cytotoxic activity.
3. Materials and Methods
3.1. Chemistry
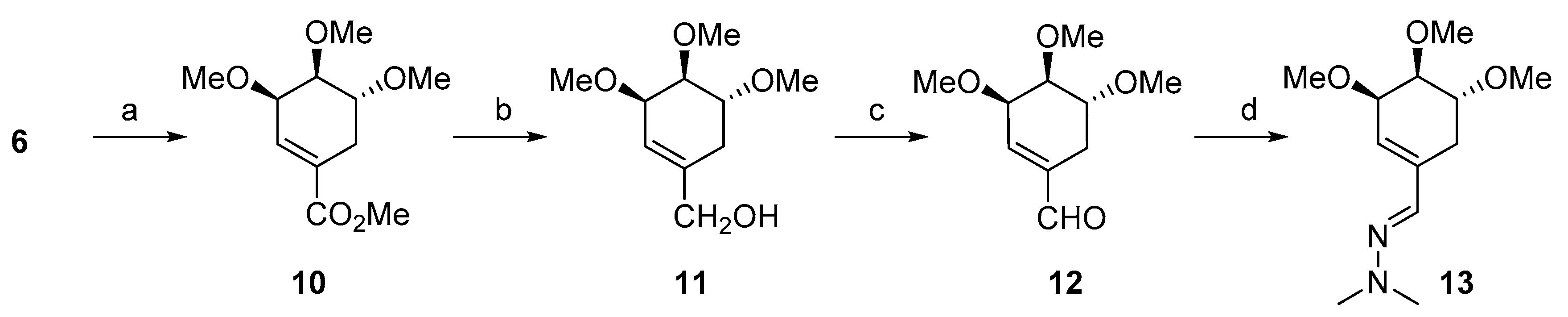
3.1.1. Synthesis of ((3R,4S,5R)-3,4,5-Trimethoxycyclohex-1-en-1-yl) methanol (11)
3.1.2. Synthesis of (3R,4S,5R)-3,4,5-Trimethoxycyclohex-1-enecarbaldehyde (12)
3.1.3. Synthesis of (E)-1,1-Dimethyl-2-(((3R,4S,5R)-3,4,5-trimethoxycyclohex-1-en-1-yl)methylene)hydrazine (13)
3.1.4. General Procedure for the Synthesis of the Target Compounds 15–17
3.1.5. General Procedure for the Synthesis of the Compounds 19–20
3.2. Biology
Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krohn, K.; Agocs, A.; Bäuerlein, C. Total synthesis of angucyclines. XVII. First synthesis of antibiotic 100-1, a deoxydisaccharide angucycline antibiotic of the urdamycinone b-type. J. Carbohydr. Chem. 2003, 22, 579–592. [Google Scholar] [CrossRef]
- Malmierca, M.G.; González-Montes, L.; Pérez-Victoria, I.; Sialer, C.; Braña, A.F.; García Salcedo, R.; Martín, J.; Reyes, F.; Méndez, C.; Olano, C.; et al. Searching for glycosylated natural products in actinomycetes and identification of novel macrolactams and angucyclines. Front. Microbiol. 2018, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.; Thiericke, R. Angucycline group antibiotics. Nat. Prod. Rep. 1992, 9, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.P.; Liu, B.; Wang, H.P.; Yang, S.X.; Zhang, H.Y.; Wang, Y.P.; Ji, N.Y.; Qin, S.; Laatsch, H. Kiamycin, a unique cytotoxic angucyclinone derivative from a marine Streptomyces sp. Mar. Drugs 2012, 10, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.K.; Liu, S.B.; Jiao, R.H.; Wang, T.; Tan, R.X.; Ge, H.M. Angucyclines from an insect-derived actinobacterium Amycolatopsis sp. Hca1 and their cytotoxic activity. Bioorg. Med. Chem. Lett. 2012, 22, 7490–7493. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, G.; Li, J.; Huang, H.; Zhang, X.; Zhang, H.; Ju, J. Cytotoxic and antibacterial angucycline- and prodigiosin- analogues from the deep-sea derived Streptomyces sp. Scsio 11594. Mar Drugs 2015, 13, 1304. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Rateb, M.E.; Teng, Q.; Yang, D.; Rudolf, J.D.; Zhu, X.; Huang, Y.; Zhao, L.-X.; Jiang, Y.; Li, X.; et al. Angucyclines and angucyclinones from Streptomyces sp. Cb01913 featuring c-ring cleavage and expansion. J. Nat. Prod. 2015, 78, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Kaliappan, K.P.; Ravikumar, V. Angucyclinone antibiotics: Total syntheses of YM-181741, (+)-ochromycinone, (+)-rubiginone B2, (−)-tetrangomycin, and MM-47755. J. Org. Chem. 2007, 72, 6116–6126. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.H.; Johnson, A.W. The structure of ochromycinone. Tetrahedron Lett. 1967, 8, 1449–1452. [Google Scholar] [CrossRef]
- Oka, M.; Kamei, H.; Hamagishi, Y.; Tomita, K.; Miyaki, T.; Konishi, M.; Oki, T. Chemical and biological properties of rubiginone, a complex of new antibiotics with vincristine-cytotoxicity potentiating activity. J. Antibiot. 1990, 43, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.; Zeeck, A. Metabolic products of microorganisms. 240. Urdamycins, new angucycline antibiotics from streptomyces fradiae. II. Structural studies of urdamycins B to F. J. Antibiot. 1987, 40, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Puder, C.; Zeeck, A.; Beil, W. New biologically active rubiginones from Streptomyces sp. J. Antibiot. 2000, 53, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Park, H.B.; Lee, J.K.; Lee, K.R.; Kwon, H.C. Angumycinones a and b, two new angucyclic quinones from Streptomyces sp. KMC004 isolated from acidic mine drainage. Tetrahedron Lett. 2014, 55, 63–66. [Google Scholar]
- Cone, M.C.; Hassan, A.M.; Gore, M.P.; Gould, S.J.; Borders, D.B.; Alluri, M.R. Detection of phenanthroviridin aglycon in a UV mutant of Streptomyces murayamaensis. J. Org. Chem. 1994, 59, 1923–1924. [Google Scholar] [CrossRef]
- Gore, M.P.; Gould, S.J.; Weller, D.D. Total synthesis of phenanthroviridin aglycon: The first naturally-occurring benzo[b]phenanthridine. J. Org. Chem. 1991, 56, 2289–2291. [Google Scholar] [CrossRef]
- Habbu, P.; Warad, V.; Shastri, R.; Madagundi, S.; Kulkarni, V.H. Antimicrobial metabolites from marine microorganisms. Chin. J. Nat. Med. 2016, 14, 101–116. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Z.; Li, S.; Lu, Y.; Chen, Y.; Zhang, H.; Zhang, G.; Zhu, Y.; Liu, J.; Zhang, C. Fluostatins I-K from the south china sea-derived Micromonospora rosaria SCSIO N160. J. Nat. Prod. 2012, 75, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Doull, J.L.; Singh, A.K.; Hoare, M.; Ayer, S.W. Conditions for the production of jadomycin-B by Streptomyces-venezuelae isp5230—Effects of heat-shock, ethanol treatment and phage infection. J. Ind. Microbiol. 1994, 13, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Ayer, S.W.; Mcinnes, A.G.; Thibault, P.; Walter, J.A.; Doull, J.L.; Parnell, T.; Vining, L.C. Jadomycin, a novel 8H-benz[b]oxazolo[3,2-f]phenanthridine antibiotic from Streptomyces-venezuelae isp5230. Tetrahedron Lett. 1991, 32, 6301–6304. [Google Scholar] [CrossRef]
- Hall, S.R.; Toulany, J.; Bennett, L.G.; Martinez-Farina, C.F.; Robertson, A.W.; Jakeman, D.L.; Goralski, K.B. Jadomycins inhibit type II topoisomerases and promote DNA damage and apoptosis in multidrug-resistant triple-negative breast cancer cells. J. Pharmacol. Exp. Ther. 2017, 363, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Hall, S.R.; Dupuis, S.N.; Graham, C.L.; Jakeman, D.L.; Goralski, K.B. Jadomycins are cytotoxic to ABCB1-, ABCC1-, and ABCG2-overexpressing MCF7 breast cancer cells. Anti Cancer Drugs 2014, 25, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Zhang, X.; Liu, H.; Han, H.; Luo, Y.; Wang, Q.; Geng, M.; Yang, K. Evaluation of the cytotoxic activity of new jadomycin derivatives reveals the potential to improve its selectivity against tumor cells. J. Antibiot. 2012, 65, 449–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef] [PubMed]
- Kesenheimer, C.; Kalogerakis, A.; Meißner, A.; Groth, U. The cobalt way to angucyclinones: Asymmetric total synthesis of the antibiotics (+)-rubiginone B2, (−)-tetrangomycin, and (−)-8-O-methyltetrangomycin. Chem. Eur. J. 2010, 16, 8805–8821. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K.; Böker, N.; Flörke, U.; Freund, C. Synthesis of angucyclines. 8. Biomimetic-type synthesis of rabelomycin, tetrangomycin, and related ring B aromatic angucyclinones. J. Org. Chem. 1997, 62, 2350–2356. [Google Scholar] [CrossRef] [PubMed]
- Mal, D.; Dey, S. Synthesis of chlorine-containing angucycline BE-23254 and its analogs. Tetrahedron 2006, 62, 9589–9602. [Google Scholar] [CrossRef]
- Landells, J.S.; Larsen, D.S.; Simpson, J. Remote stereochemical control in asymmetric diels-alder reactions: Synthesis of the angucycline antibiotics, (−)-tetrangomycin and MM 47755. Tetrahedron Lett. 2003, 44, 5193–5196. [Google Scholar] [CrossRef]
- Carreno, M.C.; Urbano, A.; Di Vitta, C. Short and efficient enantioselective total synthesis of angucyclinone type antibiotics (+)-rubiginone B-2 and (+)-ochromycinone. Chem. Commun. 1999, 817–818. [Google Scholar] [CrossRef]
- Vu, N.Q.; Dujardin, G.; Collet, S.C.; Raiber, E.-A.; Guingant, A.Y.; Evain, M. Synthesis of 5-aza-analogues of angucyclines: Manipulation of the 2-deoxy-C-glycoside subunit. Tetrahedron Lett. 2005, 46, 7669–7673. [Google Scholar] [CrossRef]
- Sissouma, D.; Dequirez, G.; Collet, S.; Guingant, A. Preparation of a chiral azadiene for the synthesis of 5-aza analogues of angucyclinones. Tetrahedron Lett. 2011, 52, 2336–2339. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Gonzalez, M.F.; Valderrama, C. Studies on quinones. Part 32. Regioselective synthesis of benz[b]phenantridines related to phenantroviridone. Tetrahedron 1999, 55, 6039–6050. [Google Scholar] [CrossRef]
- Cuellar, M.A.; Quinones, N.; Vera, V.; Salas, C.O.; Estevez, J.C.; Estevez, R.J. Preliminary studies on the synthesis of (−)-shikimic acid based 1,2,3,4-tetrahydrobenzo[b]phenanthridine-7,12-diones. Synlett 2015, 26, 552–556. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Vasquez, D. Design and synthesis of angucyclinone ab-pyrido[2,3-d] pyrimidine analogues. Tetrahedron Lett. 2008, 49, 703–706. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Colonelli, P.; Vasquez, D.; Gonzalez, M.F.; Rodriguez, J.A.; Theoduloz, C. Studies on quinones. Part 44: Novel angucyclinone N-heterocyclic analogues endowed with antitumoral activity. Bioorg. Med. Chem. 2008, 16, 10172–10181. [Google Scholar] [CrossRef] [PubMed]
- Jeso, V.; Iqbal, S.; Hernandez, P.; Cameron, M.D.; Park, H.; LoGrasso, P.V.; Micalizio, G.C. Synthesis of benzoquinone ansamycin-inspired macrocyclic lactams from shikimic acid. Angew. Chem. Int. Ed. Engl. 2013, 52, 4800–4804. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, K.; Espinosa-Bustos, C.; Soto-Delgado, J.; Tapia, R.A.; Varela, J.; Birriel, E.; Segura, R.; Pizarro, J.; Cerecetto, H.; Gonzalez, M.; et al. New aryloxy-quinone derivatives as potential antichagasic agents: Synthesis, trypanosomicidal activity, electrochemical properties, pharmacophore elucidation and 3D-QSAR analysis. RSC Adv. 2015, 5, 65153–65166. [Google Scholar] [CrossRef]
- Cuellar, M.A.; Alegria, L.K.; Prieto, Y.A.; Cortes, M.J.; Tapia, R.A.; Preite, M.D. Hetero-diels-alder reaction of halogenated quinones with a polygodial-derived azadiene. Tetrahedron Lett. 2002, 43, 2127–2131. [Google Scholar] [CrossRef]
- Nebois, P.; do Nascimento, S.C.; Boitard, M.; Bartoli, M.H.; Fillion, H. Synthesis and in vitro cytotoxic activity of aza- and diazaanthraquinone derivatives. Pharmazie 1994, 49, 819–821. [Google Scholar] [PubMed]
- Poumaroux, A.; Bouaziz, Z.; Fillion, H.; Domard, M.; Giraud, J.; Petavy, A.F. Regiospecific synthesis of pyrido[3,4-b]- and pyrido[4,3-b]carbazole-5,11-dione derivatives. Evaluation of their in vitro antifungal or antiprotozoological activities. Chem. Pharm. Bull. 1999, 47, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Salas, C.O.; Faundez, M.; Morello, A.; Maya, J.D.; Tapia, R.A. Natural and synthetic naphthoquinones active against Trypanosoma cruzi: An initial step towards new drugs for chagas disease. Curr. Med. Chem. 2011, 18, 144–161. [Google Scholar] [CrossRef] [PubMed]
- Sieveking, I.; Thomas, P.; Estevez, J.C.; Quinones, N.; Cuellar, M.A.; Villena, J.; Espinosa-Bustos, C.; Fierro, A.; Tapia, R.A.; Maya, J.D.; et al. 2-phenylaminonaphthoquinones and related compounds: Synthesis, trypanocidal and cytotoxic activities. Bioorg. Med. Chem. 2014, 22, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Values (μM) a | |||||
|---|---|---|---|---|---|
| Compound | Structure | PC-3 | HT-29 | MCF-7 | CCD841-CoN |
| 7a |  | 0.7 ± 0.1 b (3.3) | 0.2 ± 0.05 (11.5) | 16.0 ± 1.0 (0.1) | 2.3 ± 0.3 |
| 7b |  | 2.4 ± 0.9 (0.3) | 2.1 ± 1.2 (0.4) | 12.1 ± 4.0 (0.1) | 0.8 ± 0.1 |
| 8 | 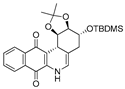 | 5.7 ± 1.3 (2.4) | 7.8 ± 0.9 (1.8) | 4.2 ± 4.2 (3.3) | 13.8 ± 1.0 |
| 9a | 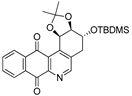 | 7.6 ± 1.2 (0.4) | 3.7 ± 0.8 (0.8) | 17.8 ± 3.0 (0.2) | 2.8 ± 0.4 |
| 9b | 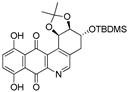 | 13.1 ± 2.0 (1.2) | 15.0 ± 4.2 (1.1) | 28.3 ± 4.0 (0.6) | 15.6 ± 2.1 |
| 15 | 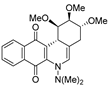 | 34.8 ± 0.3 (2.3) | 29.2 ± 6.8 (2.8) | 78.1 ± 6.9 (1.0) | 81.6 ± 15.8 |
| 16a |  | 23.6 ± 3.7 (4.9) | 42.9 ± 8.4 (2.7) | 118.1 ± 10.8 (1.0) | 115.7 ± 17.8 |
| 16b | 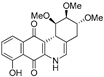 | 6.6 ± 1.1 (2.3) | 5.3 ± 0.8 (2.9) | 11.5 ± 3.1 (1.3) | 15.5 ± 3.1 |
| 16c | 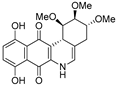 | 6.4 ± 0.8 (1.4) | 4.0 ± 0.3 (2.2) | 13.5 ± 0.9 (0.7) | 8.8 ± 1.3 |
| 17 | 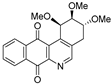 | 3.9 ± 0.6 (3.0) | 3.3 ± 0.9 (3.6) | 11.9 ± 1.5 (1.0) | 11.8 ± 1.3 |
| 19a | 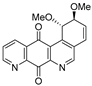 | 0.08 ± 0.01 (0.5) | 0.10 ± 0.01 (0.4) | 0.01± 0.005 (4.0) | 0.04 ± 0.01 |
| 19b | 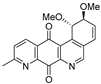 | 0.42 ± 0.04 (0.7) | 0.39 ± 0.04 (0.8) | 0.19 ± 0.04 (1.6) | 0.30 ± 0.02 |
| 19c |  | 4.0 ± 0.4 (4.4) | 21.0 ± 3.6 (0.8) | 12.8 ± 1.4 (1.4) | 17.4 ± 2.0 |
| 20 | 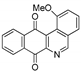 | 7.8 ± 1.0 (3.0) | 3.7 ± 1.2 (6.4) | 14.0 ± 1.7 (1.7) | 23.6 ± 3.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiñones, N.; Hernández, S.; Espinoza Catalán, L.; Villena, J.; Brito, I.; Cabrera, A.R.; Salas, C.O.; Cuellar, M.A. (-)-Shikimic Acid as a Chiral Building Block for the Synthesis of New Cytotoxic 6-Aza-Analogues of Angucyclinones. Molecules 2018, 23, 1422. https://doi.org/10.3390/molecules23061422
Quiñones N, Hernández S, Espinoza Catalán L, Villena J, Brito I, Cabrera AR, Salas CO, Cuellar MA. (-)-Shikimic Acid as a Chiral Building Block for the Synthesis of New Cytotoxic 6-Aza-Analogues of Angucyclinones. Molecules. 2018; 23(6):1422. https://doi.org/10.3390/molecules23061422
Chicago/Turabian StyleQuiñones, Natalia, Santiago Hernández, Luis Espinoza Catalán, Joan Villena, Ivan Brito, Alan R. Cabrera, Cristian O. Salas, and Mauricio A. Cuellar. 2018. "(-)-Shikimic Acid as a Chiral Building Block for the Synthesis of New Cytotoxic 6-Aza-Analogues of Angucyclinones" Molecules 23, no. 6: 1422. https://doi.org/10.3390/molecules23061422