Molecular Identification and Taxonomic Implication of Herbal Species in Genus Corydalis (Papaveraceae)

 and
and
Abstract
:1. Introduction
2. Results
2.1. Sequence Analyses of ITS Region
2.2. DNA Barcoding Analyses Using Plastid Regions
2.2.1. Plastid Region Test for Screening Efficient DNA Barcodes
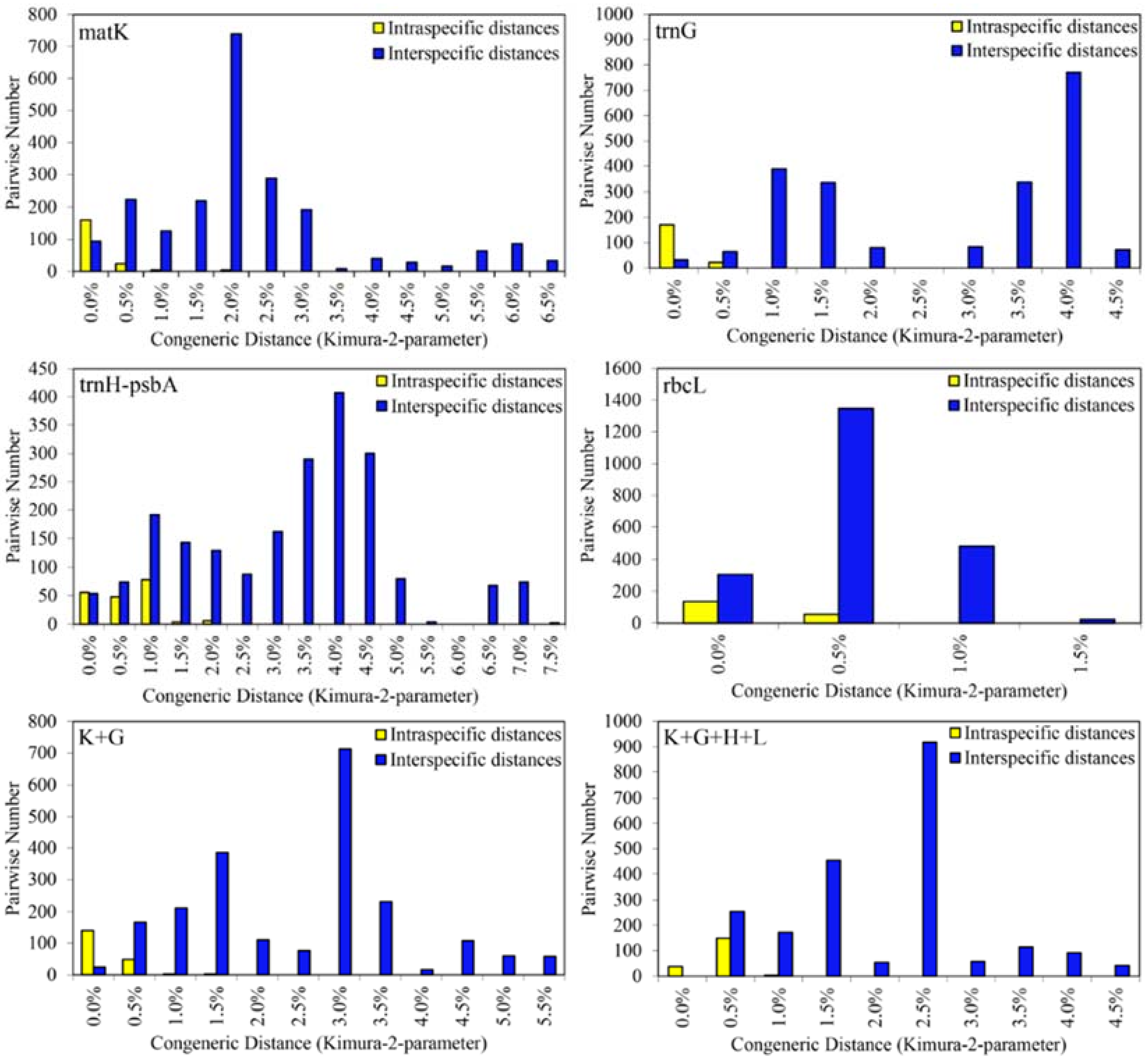
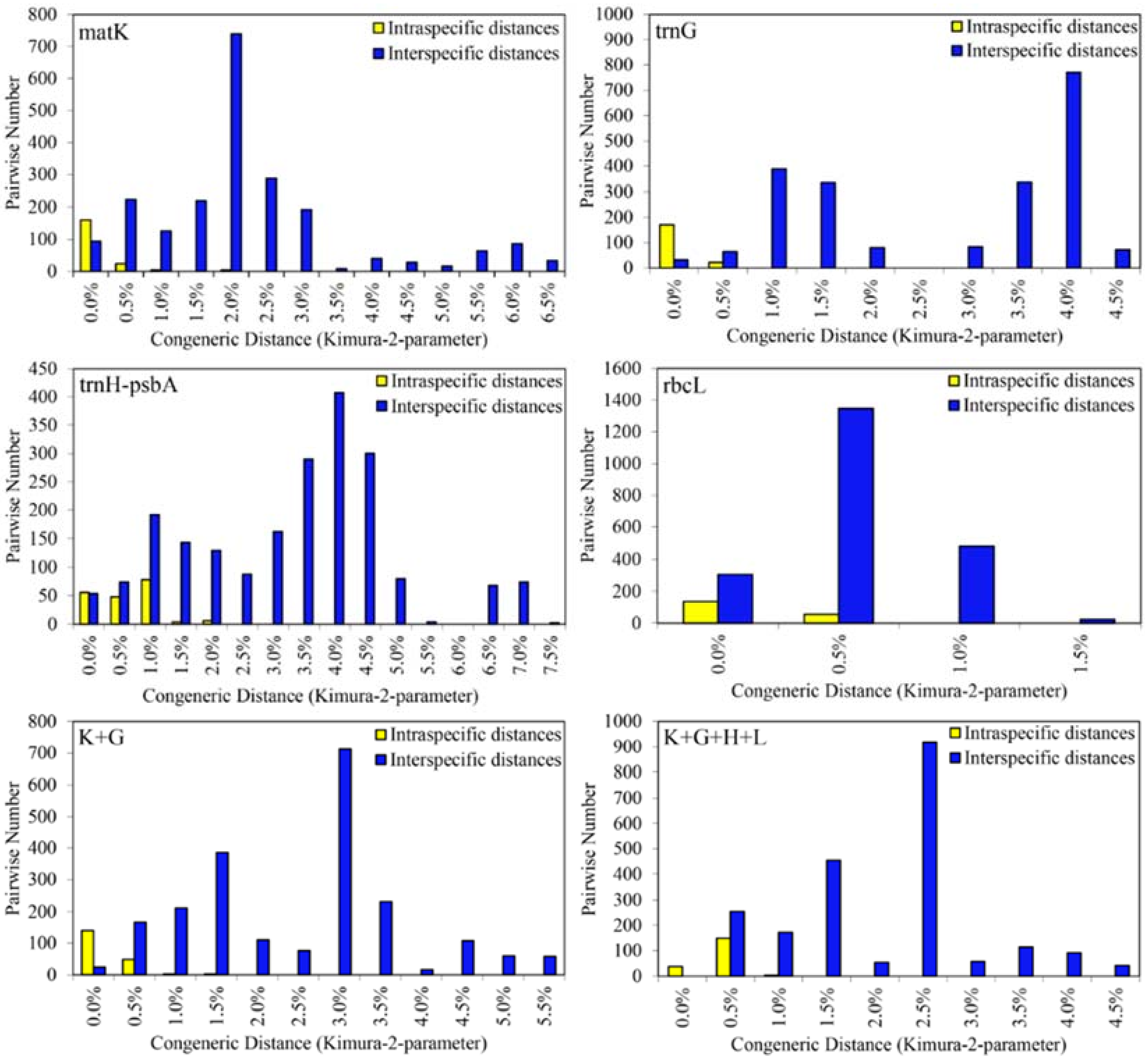
2.2.2. Barcoding Gap in Sect. Corydalis
2.2.3. Species Discrimination
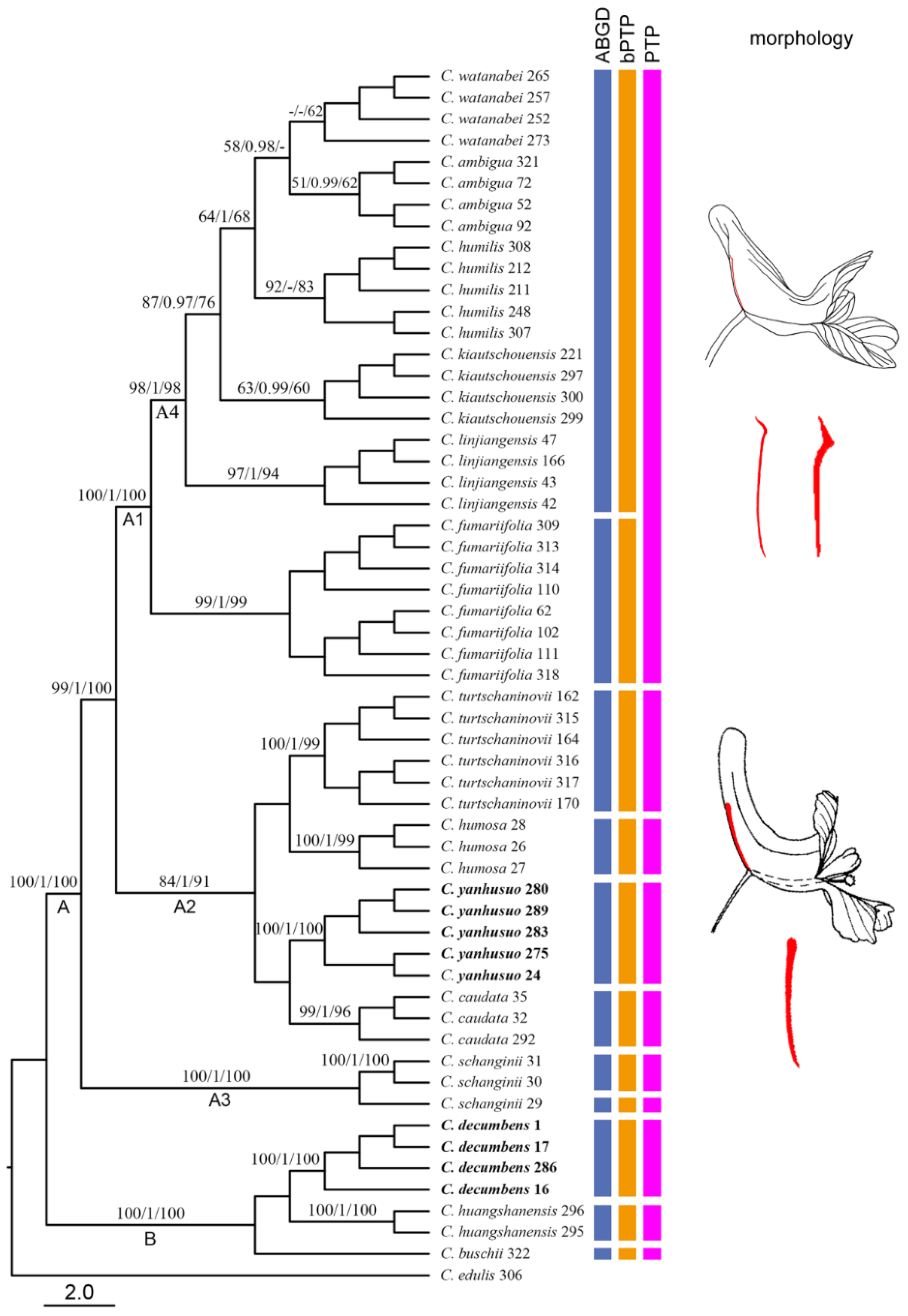
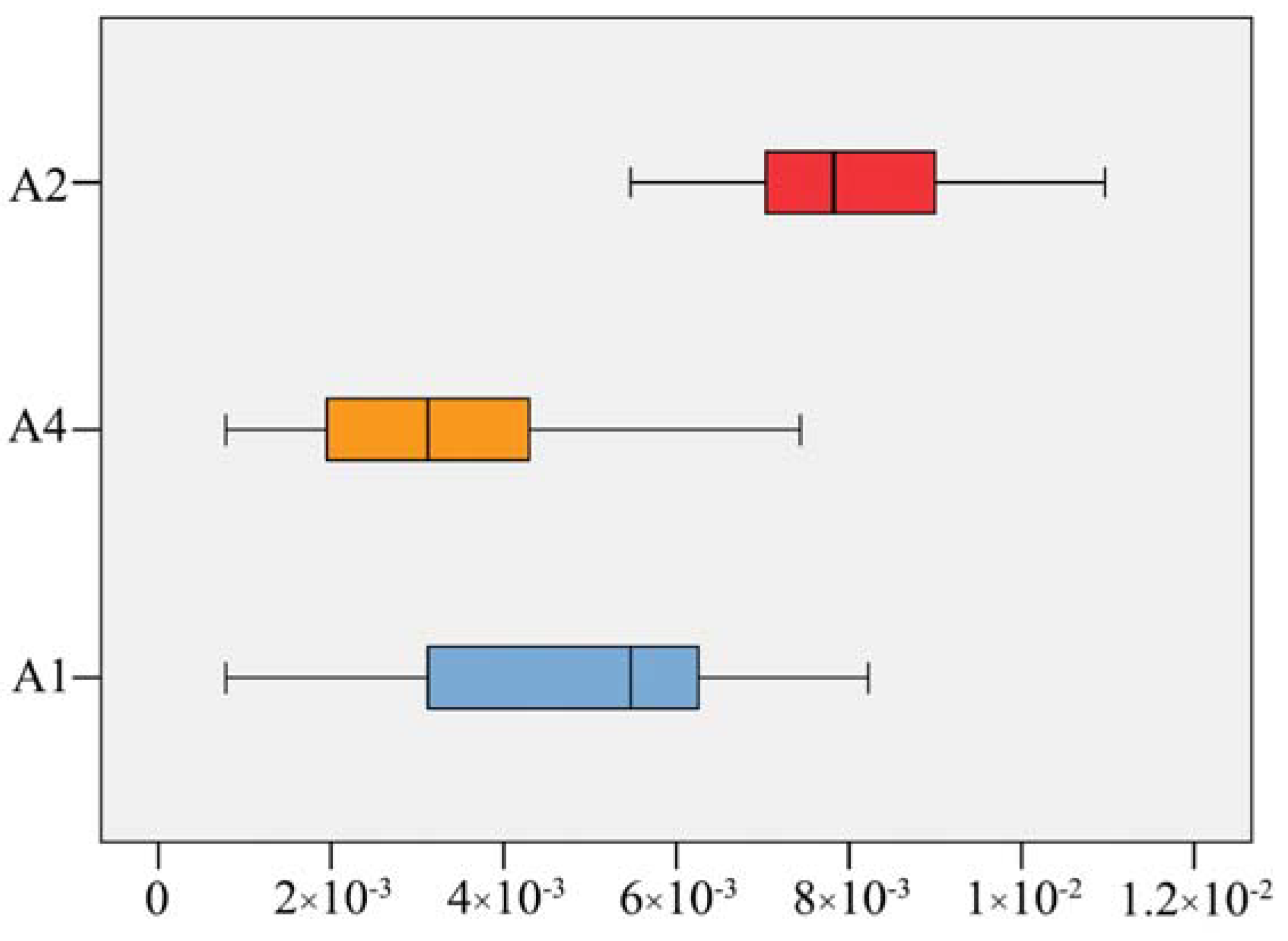
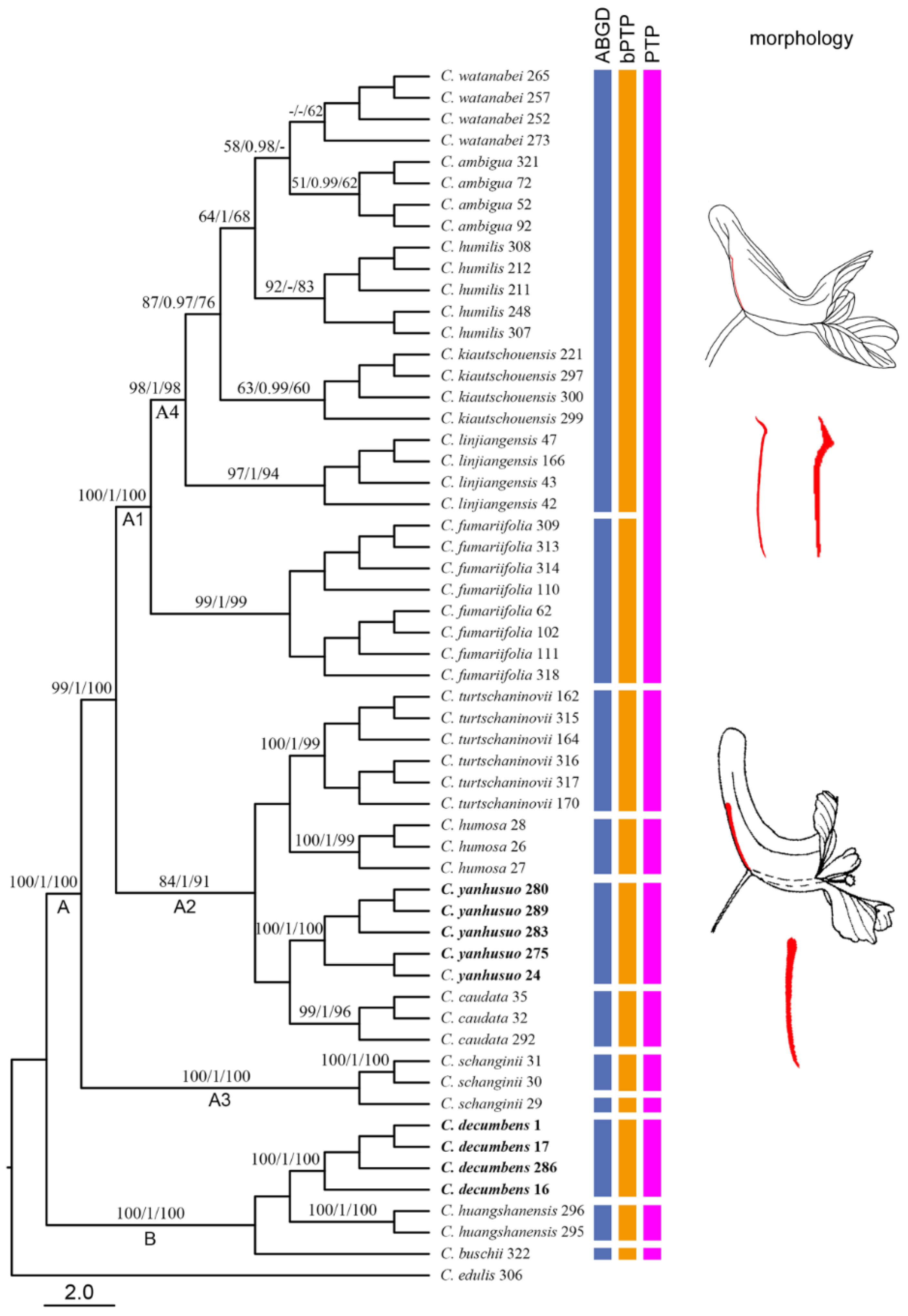
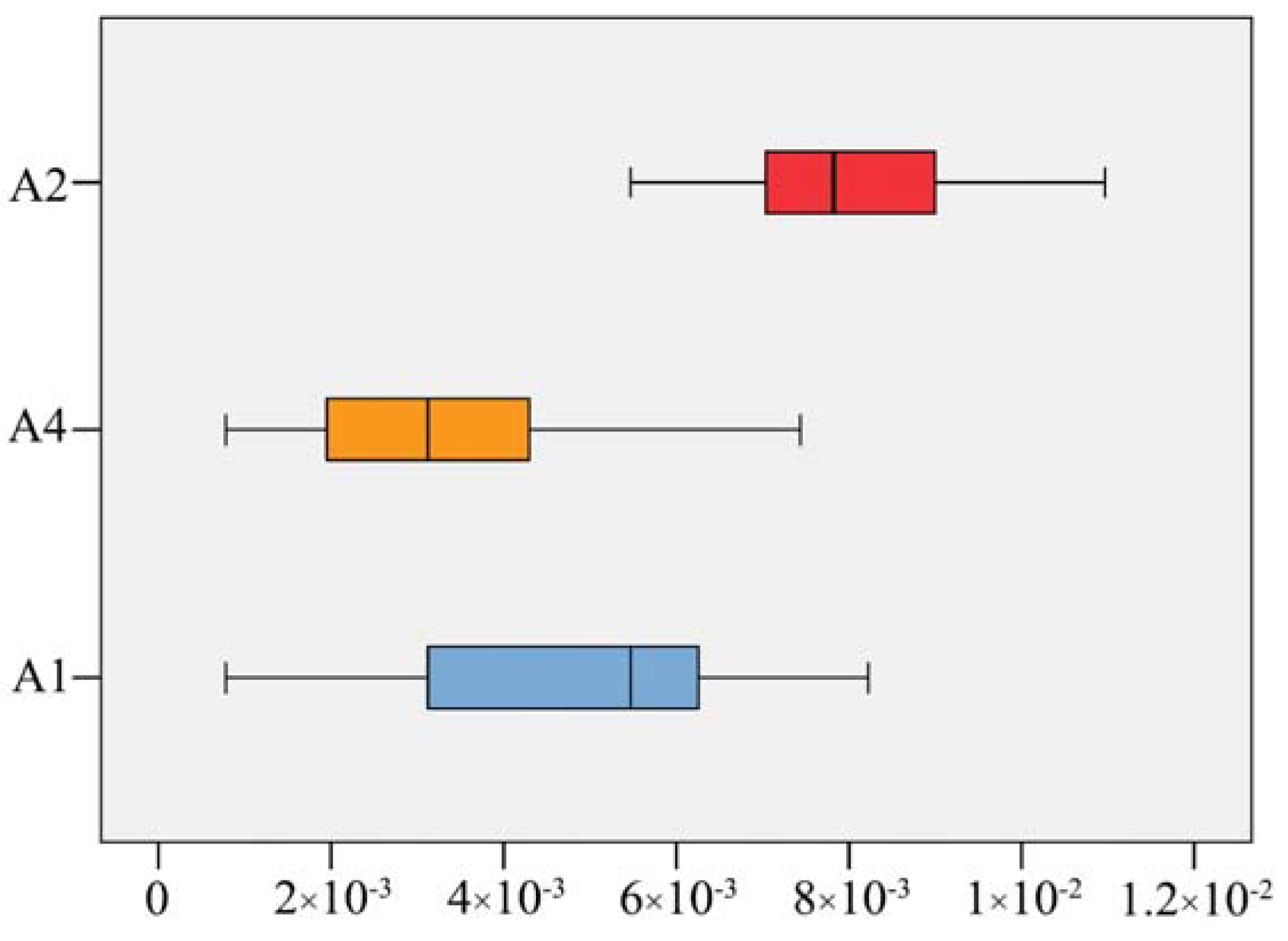
2.3. Molecular Delimitation and Classical Taxonomy
3. Discussion
3.1. Multi-Copy Nature of ITS in Corydalis
3.2. matK Is a Promising DNA Barcode for Corydalis
3.3. Taxonomic Implications of Molecular Identification in Corydalis
4. Materials and Methods
4.1. Taxonomic Sampling
4.2. DNA Extraction, Amplification, and Sequencing
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, M.L.; Su, Z.Y.; Magnus, L. Corydalis. In Flora of China; Wu, Z.Y., Peter, H.R., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2008; Volume 7, pp. 295–428. [Google Scholar]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2015; Volume 1, pp. 139–140. [Google Scholar]
- Committee on Japanese Pharmacopoeia. The Japanese Pharmacopoeia, 17th ed.; The Minister of Health, Labour and Welfare: Tokyo, Japan, 2016; pp. 1840–1841. [Google Scholar]
- Ministry of Food and Drug Safety. The Korean Pharmacopoeia, 10th ed.; Ministry of Food and Drug Safety: Osong, Korea, 2012; p. 1284. [Google Scholar]
- Su, Z.Y.; Wu, C.Y. A study on the classification, distribution, evolutionary trends and uses of Chinese Corydalis subgen, Capuites DC. Acta Bot. Yunnanica 1985, 7, 253–276. [Google Scholar]
- Wang, X.; Geng, Y.; Li, F.; Shi, X.; Liu, J. Large-scale separation of alkaloids from Corydalis decumbens by pH-zone-refining counter-current chromatography. J. Chromatogr. A 2012, 17, 14968–14974. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.D.; Zhou, P.; Bai, G.G.; Li, X.; Chen, J.W. Discussion of HPLC fingerprint of traditional Chinese medicine of Corydalis yanhusuo and its preparation. Chin. J. Chin. Mater. Med. 2015, 40, 2470–2473. [Google Scholar]
- Ding, B.; Zhou, T.; Fan, G.; Hong, Z.; Wu, Y. Qualitative and quantitative determination of ten alkaloids in traditional Chinese medicine Corydalis yanhusuo WT Wang by LC–MS/MS and LC–DAD. J. Pharm. Biomed. 2007, 45, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, W.; Zhou, J.; Gao, X.; Liu, Z.; Han, N.; Yin, J. Study on the discrimination between Corydalis Rhizoma and its adulterants based on HPLC-DAD-Q-TOF-MS associated with chemometric analysis. J. Chromatogr. B 2018, 1090, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Pérezgutiérrez, M.A.; Romerogarcía, A.T.; Salinas, M.J.; Blanca, G.; Fernández, M.C.; Suárez-Santiago, V.N. Phylogeny of the tribe Fumarieae (Papaveraceae s.l.) based on chloroplast and nuclear DNA sequences: Evolutionary and biogeographic implications. Am. J. Bot. 2012, 99, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Gutiérrez, M.A.; Romero-García, A.T.; Fernández, M.C.; Blanca, G.; Salinas-Bonillo, M.J.; Suárez-Santiago, V.N. Evolutionary history of fumitories (subfamily Fumarioideae, Papaveraceae): An old story shaped by the main geological and climatic events in the Northern Hemisphere. Mol. Phylogenet. Evol. 2015, 88, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Hoot, S.B.; Wefferling, K.M.; Wulff, J.A. Phylogeny and character evolution of Papaveraceae sl (Ranunculales). Syst. Bot. 2015, 40, 474–488. [Google Scholar] [CrossRef]
- Lidén, M.; Fukuhara, T.; Axberg, T. Phylogeny of Corydalis, ITS and morphology. In Systematics and Evolution of the Ranunculiflorae; Jensen, U., Kadereit, J.W., Eds.; Springer: Vienna, Austria, 1995; pp. 183–188. [Google Scholar]
- Lidén, M.; Fukuhara, T.; Rylander, J.; Oxelman, B. Phylogeny and classification of Fumariaceae, with emphasis on Dicentra sl, based on the plastid generps16 intron. Plant Syst. Evol. 1997, 206, 411–420. [Google Scholar] [CrossRef]
- Leliaert, F.; Verbruggen, H.; Vanormelingen, P.; Steen, F.; Lópezbautista, J.M.; Zuccarello, G.C.; Clerck, O.D. DNA-based species delimitation in algae. Eur. J. Phycol. 2014, 49, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.Q.; Humphreys, A.M.; Fontaneto, D.; Barraclough, T.G. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 2014, 5, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, C.P.B.; Li, D.Z.; Gao, L.M.; Kong, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L.; Yang, J.B.; et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Li, M.; Cao, Y.; Gao, Y.; Zhang, W. Molecular thresholds of ITS2 and their implications for molecular evolution and species identification in seed plants. Sci. Rep. 2017, 7, 17316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.W. Systematics of Corydalis DC. (Fumariaceae). Ph.D. Thesis, Institute of Botany, the Chinese Academy of Sciences, Beijing, China, 2006. [Google Scholar]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Sang, T.; Crawford, D.; Stuessy, T. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 1997, 84, 1120. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.A.; Simpson, B.B. Paraphyly of Tarasa (Malvaceae) and Diverse Origins of the Polyploid Species. Syst. Bot. 2003, 28, 723–737. [Google Scholar]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.S.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, B.G. Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: An example from the compositae. Mol. Phylogenet. Evol. 1992, 1, 3. [Google Scholar] [CrossRef]
- Hou, D.; Song, J.; Shi, L.; Ma, X.; Xin, T.; Han, J.; Xiao, W.; Sun, Z.; Cheng, R.; Yao, H. Stability and Accuracy Assessment of Identification of Traditional Chinese Materia Medica Using DNA Barcoding: A Case Study on Flos Lonicerae Japonicae. Biomed. Res. Int. 2013, 2013, 549037. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual, 3nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403. [Google Scholar] [CrossRef]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K. DNA Barcoding and Taxonomy in Diptera: A Tale of High Intraspecific Variability and Low Identification Success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swofford, D. PAUP. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688. [Google Scholar] [CrossRef] [PubMed]
- Fredrik, R.; Maxim, T.; Paul, V.D.M.; Daniel, L.A.; Aaron, D.; Sebastian, H.; Bret, L.; Liang, L.; Marc, A.S.; John, P.H. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Statistic | K | G | L | H | K + G | K + G + H | K + G + H + L |
|---|---|---|---|---|---|---|---|
| Amplification success rate (%) | 100 | 100 | 100 | 100 | |||
| Sequencing success rate (%) | 100 | 100 | 100 | 100 | |||
| Length range (bp) | 834–849 | 692–681 | 688 | 333–456 | 1526–1552 | 1872–1888 | 2562–2676 |
| Aligned length (bp) | 859 | 730 | 688 | 468 | 1589 | 2057 | 2745 |
| No. of variable characters (%) | 151 (17.58) | 75 (10.27) | 37 (5.38) | 91 (19.44) | 226 (14.22) | 317 (15.41) | 354 (12.90) |
| No. of parsimony information characters (%) | 117 (13.62) | 55 (7.53) | 28 (4.07) | 67 (14.32) | 172 (10.82) | 239 (11.62) | 267 (9.73) |
| CI | 0.9257 | 0.9011 | 0.7872 | 0.8000 | 0.9071 | 0.8544 | 0.8330 |
| RI | 0.9812 | 0.9803 | 0.8834 | 0.9386 | 0.9783 | 0.9623 | 0.9547 |
| Species identification rate (%) | 78.57 | 64.29 | 50.00 | 42.86 | 85.71 | 92.86 | 100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Li, M.; Zhao, F.; Chu, S.; Zha, L.; Xu, T.; Peng, H.; Zhang, W. Molecular Identification and Taxonomic Implication of Herbal Species in Genus Corydalis (Papaveraceae). Molecules 2018, 23, 1393. https://doi.org/10.3390/molecules23061393
Jiang L, Li M, Zhao F, Chu S, Zha L, Xu T, Peng H, Zhang W. Molecular Identification and Taxonomic Implication of Herbal Species in Genus Corydalis (Papaveraceae). Molecules. 2018; 23(6):1393. https://doi.org/10.3390/molecules23061393
Chicago/Turabian StyleJiang, Lu, Meihui Li, Fengxi Zhao, Shanshan Chu, Liangping Zha, Tao Xu, Huasheng Peng, and Wei Zhang. 2018. "Molecular Identification and Taxonomic Implication of Herbal Species in Genus Corydalis (Papaveraceae)" Molecules 23, no. 6: 1393. https://doi.org/10.3390/molecules23061393