Knowledge-Based Neuroendocrine Immunomodulation (NIM) Molecular Network Construction and Its Application

 ,
,
Abstract
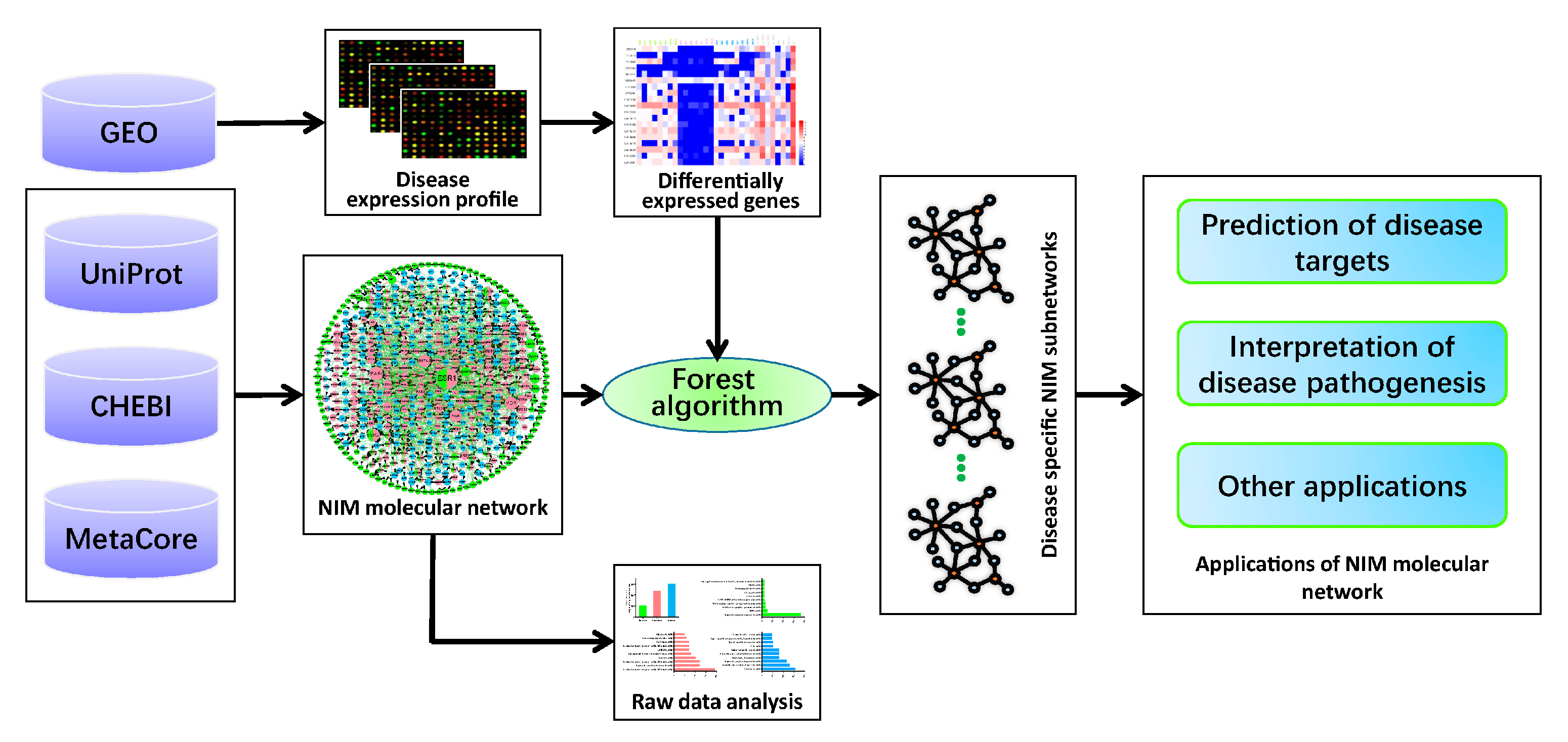
:1. Introduction
2. Results
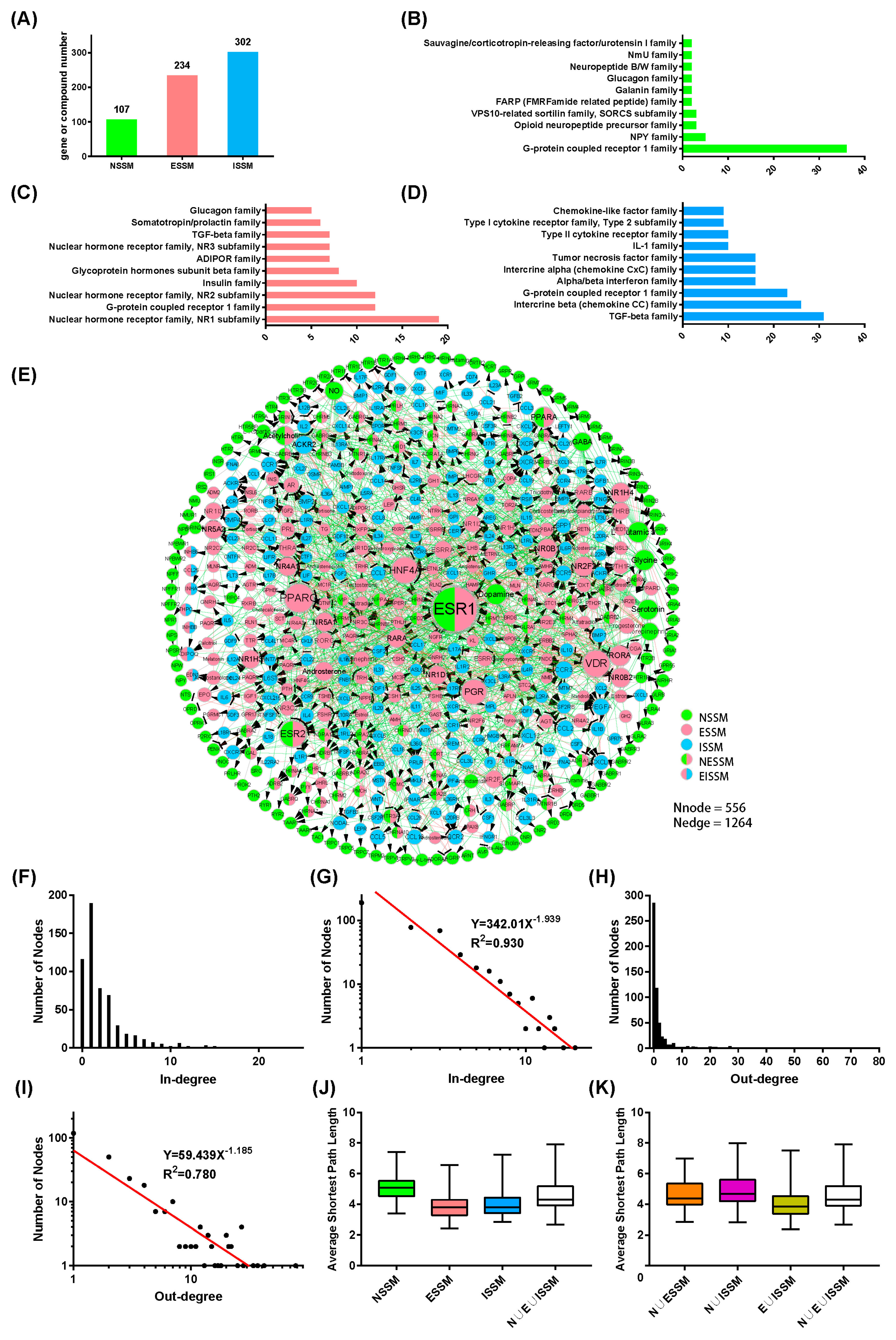
2.1. The Construction of the NIM Molecular Network
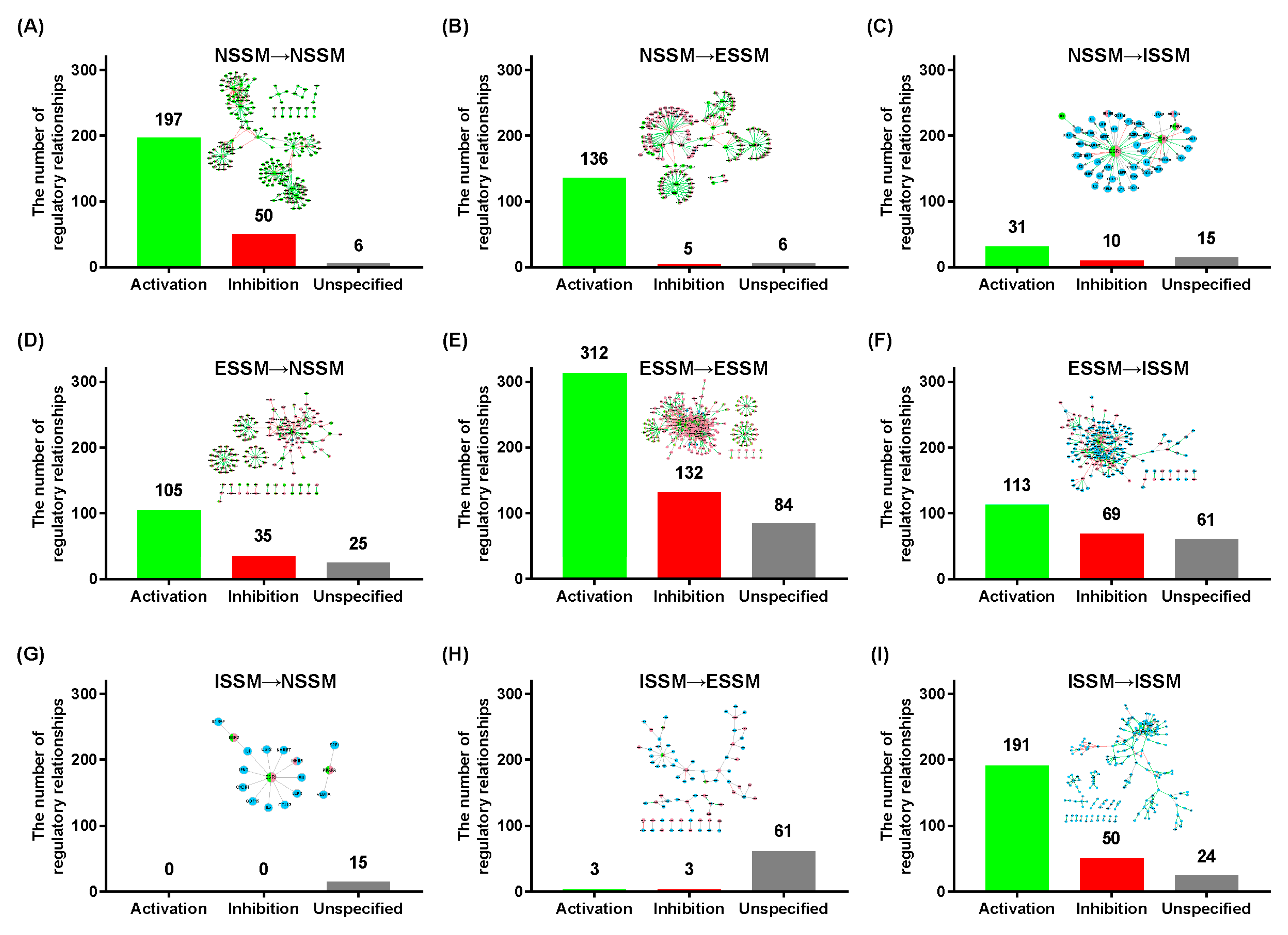
2.2. Analysis of the NIM Molecular Network Characteristics
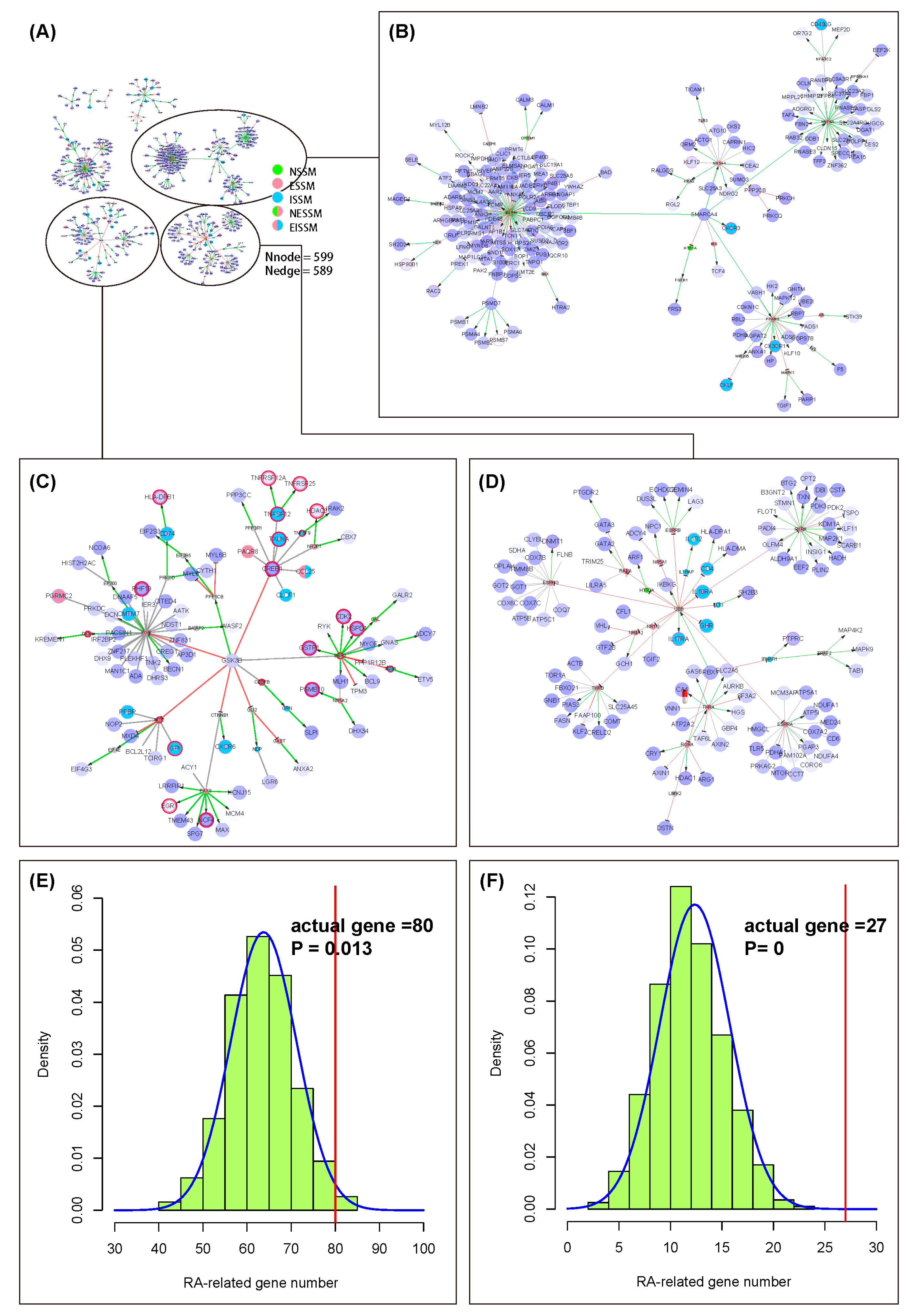
2.3. Case One of Applying the NIM Molecular Network: Rheumatoid Arthritis
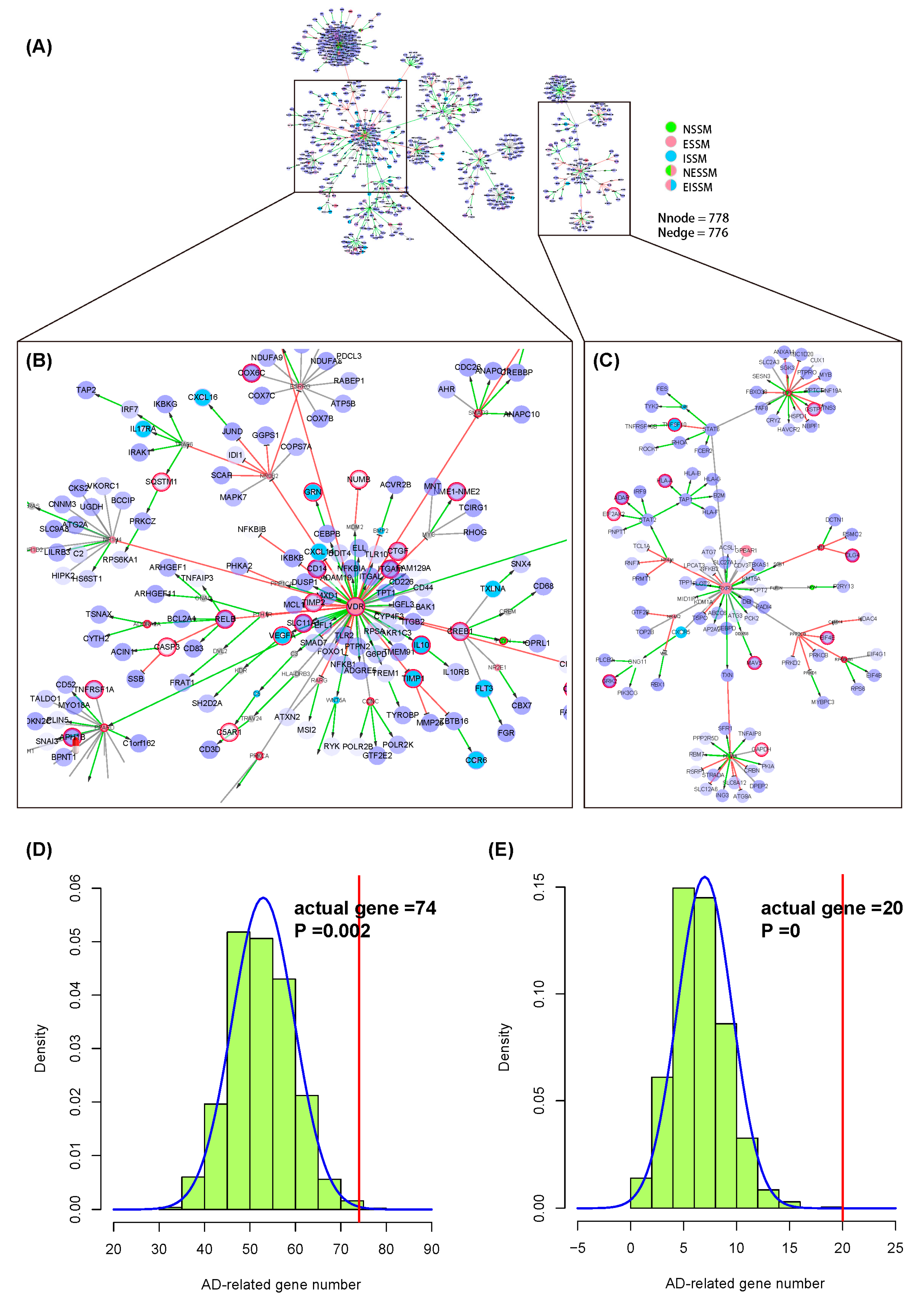
2.4. Case Two of Applying the NIM Molecular Network: Alzheimer’s Disease
3. Discussion
4. Materials and Methods
4.1. Construction of the NIM Signaling Molecule Dataset
4.2. Protein Family Analysis of NIM Signaling Molecules
4.3. Tissue Specificity Analysis of NIM Signaling Molecules
4.4. Construction and Analysis of the Network of NIM Signaling Molecules
4.5. Construction of the Disease-Specific NIM Subnetwork
4.6. Permutation Test
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabasi, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed]
- Lage, K.; Karlberg, E.O.; Storling, Z.M.; Olason, P.I.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tumer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Moctezuma, J.; Dominguez-Salazar, E.; Gomez-Gonzalez, B. Beyond the borders: The gates and fences of neuroimmune interaction. Front. Integr. Neurosci. 2014, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Sturmberg, J.P.; Bennett, J.M.; Martin, C.M.; Picard, M. ‘Multimorbidity’ as the manifestation of network disturbances. J. Eval. Clin. Pract. 2017, 23, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; He, Y.; Park, J.S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; Plambeck, K.E.; Udeochu, J.; et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology. Nat. Biotechnol. 2007, 25, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Bailly-Bechet, M.; Braunstein, A.; Pagnani, A.; Weigt, M.; Zecchina, R. Inference of sparse combinatorial-control networks from gene-expression data: A message passing approach. BMC Bioinform. 2010, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Akavia, U.D.; Litvin, O.; Kim, J.; Sanchez-Garcia, F.; Kotliar, D.; Causton, H.C.; Pochanard, P.; Mozes, E.; Garraway, L.A.; Pe’er, D. An integrated approach to uncover drivers of cancer. Cell 2010, 143, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhang, Y.; Wang, Y.; Wang, B.; Jin, J.; Wang, X. A novel multilayer correlation maximization model for improving cca-based frequency recognition in ssvep brain-computer interface. Int. J. Neural. Syst. 2018, 28, 1750039. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Waytowich, N.R.; Krusienski, D.J.; Zhou, G.; Jin, J.; Wang, X.; Cichocki, A. Discriminative feature extraction via multivariate linear regression for ssvep-based bci. IEEE Trans. Neural. Syst. Rehabil. Eng. 2016, 24, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Zhou, G.X.; Jin, J.; Wang, B.; Wang, X.Y.; Cichocki, A. Multi-kernel extreme learning machine for eeg classification in brain-computer interfaces. Exp. Syst. Appl. 2018, 96, 302–310. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, G.; Jin, J.; Zhao, Q.; Wang, X.; Cichocki, A. Sparse bayesian classification of eeg for brain-computer interface. IEEE Trans. Neural. Netw. Learn. Syst. 2016, 27, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.; Zhang, L. An adaptive neural network approach for operator functional state prediction using psychophysiological data. Integr. Comput. Aided Eng. 2015, 23, 81–97. [Google Scholar] [CrossRef]
- Zhou, G.X.; Zhao, Q.B.; Zhang, Y.; Adali, T.; Xie, S.L.; Cichocki, A. Linked component analysis from matrices to high-order tensors: Applications to biomedical data. Proc. IEEE 2016, 104, 310–331. [Google Scholar] [CrossRef]
- Joehanes, R. Network analysis of gene expression. Methods Mol. Biol. 2018, 1783, 325–341. [Google Scholar] [PubMed]
- Averesch, N.; Martínez, V.; Nielsen, L.; Krömer, J. Toward synthetic biology strategies for adipic acid production: An in silico tool for combined thermodynamics and stoichiometric analysis of metabolic networks. ACS Synth. Biol. 2018, 7, 490–509. [Google Scholar] [CrossRef] [PubMed]
- Goehler, H.; Lalowski, M.; Stelzl, U.; Waelter, S.; Stroedicke, M.; Worm, U.; Droege, A.; Lindenberg, K.; Knoblich, M.; Haenig, C.; et al. A protein interaction network links git1, an enhancer of huntingtin aggregation, to huntington’s disease. Mol. Cell 2004, 15, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.; Segal, E.; Koller, D.; Kim, S. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Besedovsky, H.; Sorkin, E. Network of immune-neuroendocrine interactions. Clin. Exp. Immunol. 1977, 27, 1–12. [Google Scholar] [PubMed]
- Li, Y.; Ma, T.; Lu, L.; Li, S. Networking Drugs and Diseases in the Context of Neuro-Endocrine-Immune System. In Proceedings of the IEEE International Biomedical Engineering and Informatics (BMEI), Yantai, China, 16–18 October 2010. [Google Scholar]
- Trombetta, A.C.; Meroni, M.; Cutolo, M. Steroids and autoimmunity. Front. Horm. Res. 2017, 48, 121–132. [Google Scholar] [PubMed]
- Syzon, O.; Voznyak, I.; Dashko, M. Features of some clinical examination parameters in patients with psoriatic arthritis. Wiad. Lek. 2017, 70, 205–207. [Google Scholar]
- Malagoli, D.; Ottaviani, E. Cross-talk among immune and neuroendocrine systems in molluscs and other invertebrate models. Horm. Behav. 2017, 88, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Malagoli, D.; Mandrioli, M.; Tascedda, F.; Ottaviani, E. Circulating phagocytes: The ancient and conserved interface between immune and neuroendocrine function. Biol. Rev. Camb. Philos. Soc. 2017, 92, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Jara, L.J.; Medina, G.; Saavedra, M.A.; Vera-Lastra, O.; Torres-Aguilar, H.; Navarro, C.; Vazquez Del Mercado, M.; Espinoza, L.R. Prolactin has a pathogenic role in systemic lupus erythematosus. Immunol. Res. 2017, 65, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.L.; Li, S.; Li, Y.D. Dbnei: A specific database for neuro-endocrine-immune interactions. Neuroendocrinol. Lett. 2006, 27, 53–59. [Google Scholar] [PubMed]
- Zhang, J.; Ma, T.; Li, Y.D.; Li, S. Dbnei2.0: Building multilayer network for drug-nei-disease. Bioinformatics 2008, 24, 2409–2411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, S. Modeling of neuro-endocrine-immune network via subject oriented literature mining. In Proceedings of the Fourth International Conference on Bioinformatics of Genome Regulation and Structure, Novosibirsk, Russia, 25–30 July 2004; Volume 2, pp. 167–170. [Google Scholar]
- Wu, L.J.; Li, S. Combined literature mining and gene expression analysis for modeling neuro-endocrine-immune interactions. In Advances in Intelligent Computing; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3645, pp. 31–40. [Google Scholar]
- Zhai, X.; Feng, X.C.; Liu, J.W.; Gao, K.; Jia, Z.H.; Zhao, H.H.; Wang, J.; Wang, W.; Chen, J.X. Neuro-endocrine-immune biological network construction of qi deficiency pattern and qi stagnation pattern in traditional chinese medicine. J. Biol. Syst. 2015, 23, 305–321. [Google Scholar] [CrossRef]
- Teixeira, V.H.; Olaso, R.; Martin-Magniette, M.L.; Lasbleiz, S.; Jacq, L.; Oliveira, C.R.; Hilliquin, P.; Gut, I.; Cornelis, F.; Petit-Teixeira, E. Transcriptome analysis describing new immunity and defense genes in peripheral blood mononuclear cells of rheumatoid arthritis patients. PLoS ONE 2009, 4, e6803. [Google Scholar] [CrossRef] [PubMed]
- Sood, S.; Gallagher, I.J.; Lunnon, K.; Rullman, E.; Keohane, A.; Crossland, H.; Phillips, B.E.; Cederholm, T.; Jensen, T.; van Loon, L.J.; et al. A novel multi-tissue rna diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 2015, 16, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeang, C.H.; Ideker, T.; Jaakkola, T. Physical network models. J. Comput. Biol. 2004, 11, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Sharan, R.; Ideker, T. Modeling cellular machinery through biological network comparison. Nat. Biotechnol. 2006, 24, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Ourfali, O.; Shlomi, T.; Ideker, T.; Ruppin, E.; Sharan, R. Spine: A framework for signaling-regulatory pathway inference from cause-effect experiments. Bioinformatics 2007, 23, i359–i366. [Google Scholar] [CrossRef] [PubMed]
- Bailly-Bechet, M.; Borgs, C.; Braunstein, A.; Chayes, J.; Dagkessamanskaia, A.; Francois, J.M.; Zecchina, R. Finding undetected protein associations in cell signaling by belief propagation. Proc. Natl. Acad. Sci. USA 2011, 108, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Wuchty, S.; Przytycka, T.M. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput. Biol. 2011, 7, e1001095. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Smoly, I.Y.; Rapaport, G.; Lindquist, S.; Fraenkel, E.; Yeger-Lotem, E. Responsenet: Revealing signaling and regulatory networks linking genetic and transcriptomic screening data. Nucleic Acids Res. 2011, 39, W424–W429. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Braunstein, A.; Pagnani, A.; Huang, S.S.; Chayes, J.; Borgs, C.; Zecchina, R.; Fraenkel, E. Simultaneous reconstruction of multiple signaling pathways via the prize-collecting steiner forest problem. J. Comput. Biol. 2013, 20, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Barer, F.; Patoka, R.; Amital, H.; Reitblat, T.; Reitblat, A.; Ophir, J.; Konfino, I.; et al. The anti-inflammatory target a(3) adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and crohn’s disease. Cell. Immunol. 2009, 258, 115–122. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, A.N.; Bresnihan, B.; FitzGerald, O.; Murphy, E.P. Cyclooxygenase 2-derived prostaglandin e2 production by corticotropin-releasing hormone contributes to the activated camp response element binding protein content in rheumatoid arthritis synovial tissue. Arthr. Rheum. 2004, 50, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Meusch, U.; Klingner, M.; Mathar, C.; Malysheva, O.; Baerwald, C.; Rossol, M.; Wagner, U. Brief report: Autocrine cytokine-mediated deficiency of trail-induced monocyte apoptosis in rheumatoid arthritis. Arthr. Rheumatol. 2015, 67, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- De Hair, M.J.; van de Sande, M.G.; Ramwadhdoebe, T.H.; Hansson, M.; Landewe, R.; van der Leij, C.; Maas, M.; Serre, G.; van Schaardenburg, D.; Klareskog, L.; et al. Features of the synovium of individuals at risk of developing rheumatoid arthritis: Implications for understanding preclinical rheumatoid arthritis. Arthr. Rheumatol. 2014, 66, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Dharmapatni, A.A.; Smith, M.D.; Crotti, T.N.; Holding, C.A.; Vincent, C.; Weedon, H.M.; Zannettino, A.C.; Zheng, T.S.; Findlay, D.M.; Atkins, G.J.; et al. Tweak and Fn14 expression in the pathogenesis of joint inflammation and bone erosion in rheumatoid arthritis. Arthr. Res. Ther. 2011, 13, R51. [Google Scholar] [CrossRef] [PubMed]
- Dziedziejko, V.; Kurzawski, M.; Safranow, K.; Drozdzik, M.; Chlubek, D.; Pawlik, A. Oestrogen receptor polymorphisms in female patients with rheumatoid arthritis. Scand. J. Rheumatol. 2011, 40, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.C.; Choi, S.J.; Ji, J.D.; Song, G.G. Associations between vitamin d receptor polymorphisms and susceptibility to rheumatoid arthritis and systemic lupus erythematosus: A meta-analysis. Mol. Biol. Rep. 2011, 38, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.C.; Song, G.G. Meta-analysis of associations between the peroxisome proliferator-activated receptor-gamma Pro12Ala polymorphism and susceptibility to nonalcoholic fatty liver disease, rheumatoid arthritis, and psoriatic arthritis. Genet. Test. Mol. Biomark. 2014, 18, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, M.; Hatori, M.; Suzuki, T.; Miki, Y.; Darnel, A.D.; Tazawa, C.; Sawai, T.; Uzuki, M.; Tanaka, Y.; Kokubun, S.; et al. Sex steroid receptors in rheumatoid arthritis. Clin. Sci. 2004, 106, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ince-Askan, H.; Dolhain, R.J. Pregnancy and rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2015, 29, 580–596. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Infante, J.; Llorca, J.; Rodriguez, E.; Berciano, J.; Combarros, O. Association between glycogen synthase kinase-3β genetic polymorphism and late-onset alzheimer’s disease. Dement. Geriatr. Cognit. Disord. 2006, 21, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Ohana, G.; Barer, F.; Ochaion, A.; Erlanger, A.; Madi, L. An agonist to the a3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of gsk-3β and nf-kappa b. Oncogene 2004, 23, 2465–2471. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3β (gsk3β) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Mazzon, E.; Di Paola, R.; Muia, C.; Crisafulli, C.; Dugo, L.; Collin, M.; Britti, D.; Caputi, A.P.; Thiemermann, C. Glycogen synthase kinase-3β inhibition attenuates the degree of arthritis caused by type ii collagen in the mouse. Clin. Immunol. 2006, 120, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived asc specks cross-seed amyloid-β in alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Akiyama, H.; Itagaki, S.; McGeer, E.G. Immune system response in alzheimer’s disease. Can. J. Neurol. Sci. 1989, 16, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Kroll, T.G.; Sarraf, P.; Pecciarini, L.; Chen, C.J.; Mueller, E.; Spiegelman, B.M.; Fletcher, J.A. Pax8-ppargamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 2000, 289, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Laczmanski, L.; Jakubik, M.; Bednarek-Tupikowska, G.; Rymaszewska, J.; Sloka, N.; Lwow, F. Vitamin d receptor gene polymorphisms in alzheimer’s disease patients. Exp. Gerontol. 2015, 69, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Santpere, G.; Nieto, M.; Puig, B.; Ferrer, I. Abnormal sp1 transcription factor expression in alzheimer disease and tauopathies. Neurosci. Lett. 2006, 397, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Bartolotti, N.; Bennett, D.A.; Lazarov, O. Reduced pcreb in alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 2016, 21, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at ms4a4/ms4a6e, cd2ap, cd33 and epha1 are associated with late-onset alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liang, B.; Hao, Y.; Zhou, W. Estrogen receptor alpha gene polymorphisms and risk of alzheimer’s disease: Evidence from a meta-analysis. Clin. Interv. Aging 2014, 9, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Song, G.G. PPARγ pro12Ala and His447His polymorphisms and susceptibility to Alzheimer’s disease: A meta-analysis. Genet. Mol. Res. 2015, 14, 7248–7257. [Google Scholar] [CrossRef] [PubMed]
- Pirskanen, M.; Hiltunen, M.; Mannermaa, A.; Helisalmi, S.; Lehtovirta, M.; Hanninen, T.; Soininen, H. Estrogen receptor β gene variants are associated with increased risk of alzheimer’s disease in women. Eur. J. Hum. Genet. 2005, 13, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Helisalmi, S.; Vepsalainen, S.; Hiltunen, M.; Koivisto, A.M.; Salminen, A.; Laakso, M.; Soininen, H. Genetic study between sirt1, ppard, pgc-1alpha genes and alzheimer’s disease. J. Neurol. 2008, 255, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Feart, C.; Helmer, C.; Merle, B.; Herrmann, F.; Annweiler, C.; Dartigues, J.; Delcourt, C.; Samieri, C. Associations of lower vitamin d concentrations with cognitive decline and long-term risk of dementia and alzheimer’s disease in older adults. Alzheimers Dement 2017, 13, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.; Dumas, K.; Bhaumik, D.; Schilling, B.; Davis, S.; Oron, T.; Sorensen, D.; Lucanic, M.; Brem, R.; Melov, S.; et al. Vitamin D promotes protein homeostasis and longevity via the stress response pathway genes skn-1, ire-1, and xbp-1. Cell. Rep. 2016, 17, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Mokry, L.; Ross, S.; Morris, J.; Manousaki, D.; Forgetta, V.; Richards, J. Genetically decreased vitamin d and risk of alzheimer disease. Neurology 2016, 87, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Byberg, L.; Karlström, B.; Cederholm, T.; Melhus, H.; Sjögren, P.; Kilander, L. Vitamin D is not associated with incident dementia or cognitive impairment: An 18-y follow-up study in community-living old men. Am. J. Clin. Nutr. 2017, 105, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Luedecking-Zimmer, E.; DeKosky, S.T.; Nebes, R.; Kamboh, M.I. Association of the 3′ UTR transcription factor lbp-1c/cp2/lsf polymorphism with late-onset alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 117, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Tabunoki, H.; Arima, K. Molecular network analysis suggests aberrant creb-mediated gene regulation in the alzheimer disease hippocampus. Dis. Markers 2009, 27, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Dresner-Pollak, R.; Kinnar, T.; Friedlander, Y.; Sharon, N.; Rosenmann, H.; Pollak, A. Estrogen receptor β gene variant is associated with vascular dementia in elderly women. Genet. Test. Mol. Biomarkers 2009, 13, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Ishunina, T.A.; van Heerikhuize, J.J.; Ravid, R.; Swaab, D.F. Estrogen receptors and metabolic activity in the human tuberomamillary nucleus: Changes in relation to sex, aging and alzheimer’s disease. Brain Res. 2003, 988, 84–96. [Google Scholar] [CrossRef]
- Savaskan, E.; Olivieri, G.; Meier, F.; Ravid, R.; Muller-Spahn, F. Hippocampal estrogen β-receptor immunoreactivity is increased in alzheimer’s disease. Brain Res. 2001, 908, 113–119. [Google Scholar] [CrossRef]
- Forsell, C.; Enmark, E.; Axelman, K.; Blomberg, M.; Wahlund, L.O.; Gustafsson, J.A.; Lannfelt, L. Investigations of a CA repeat in the oestrogen receptor β gene in patients with alzheimer’s disease. Eur. J. Hum. Genet. 2001, 9, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, J.; Heun, R.; Lutjohann, D.; Jessen, F.; Maier, W.; Kolsch, H. Ppard haplotype influences cholesterol metabolism but is no risk factor of alzheimer’s disease. Neurosci. Lett. 2006, 408, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Ridolfi, E.; Fenoglio, C.; Ghezzi, L.; Vimercati, R.; Clerici, F.; Marcone, A.; Gallone, S.; Serpente, M.; Cantoni, C.; et al. Expression of the transcription factor Sp1 and its regulatory hsa-miR-29b in peripheral blood mononuclear cells from patients with Alzheimer’s disease. J. Alzheimer Dis. 2013, 35, 487–494. [Google Scholar]
- Wang, X.; Tan, L.; Lu, Y.; Peng, J.; Zhu, Y.; Zhang, Y.; Sun, Z. Microrna-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 2015, 589, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Kolsch, H.; Lutjohann, D.; Jessen, F.; Popp, J.; Hentschel, F.; Kelemen, P.; Friedrichs, S.; Maier, T.A.; Heun, R. Rxra gene variations influence alzheimer’s disease risk and cholesterol metabolism. J. Cell. Mol. Med. 2009, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cheng, X.; Zhang, Y. Effect of liuwei dihuang decoction, a traditional chinese medicinal prescription, on the neuroendocrine immunomodulation network. Pharmacol. Ther. 2016, 162, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- De Matos, P.; Adams, N.; Hastings, J.; Moreno, P.; Steinbeck, C. A database for chemical proteomics: Chebi. Methods Mol. Biol. 2012, 803, 273–296. [Google Scholar] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J. Systems biology and personalized medicine: A network approach to human disease. Proc. Am. Thorac. Soc. 2011, 8, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Gosline, S.J.; Kedaigle, A.; Soltis, A.R.; Gitter, A.; Fraenkel, E. Network-based interpretation of diverse high-throughput datasets through the omics integrator software package. PLoS Comput. Biol. 2016, 12, e1004879. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Entrez Gene IDs | Degree | System Category 1 |
|---|---|---|---|---|
| ESR1 | Estrogen receptor 1 | 2099 | 98 | nervous; endocrine |
| PPARG | Peroxisome proliferator-activated receptor gamma | 5468 | 51 | endocrine |
| VDR | Vitamin D receptor | 7421 | 45 | endocrine |
| HNF4A | Hepatocyte nuclear factor 4 alpha | 3172 | 43 | nervous; endocrine |
| ESR2 | Estrogen receptor 2 | 2100 | 43 | endocrine |
| PGR | Progesterone receptor | 5241 | 39 | endocrine |
| l-Glutamic acid | l-Glutamic acid | / | 27 | nervous |
| Glycine | Glycine | / | 27 | nervous |
| Dopamine | Dopamine | / | 27 | nervous |
| Gene Category | Gene Symbol | Gene Name | Entrez Gene ID | Degree |
|---|---|---|---|---|
| Differentially expressed 1 and RA-related genes | CREB1 | cAMP-responsive element-binding protein 1 | 1385 | 9 |
| TNFSF10 | TNF superfamily member 10 | 8743 | 4 | |
| CD4 | CD4 molecule | 920 | 3 | |
| TNFSF12 | TNF superfamily member 12 | 8742 | 3 | |
| Non-differentially expressed and RA-related genes | ESR1 | Estrogen receptor 1 | 2099 | 95 |
| VDR | Vitamin D receptor | 7421 | 42 | |
| PPARG | Peroxisome proliferator-activated receptor gamma | 5468 | 22 | |
| ESR2 | Estrogen receptor 2 | 2100 | 14 | |
| Differentially expressed and RA-unrelated genes | GSK3B | Glycogen synthase kinase 3 beta | 2932 | 8 |
| SMARCA4 | SWI/SNF-related, matrix-associated, actin-dependent Regulator of chromatin, subfamily a, member 4 | 6597 | 7 | |
| PSMD7 | Proteasome 26S subunit, non-ATPase 7 | 5713 | 6 | |
| TGFBR1 | Transforming growth factor beta receptor 1 | 7046 | 4 | |
| Non-differentially expressed and RA-unrelated genes | HNF4A | Hepatocyte nuclear factor 4 alpha | 3172 | 32 |
| PGR | Progesterone receptor | 5241 | 27 | |
| RXRA | Retinoid X receptor alpha | 6256 | 23 | |
| ESRRA | Estrogen-related receptor alpha | 2101 | 18 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Han, L.; Zhang, X.; Wu, R.; Cheng, X.; Zhou, W.; Zhang, Y. Knowledge-Based Neuroendocrine Immunomodulation (NIM) Molecular Network Construction and Its Application. Molecules 2018, 23, 1312. https://doi.org/10.3390/molecules23061312
Wang T, Han L, Zhang X, Wu R, Cheng X, Zhou W, Zhang Y. Knowledge-Based Neuroendocrine Immunomodulation (NIM) Molecular Network Construction and Its Application. Molecules. 2018; 23(6):1312. https://doi.org/10.3390/molecules23061312
Chicago/Turabian StyleWang, Tongxing, Lu Han, Xiaorui Zhang, Rongrong Wu, Xiaorui Cheng, Wenxia Zhou, and Yongxiang Zhang. 2018. "Knowledge-Based Neuroendocrine Immunomodulation (NIM) Molecular Network Construction and Its Application" Molecules 23, no. 6: 1312. https://doi.org/10.3390/molecules23061312