Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere


1
School of Environmental and Municipal Engineering, Qingdao University of Technology, Qingdao 266033, Shandong, China
2
Qingdao Environmental Monitoring Center, Qingdao 266003, Shandong, China
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(6), 1268; https://doi.org/10.3390/molecules23061268
Submission received: 20 April 2018
/
Revised: 22 May 2018
/
Accepted: 23 May 2018
/
Published: 25 May 2018
(This article belongs to the Special Issue Theoretical Investigations of Reaction Mechanisms)
Abstract
:In this work, feasible mechanisms and pathways of the C2H5O2 + BrO reaction in the atmosphere were investigated using quantum chemistry methods, i.e., QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p) levels of theory. Our result indicates that the title reaction occurs on both the singlet and triplet potential energy surfaces (PESs). Kinetically, singlet C2H5O3Br and C2H5O2BrO were dominant products under the atmospheric conditions below 300 K. CH3CHO2 + HOBr, CH3CHO + HOBrO, and CH3CHO + HBrO2 are feasible to a certain extent thermodynamically. Because of high energy barriers, all products formed on the triplet PES are negligible. Moreover, time-dependent density functional theory (TDDFT) calculation implies that C2H5O3Br and C2H5O2BrO will photolyze under the sunlight.
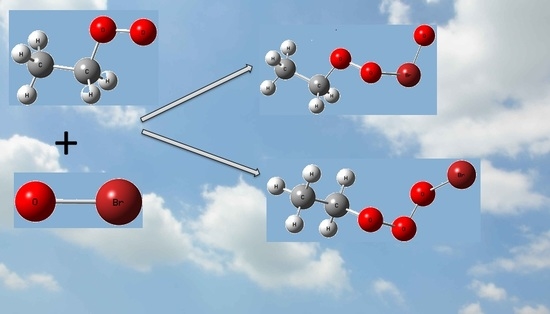
1. Introduction
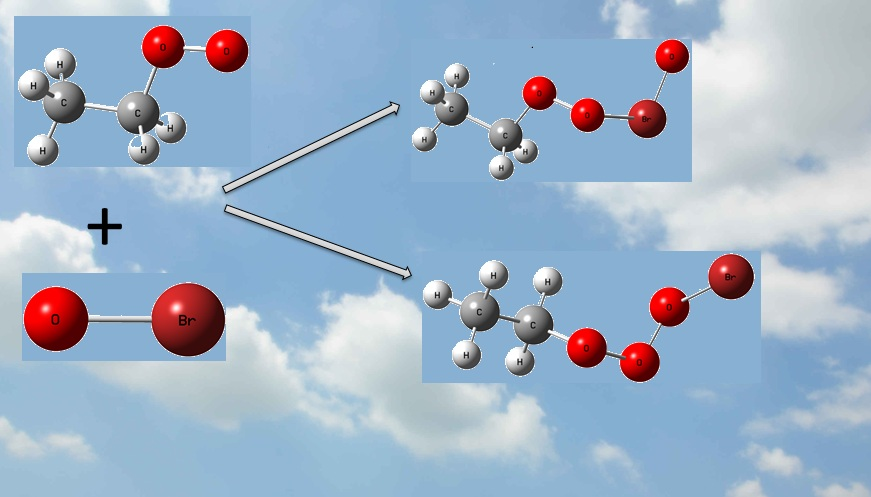
With more and more attention paid to atmospheric environments, researchers focused on the reactions that will increase or produce pollution. Observation indicates halogen monoxides, i.e., XO (X = Cl, Br, and I) play important roles in the marine boundary layer (MBL) and lower stratosphere [1,2]. As catalysts in the ozone destruction processes, with higher efficiency than ClO, the reactions of BrO with peroxy radicals RO2 (R is organic group), such as HO2 and CH3O2, have been investigated extensively by experimental and theoretical methods [3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Experimentally, the rate constants of BrO reacting with HO2 [8,9,10], CH3O2 [11,12,15] and C2H5O2 [16] radicals were determined by several groups. For the reactions of HO2 and CH3O2 with BrO, mechanisms and reaction pathways were investigated theoretically [11,12,13,14,16]; however, the products were not confirmed for the C2H5O2 + BrO reaction from experiments, and no literature is available from theoretical investigations yet. The following channels were proposed by Sakamoto [16]
C2H5O2 + BrO → C2H5O + BrOO ΔrH = 0.2 kJ/mol
→ C2H5O + OBrO ΔrH = 56.1 kJ/mol
→ CH3CHO + HOOBr ΔrH = −222.1 kJ/mol
→ CH3CHO + HBr + O2 ΔrH = −294.4 kJ/mol
Due to distinct advantages over experimental methodology, quantum chemistry is popular as a useful tool to explore mechanisms and feasible products in many atmospheric reactions [17,18,19,20]. By quantum chemistry methods, the C2H5O2 + BrO reaction in the atmosphere was explored at the molecular level to address the mechanisms, channels and products. Moreover, it is expected to provide useful information and new insights into the atmospheric chemistry of C2H5O2 with BrO.
2. Results
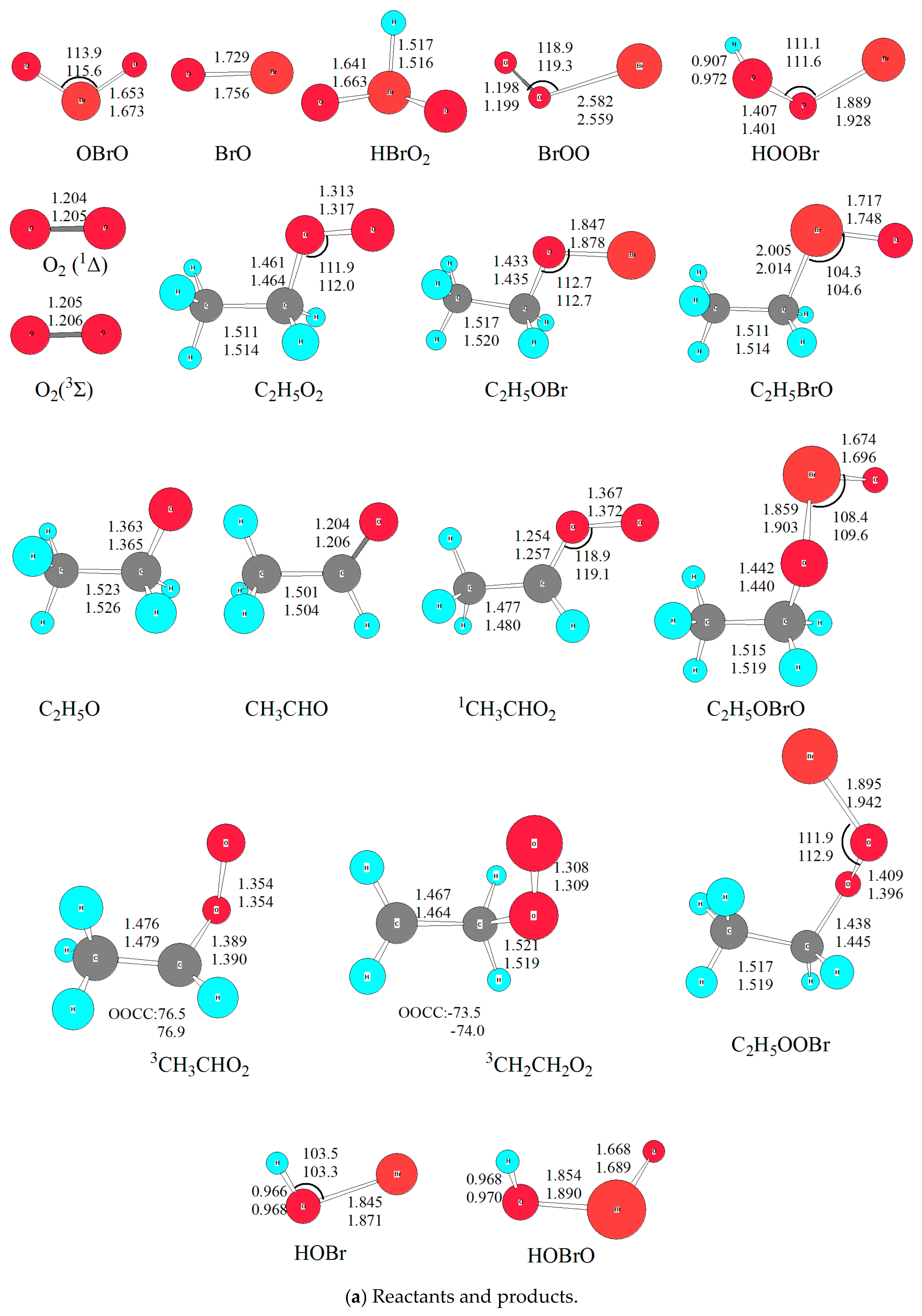
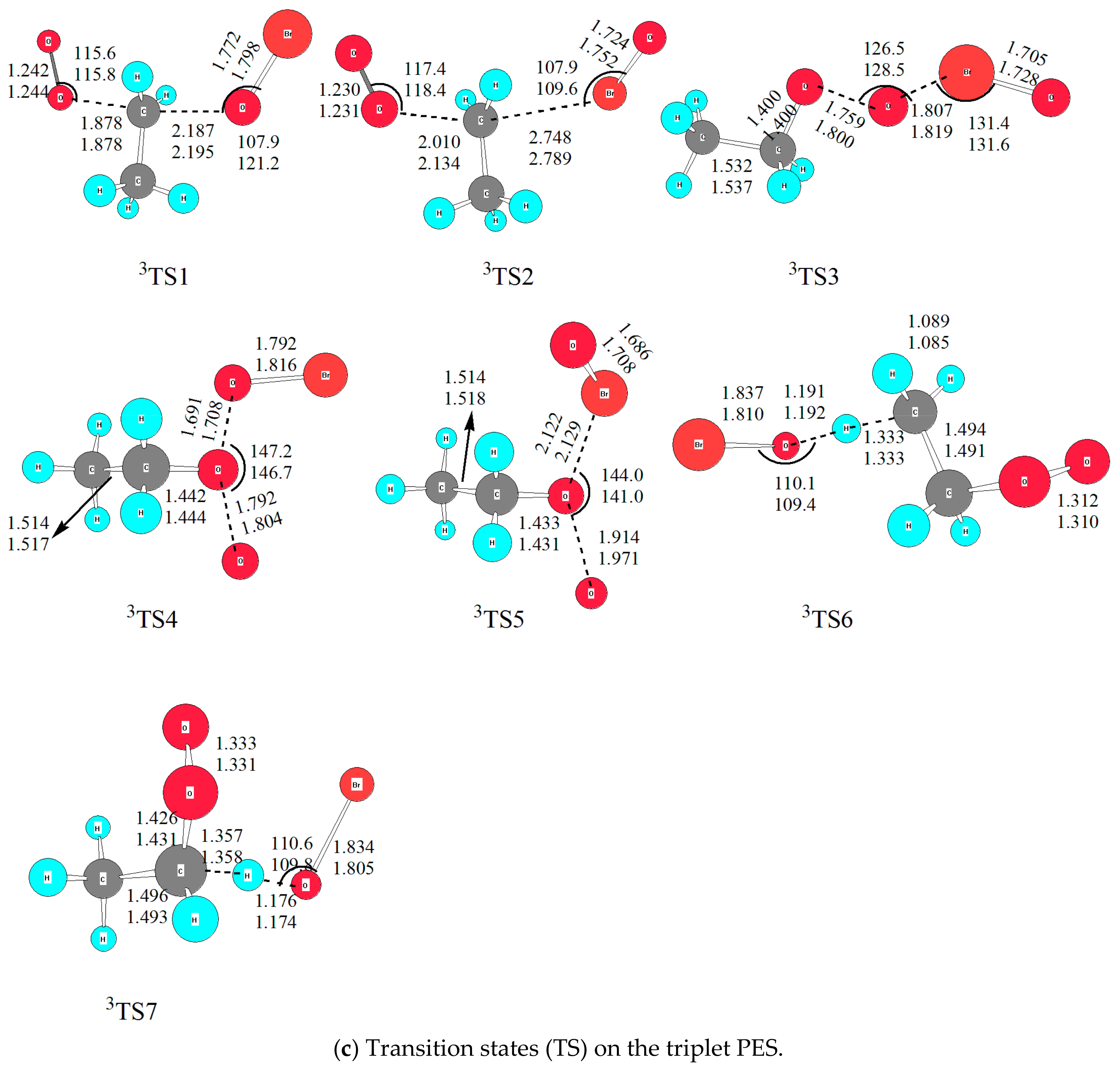
Optimized geometries of all reactants, products, intermediates (IM) and transition states (TS) involved in the title reaction were shown in Figure 1. The energetic profiles of the singlet and triplet PESs at the level of QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p) was depicted in Figure 2. Moreover, 3 is superscripted to differentiate triplet species from the singlet ones. The reaction enthalpy (ΔH) of various channels in the C2H5O2 + BrO reaction obtained from the present work and available references are listed in Table 1. The zero-point energy correction (ZPE) and relative energy (ΔE) and reaction enthalpy (ΔH) at different levels of theory are displayed in Table 2. Before reaction mechanisms and channels are discussed, it is cautious and meaningful to check the validity of the current theoretical level to ensure that our computational result is conceivable and reliable for the title reaction.
2.1. Reliability of Theoretical Methods
The reaction enthalpies (ÿH) of four possible channels was proposed by Sakamoto [16]. In order to check the reliability of methods, the reaction enthalpies (ΔH) for all proposed channels was calculated at four levels of theory, i.e., G4 [21], QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(d,p), QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p), and QCISD(T)/6-311++G(2df,2p)//MP2/6-311++G(2df,2p) [22,23,24,25,26]. The result is listed in Table 1, from which it could be found that relative energy (∆E) and reaction enthalpy (∆H) varies from different levels of theory. Although it is common that the MP2 [27] method provides high quality quantitative prediction for many systems; unfortunately, for the title reaction involving open-shell electrons, it really does not perform well. Since it is known that multireference methods, such as Complete Active Space Self Consistent Field (CASSCF) [28], are more accurate to many atmosphere systems, especially for photo-chemical processes [29,30]. However, due to limited computational resources, we chose QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p) for single point energy calculation, as used in previous study for the CH3O2 + BrO reaction [14]. On the other hand, compared with other levels of theory, the density functional (DFT) method was found to be sufficiently accurate for predicting reliable geometries of the stationary points; at the same time, it is not expensive computationally for scanning PES [14,17,20]. Thus, B3LYP functional was used in this work with two basis sets, i.e., 6-311++G(d,p) and 6-311++G(2df,2p) for all stationary points to check the influence of basis sets on geometry. The optimized geometry was depicted in Figure 1, from which it could be seen that the geometrical parameters from both basis sets are close to each other, indicating that polarization functions have no significant influence on the title reaction. Considering the Br atom involved, the geometry obtained from 6-311++G(2df,2p) basis set was used in the discussion unless otherwise stated. Besides, to check the optimized geometry from B3LYP functional, M062X [31] functional was employed as previously used for the CH3O2 + BrO reaction. The details are shown in Section 2.2.2. Therefore, it is conceivable to discuss the reaction channels based on the QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p) calculations.
2.2. Reaction Channels of the C2H5O2 + BrO Reaction
In order to give a clear and distinct description of the reaction mechanisms and pathways, we will discuss the formation of intermediates firstly, and the separate reaction channels on the singlet and triplet PESs subsequently.
2.2.1. The Formation of Intermediates in the C2H5O2 + BrO Reaction
On the singlet PES with two reactive sites of BrO attacking the reactive O atom in C2H5O2, two initial intermediates, namely C2H5O3Br (IM1) and C2H5O2BrO (IM2), are formed directly without any energy barriers. As shown in Figure 1, the newly formed O-O bond is 1.369 Å in IM1, and O-Br bond is 1.995 Å in IM2. It is worth noting that the singlet and triplet PESs intersection commonly takes place in the radical-radical reactions, especially for barrierless entrances [32,33]. However, the transition probability between the singlet and triplet surfaces was not treated explicitly in the present calculations. Our extensive attempts to calculate the single-triple crossing point for the entrance of the C2H5O2 + BrO association have not been successful due to the convergence difficulties of the multireference configuration interactions. The complete active spaces (CAS) calculations with all valence electrons are unaffordable at present. It is noted that the intersystem crossing might be significant in some small organic molecules [34,35], therefore, more rigorous treatment of the singlet-triplet transition has to be reserved for further study.
It is mentioned that several conformers of C2H5O3Br and C2H5O2BrO located according to different dihedral angle of BrOOO (OBrOO) and OOOC (BrOOC) with internal rotations of the relevant O-O and Br-O bonds, and these conformers can interconvert within a few kJ/mol energy barriers; however, the interconversion are out of our focus and not considered in this work.
Energetically, IM1 and IM2 are about 72.3 and 19.1 kJ/mol lower than the initial reactants, respectively. Therefore, C2H5O3Br and C2H5O2BrO should be formed as vibrationally hot molecules:
C2H5O2 + BrO → C2H5O3Br‡
→ C2H5O2BrO‡
With much internal energy available, the hot molecules of C2H5O3Br‡ and C2H5O2BrO‡ may experience further isomerization and dissociation before being quenched by collisions. According to our result, three conceivable isomerization scenarios are located on the singlet PES.
Firstly, IM1 and IM2 interconverts via a triangular transition state TS1. The broken O-O bond is elongated to 2.286 Å in TS1, which is about 67% longer than that in IM1; while the formed Br-O bond is stretched by 0.37 Å from its equilibrium distance in IM2. The energy barrier height for IM1 isomerizing to IM2 is about 106.7 kJ/mol, and IM2 convert to IM1 is around 53.5 kJ/mol. With so high energy barriers the interconversion between IM1 and IM2 is unfeasible in the normal atmospheric conditions with a temperature below 300 K.
Secondly, via a similar triangular structure TS2 with bromine atom migrating from the central oxygen atom to the oxygen atom in C2H5O group, IM2 (C2H5O2BrO) isomerizes to IM3 (C2H5OBrO2). Geometrically, the formed Br-O bond is 2.485 Å in TS2, and the broken O-O bond is dramatically stretched by 0.776 Å from that in IM2. While the two non-reactive Br-O bonds (around 1.66 Å) are close to their equilibrium lengths in IM3 (around 1.64 Å). Energetically, IM3 is rather stable on the singlet PES, with its relative energy (ΔE) of 83.6 kJ/mol lower than the initial reactants; and 64.5 kJ/mol lower than IM2. However, the energy barrier height for IM2 to IM3 reaches 86.4 kJ/mol; apparently, it is difficult to proceed with low temperature (e.g., T < 300 K), although IM3 is the most stable intermediate on the whole singlet PES. Nevertheless, it might happen in high temperature conditions such as combustion, which is out of consideration in the present work.
From previous studies on the CH3O2 + BrO [14], CF3O2 + IO [36] and CF3O2 + ClO [37] reactions, it is assumed that a high energy barrier for IM3 (ROBrO2) → IM4 (RBrO3) is surmounted. However, IM4 is rather unstable thermodynamically, therefore, it will not play any significant role in the overall reaction in the atmosphere. For completeness, only IM4 was optimized (C2H5BrO3) without the transition state calculated. As expected, IM4 is unstable, with its relative energy of 97.7 kJ/mol higher than the initial reactants. Based on previous theoretical results and our current calculations [13,14], for all intermediates formed in the RO2 + BrO (R = H, CH3, C2H5) reactions, the order of relative stability among the RO3Br isomers (i.e., HO3Br, CH3O3Br, and C2H5O3Br) from the most stable to the least stable structure is ROBrO2 > ROOOBr > ROOBrO > RBrO3. This implies that the substitution of alkyl group has no significant effect in RO3Br surfaces.
As for the triplet PES, in spite of many attempts, intermediates were not located at the current levels of theory.
To sum up, four intermediates are formed. Several possible dissociation reaction channels are available with abundant internal energy available from intermediates except for IM4. The details will be described in the following section.
2.2.2. The Reaction Pathways on the Singlet PES
According to our result, seven possible products and eight dissociation channels are determined, i.e., four from IM1, two from IM2 and two from IM3, respectively. To give a clear description, we will discuss the formation of products separately.
(a) C2H5O + BrOO and C2H5O + OBrO
The barrierless cleavage of the O-O bond from IM1 leads to C2H5O + BrOO. While the O-O and O-Br bonds in IM2 and IM3 dissociate to give out C2H5O + OBrO in the same way. Energetically, the relative energy of C2H5O + BrOO and C2H5O + OBrO is 14.9 and 37.5 kJ/mol, respectively. Furthermore, the reaction channels of C2H5O2 + BrO → C2H5O + BrOO and C2H5O2 + BrO → C2H5O + OBrO are endothermic by 18 and 38.8 kJ/mol, thus they are unfeasible to occur in the atmospheric conditions with low temperature (e.g., T < 300 K).
(b) CH3CHO + HOOBr
With migration of H atom in -CH2 to the O atom in -OOBr, and the relevant O-O bond fission from IM1, HOOBr + CH3CHO will be generated via TS4. The barrier energy height takes a value of 120 kJ/mol, while TS4 is 47.7 kJ/mol on the singlet PES. Moreover, the channel of C2H5O2 + BrO → CH3CHO + HOOBr is highly exothermic by 249 kJ/mol, and the product is rather stable thermodynamically. However, considering the low temperature (T < 300 K) in the atmosphere, especially in higher troposphere and lower stratosphere, CH3CHO+HOOBr are unfavorable to form kinetically at the current levels of theory.
(c) 1CH3CHO2 + HOBr
Similarly, with migration of the H atom in -CH2 to the O atom in -OBr and the relevant O-O bond fission, 1CH3CHO2 + HOBr is generated via a five-membered-ring structure TS5 with a barrier height of 123 kJ/mol. Here it is mentioned that HOBr was presumed in the CH3O2 + BrO reaction by Shallcross via a much lower energy barrier (around 62 kJ/mol and the transition state is −3.2 kJ/mol) at the CASPT2-F12/AVDZ//M06-2X/AVDZ levels of theory [15]. In order to check the deviation between our present computational results with Shallcross’s, we performed the optimization of several significant intermediates and transition states with M062X functional from DFT methods. The optimized geometrical parameters were listed in Figure 1, from which it could be seen that the bond length and bond angle are close at the B3LYP and M062X methods. Thus, the single point energy deviations come from the employed methods. Regrettably, multiconfigurational methods were not affordable at the moment due to limited computational resource.
(d) CH3CHO + HBrO2
Starting from IM2, CH3CHO + HBrO2 (ΔE = −14.8 kJ/mol) is obtained with migration of one H atom from -CH2 to Br atom and cleavage of the O-O bond simultaneously via TS6 while the relative energy is about 5 kJ/mol lower than that of TS4. Although the relative energy is close (within 8 kJ/mol) among TS4, TS5 and TS6, the energy barrier height of TS6 is 61.9 kJ/mol, which is much lower than that of TS4 (120 kJ/mol) and TS5 (123 kJ/mol). Thus, the channel via C2H5O2 + BrO → IM2 → TS6 → CH3CHO + HBrO2 will be more feasible to occur kinetically than the channels via C2H5O2 + BrO → IM2 → TS4 → CH3CHO + HOOBr or C2H5O2 + BrO → IM2 → TS5 → 1CH3CHO2 + HOBr. However, all the channels are unfavorable at low temperature in the typical atmospheric conditions (T < 300 K).
(e) CH3CHO + HOBrO
From IM3, H atom in -CH2 moves to O atom in OBrO forming a rather stable product CH3CHO + HOBrO, and this process is exothermic by 227.8 kJ/mol. Although TS7 (ΔE = −17.6 kJ/mol) is the lowest transition state on the singlet PES, the channel via C2H5O2 + BrO → IM2 → TS2 → IM3 → TS7 → CH3CHO + HOBrO will make a minor contribution to the overall reaction due to the high energy barrier of TS2 (ΔE = 67.3 kJ/mol).
(f) C2H5OBr + 1O2
From IM1, a concerted process occurs via TS3 with the original C-O and O-O bonds stretched to be 2.386 and 1.786 Å, respectively; and the new C-O bond formed with the equilibrium distance to be 2.320 Å. The cleavage of stable C-O and O-O bond is rather tough with the energy barrier height reaching to 273.6 kJ/mol.
Besides the elimination channel from IM1, C2H5OBr + 1O2 could be formed via the substitution mechanism as well on the singlet PES, with O atom in BrO attacking the C center in -CH2 group via a rather high energy barrier of TS8, which relative energy is around 237.6 kJ/mol. Evidently, both channels have no possibility of occurring kinetically in the normal atmospheric conditions, and are negligible to the overall reaction although the formation of the product is exothermic.
In summary, on the singlet PES C2H5O3Br and C2H5O2BrO will be dominant products. Other minor products include CH3CHO + HOOBr via IM1 and TS4, CH3CHO + HBrO2 via IM2 and TS6, and 1CH3CHO2 + HOBr via IM1 and TS5. Considering the typical limitation of 20 kJ/mol for atmospheric reactions, these sub-dominant channels are of no significance kinetically, although their formations should be feasible thermodynamically.
2.2.3. The Substitution and Abstraction Channels on the Triplet PES
On the triplet PES, no intermediate was located with many attempts, thus the channels are much simpler than that on the singlet PES. According to our result, both substitution and direct abstraction mechanisms were determined leading to seven products. As shown in Figure 2, surmounting 3TS1, 3TS2, 3TS3, 3TS4 and 3TS5, C2H5OBr + 3O2, C2H5BrO + 3O2, C2H5O + OBrO, C2H5OOBr + O(3P) and C2H5OBrO + O(3P) are generated, and the relative energy is −190.5, −3.3, 14.9, 119.1 and 150.2 kJ/mol, respectively. The energy barriers of five transition states are 95.7, 142.4, 151.8, 193.2 and 210.6 kJ/mol, respectively. Obviously, with so high barriers it is conceivable that all channels play no important roles to the title reaction under the atmospheric conditions below 300 K.
Here it is mentioned that the formation of HOBr was located via direct hydrogen-abstraction channels with H atom in -CH3 or -CH2 group was abstracted. The corresponding transition states are 3TS6 and 3TS7, with similar relative energies, i.e., 55.4 and 49.6 kJ/mol, respectively, which are modest to happen in higher temperature conditions. The products in the two channels are 3CH2CH2O2 and 3CH3CHO2, which are much more unstable than their singlet species. The formations of 3CH3CHO2 + HOBr and 3CH2CH2O2 + HOBr are endothermic by 17.1 and 21.3 kJ/mol, therefore, the channels are unfavorable thermodynamically.
To sum up, from the above discussions it is concluded that all substitution and abstraction channels on both the singlet and triplet PES are of on significance to the C2H5O2 + BrO reaction below 300 K. C2H5O3Br and C2H5O2BrO are dominant to the overall reaction. Thermodynamically, the subsequent dissociation from intermediates leading to CH3CHO + HBrO2, CH3CHO + HOOBr, CH3CHO + HOBrO and CH3CHOO + HOBr are favorable. However, with high energy barriers involved, these products are difficult to be formed kinetically in the atmospheric conditions below 300 K.
2.2.4. Vertical Excitation Energy TV of C2H5O3Br, C2H5O2BrO and C2H5OBrO2
It is known that the photo-oxidation of compounds containing bromine is significant for Br atmospheric chemistry, therefore their photolysis might influence the stratosphere and troposphere. In order to obtain new insights of photolytic information into the Br-containing compounds, the vertical excitation energy (TV) of the first five excited states for C2H5O3Br, C2H5O2BrO and C2H5OBrO2 was calculated by the TDDFT method [38] employing B3LYP/6-311++G(2df,2p), and the results including wavelength (λ), excitation energy (TV) and oscillator strength (f) are listed in Table 3.
Known most ultraviolet (UV) light is absorbed and only is 7% left when solar radiation reaches to the surface of Earth, therefore, compounds will be considered to photolyze if the TV value is smaller than 4.13 eV (about 300 nm of threshold in the visible light). From Table 3 it is seen that the TV value of the first two/three excited states of C2H5O3Br and C2H5O2BrO take values smaller than 4.13 eV, and their oscillator strength is not null, implying that the C2H5O3Br and C2H5O2BrO photolyze under the sunlight. Checking the occupied and virtual molecular orbitals it is found that the most contribution comes from HOMO-1 to LUMO with an npO → π* transition associated in both C2H5O3Br and C2H5O2BrO. Therefore, it is speculated that the processes C2H5O3Br → C2H5O2 + BrO and C2H5O2BrO → C2H5O2 + BrO occur after absorption of sunlight. On the other hand, the TV value of all excited states for C2H5OBrO2 is larger than 4.13 eV, thus it will not be the source of reactive bromine species in the troposphere.
3. Materials and Methods
All calculations were carried out using GAUSSIAN 09 program package [39]. The geometries of reactants (R), products (P), intermediates (IM), and transition states (TS) involved in the title reaction were optimized using B3LYP [23] and M062X [31] functionals from DFT methods, and two basis [24,25,26] sets, namely, 6-311++G(d,p) and 6-311++G(2df,2p). In order to obtain more reliable relative energy of each stationary point on the potential energy surfaces (PES), single-point energy is refined by QCISD(T)/6-311++G(2df,2p) [22] basis set based on the B3LYP/6-311++G(2df,2p) geometry and G4 method [21]. Moreover, the vertical excitation energy was calculated with TD-B3LYP/6-311++G(2df,2p) [38] level of theory.
4. Conclusions
Using quantum chemistry methods, the reaction mechanisms and pathways for the atmospheric reaction of C2H5O2 + BrO were studied in detail at the QCISD(T)//B3LYP levels of theory. The result indicates that the title reaction occurs on both the singlet and triplet PES, with addition-elimination, substitution and direct H/O-abstraction mechanisms involved. The energy barriers on the singlet PES are lower than that on the triplet PES. C2H5O3Br and C2H5O2BrO is dominant on the singlet PES. Thermodynamically, CH3CHO2 + HOBr, CH3CHO + HOBrO, and CH3CHO + HBrO2 are feasible, while they are of no significance due to high energy barriers. Moreover, C2H5O3Br and C2H5O2BrO will photolyze under the sunlight, which might be one source of Br-containing species in the atmosphere.
Author Contributions
C.L. analyzed the data, discussed, wrote and revised the manuscript, Y.T. designed the research, performed the calculations and wrote the manuscript. W.Z. and Z.F. analyzed the data and drawing geometries. X.Q. read and approved the final manuscript.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (No. 41775119) and Shandong Province Postdoctoral Special Fund for Innovative Projects (201402017). We thank Youxiang Shao for discussion and suggestions to this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Watson, R.T.; Albritton, D.L.; Anderson, S.O.; Lee-Bapty, S. Methyl bromide: Its atmospheric science, technology and economics. In Montreal Protocol Assessment Supplement; United Nations Environmental Programme: Nairobi, Kenya, 1993; p. 243. [Google Scholar]
- Montzka, S.A.; Reimann, S.; Engel, A.; Kruger, K.; O’ Doherty, S.J.; Sturges, W.T. Ozone-depleting substances (ODSs) and related chemicals. In Scientific Assessment of Ozone Depletion: 2010; Proceedings of the Global Ozone Research and Monitoring Project Report No. 52; World Meteorological Organization: Geneva, Switzerland, 2011; pp. 1–112. [Google Scholar]
- Elrod, M.J.; Meads, R.F.; Lipson, J.B.; Seeley, J.V.; Molina, M.J. Temperature dependence of the rate constant for the HO2 + BrO reaction. J. Phys. Chem. 1996, 100, 5808–5812. [Google Scholar] [CrossRef]
- Bedjanian, Y.; Riffault, V.; Poulet, G. Kinetic study of the reactions of BrO radicals with HO2 and DO2. J. Phys. Chem. A 2001, 105, 3167–3175. [Google Scholar] [CrossRef]
- Bridier, I.; Veyret, B.; Lesclaux, R. Flash photolysis kinetic study of reactions of the BrO radical with BrO and HO2. Chem. Phys. Lett. 1993, 201, 563–568. [Google Scholar] [CrossRef]
- Poulet, G.; Pirre, M.; Maguin, F.; Ramaroson, R.; LeBras, G. Role of the BrO + HO2 reaction in the stratospheric chemistry of bromine. Geophys. Res. Lett. 1992, 19, 2305–2308. [Google Scholar] [CrossRef]
- Bloss, W.J.; Rowley, D.M.; Cox, R.A.; Jones, R.L. Rate Coefficient for the BrO + HO2 Reaction at 298 K. Phys. Chem. Chem. Phys. 2002, 4, 3639–3647. [Google Scholar] [CrossRef]
- Salisbury, G.; Monks, P.S.; Bauguitte, S.; Bandy, B.J.; Penkett, S.A. A seasonal comparison of ozone photochemistry in clean and polluted air masses at Mace Head Ireland. J. Atmos. Chem. 2002, 41, 163–187. [Google Scholar] [CrossRef]
- Creasey, D.J.; Halford-Maw, P.A.; Heard, D.E.; Pilling, M.J.; Whitaker, B.J. Implementation and initial deployment of a field instrument for measurement of OH and HO2 in the troposphere by laser-induced fluorescence. J. Chem. Soc. Faraday Trans. 1997, 93, 2907–2913. [Google Scholar] [CrossRef]
- Sommariva, R.; Osthoff, H.D.; Brown, B.B.; Bates, T.S.; Ravishankara, A.R.; Trainer, M. Radicals in the marine boundary layer during NEAQS 2004: A model study of day-time and night-time sources and sinks. Atmos. Chem. Phys. 2009, 9, 3075–3093. [Google Scholar] [CrossRef]
- Aranda, A.; LeBras, G.; LaVerdet, G.L.; Poulet, G. The BrO + CH3O2 reaction: Kinetics and role in the atmospheric ozone budget. Geophys. Res. Lett. 1997, 24, 2745–2748. [Google Scholar] [CrossRef]
- Enami, S.; Yamanaka, T.; Nakayama, T.; Hashimoto, S.; Kawasaki, M.; Shallcross, D.E.; Nakano, Y.; Ishiwata, T. A gas-phase kinetic study of the reaction between bromine monoxide and methylperoxy radicals at atmospheric temperatures. J. Phys. Chem. A 2007, 111, 3342–3348. [Google Scholar] [CrossRef] [PubMed]
- Larichev, M.; Maguin, F.; LeBras, G.; Poulet, G. Kinetics and Mechanism of the BrO + HO2 Reaction. J. Phys. Chem. 1995, 99, 15911–15918. [Google Scholar] [CrossRef]
- Guha, S.; Francisco, J.S. An ab initio study of the pathways for the reaction between CH3O2 and BrO radicals. J. Chem. Phys. 2003, 118, 1779–1793. [Google Scholar] [CrossRef]
- Shallcross, D.E.; Leather, K.E.; Back, A.; Xiao, P.; Lee, E.P.F.; Ng, M.; Mok, D.K.W.; Dyke, J.M.; Hossaini, R.; Chipperfield, M.P.; et al. Reaction between CH3O2 and BrO radicals: A new source of upper troposphere lower stratosphere hydroxyl radicals. J. Chem. Phys. 2015, 119, 4618–4632. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Yamano, D.; Nakayama, T.; Hashimoto, S.; Kawasaki, M.; Wallington, T.J.; Miyano, S.; Tonokura, K.; Takahashi, K. Atmospheric chemistry of BrO radicals: Kinetics of the reaction with C2H5O2 radicals at 233–333 K. J. Phys. Chem. A 2009, 113, 10231–10237. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.X.; Hou, H.; Wang, B.S. Theoretical study of the mechanisms and kinetics of the reactions of hydroperoxy (HO2) radicals with hydroxymethylperoxy (HOCH2O2) and methoxymethylperoxy (CH3OCH2O2) radicals. Phys. Chem. Chem. Phys. 2014, 16, 22805–22814. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Ou-Yang, S.L.; Li, L. Theoretical study of ClOO + NO reaction: Mechanism and kinetics. Molecules 2017, 22, 2121. [Google Scholar] [CrossRef] [PubMed]
- De Souza, G.L.C.; Brown, A. The ground and excited states of HBrO2 [HOOBr, HOBrO, and HBr(O)O] and HBrO3 (HOOOBr and HOOBrO) isomers. Theor. Chem. Acc. 2016, 135, 178. [Google Scholar] [CrossRef]
- Vereecken, L.; Francisco, J.S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef] [PubMed]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; von Rague Schleyer, R. Efficient diffuse function-augmented basis-sets for anion calculations. Ⅲ. The 3-21+G basis set for 1st-row elements, Li-F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Binning, R.C., Jr.; Curtiss, L.A. Compact contracted basis-sets for third-row atoms: Ga-Kr. J. Comp. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth order storage: Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Wong, M.W.; Frisch, M.J.; Wiberg, K.B. Solvent effects 1. The mediation of electrostatic effects by solvents. J. Am. Chem. Soc. 1991, 113, 4776–4782. [Google Scholar] [CrossRef]
- Fang, W.H. A CASSCF study on photodissociation of acrolein in the Gas Phase. J. Am. Chem. Soc. 1999, 121, 8376–8384. [Google Scholar] [CrossRef]
- Fantacci, S.; Migani, A.; Olivucci, M. CASPT2//CASSCF and TDDFT//CASSCF mapping of the excited state isomerization path of a minimal model of the retinal chromophore. J. Phys. Chem. A 2004, 108, 1208–1213. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Comparative DFT study of van der Waals complexes: Rare-gas dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers. J. Phys. Chem. 2006, 110, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Understanding the kinetics of spin-forbidden chemical reactions. Phys. Chem. Chem. Phys. 2007, 9, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Spin-forbidden reactions: Computational insight into mechanisms and kinetics. Comp. Mol. Sci. 2014, 4, 1–14. [Google Scholar] [CrossRef]
- Schnappinger, T.; Kölle, P.; Marazzi, M.; Monari, A.; González, L.; de Vivie-Riedle, R. Ab initio molecular dynamics of thiophene: The interplay of internal conversion and intersystem crossing. Phys. Chem. Chem. Phys. 2017, 19, 25662–25670. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, M.; Wibowo, M.; Gattuso, H.; Dumont, E.; Roca-Sanjuán, D.; Monari, A. Hydrogen abstraction by photoexcited benzophenone: Consequences for DNA photosensitization. Phys. Chem. Chem. Phys. 2016, 18, 7829–7836. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Tang, Y.Z.; Wang, R.S. Atmospheric chemistry of CF3O2: A theoretical study on mechanisms and pathways of the CF3O2 + IO reaction. Phys. Chem. Chem. Phys. 2013, 15, 5936–5944. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Z.; Sun, H.F.; Sun, J.Y.; Zhang, Y.J.; Wang, R.S. Theoretical study on mechanisms and pathways of the CF3O2 + ClO reaction. Atmos. Environ. 2014, 92, 367–375. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Gill, P.W.M.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.A.; Petersson, G.A.; Pople, J.A. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
Sample Availability: not available. |
Figure 1.
The optimized geometries for all species in the C2H5O2 + BrO reaction at B3LYP/6-311++G(d,p) and B3LYP/6-311++G(2df,2p) levels. Bond distances are in Å and bond angles are in degrees. The upper one is from B3LYP/6-311++G(2df,2p); and the lower one is from B3LYP/6-311++G(d,p). (a) reactants and products; (b) intermediates (IM) and transition states (TS) on the singlet PES, with the bottom data is from M062X/6-311++G(2df,2p); (c) transition states (TS) on the triplet PES.
Figure 1.
The optimized geometries for all species in the C2H5O2 + BrO reaction at B3LYP/6-311++G(d,p) and B3LYP/6-311++G(2df,2p) levels. Bond distances are in Å and bond angles are in degrees. The upper one is from B3LYP/6-311++G(2df,2p); and the lower one is from B3LYP/6-311++G(d,p). (a) reactants and products; (b) intermediates (IM) and transition states (TS) on the singlet PES, with the bottom data is from M062X/6-311++G(2df,2p); (c) transition states (TS) on the triplet PES.
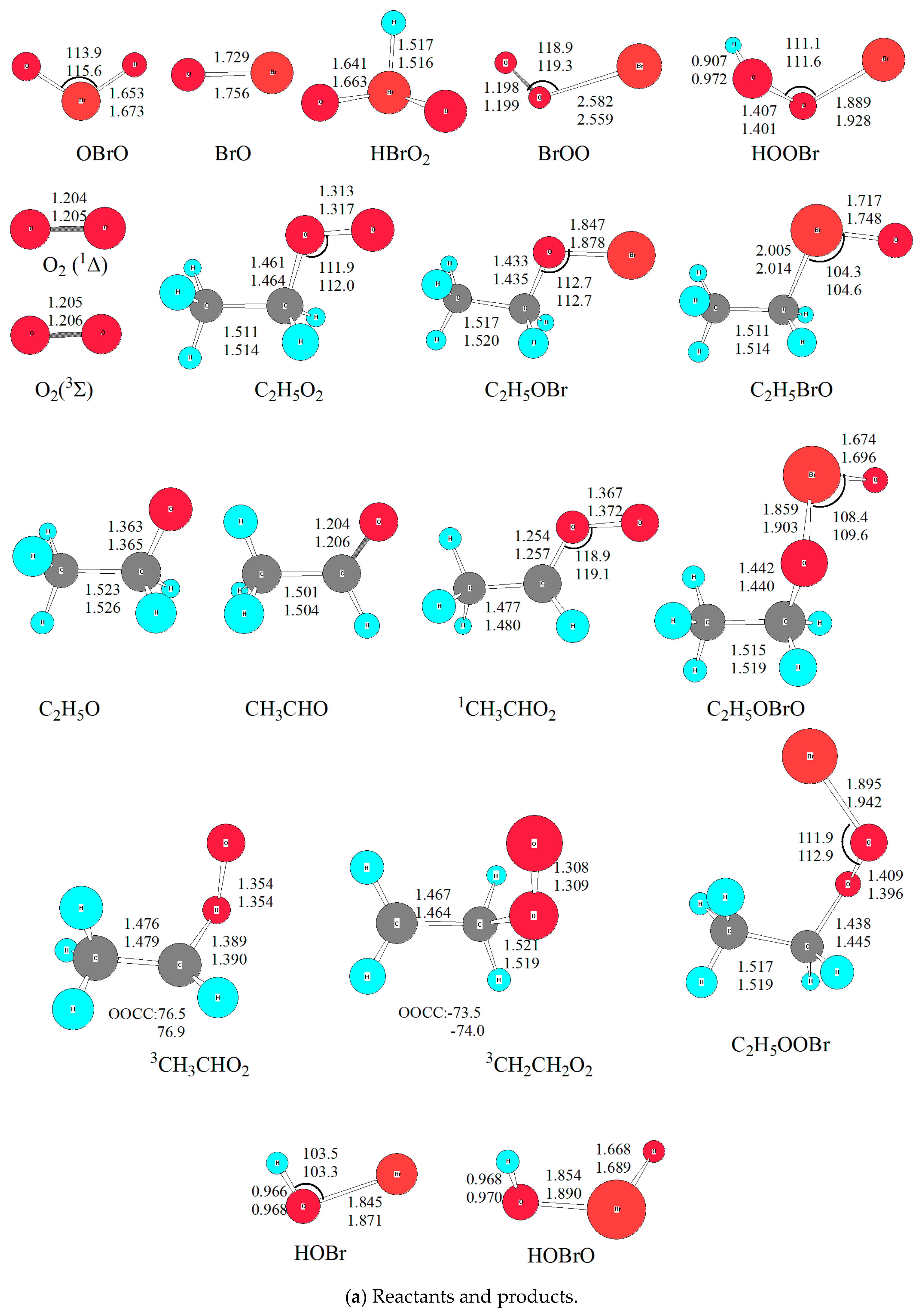
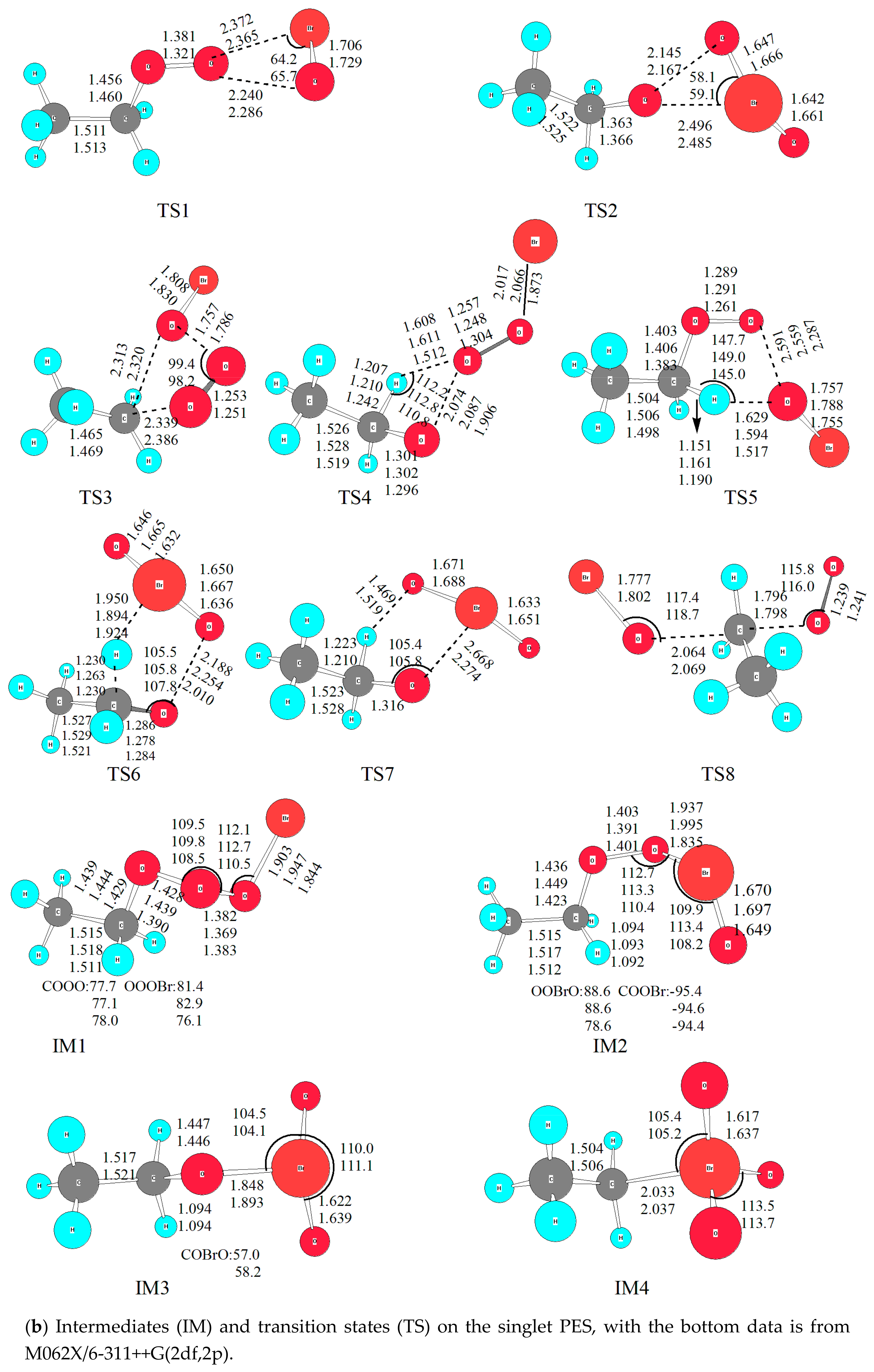
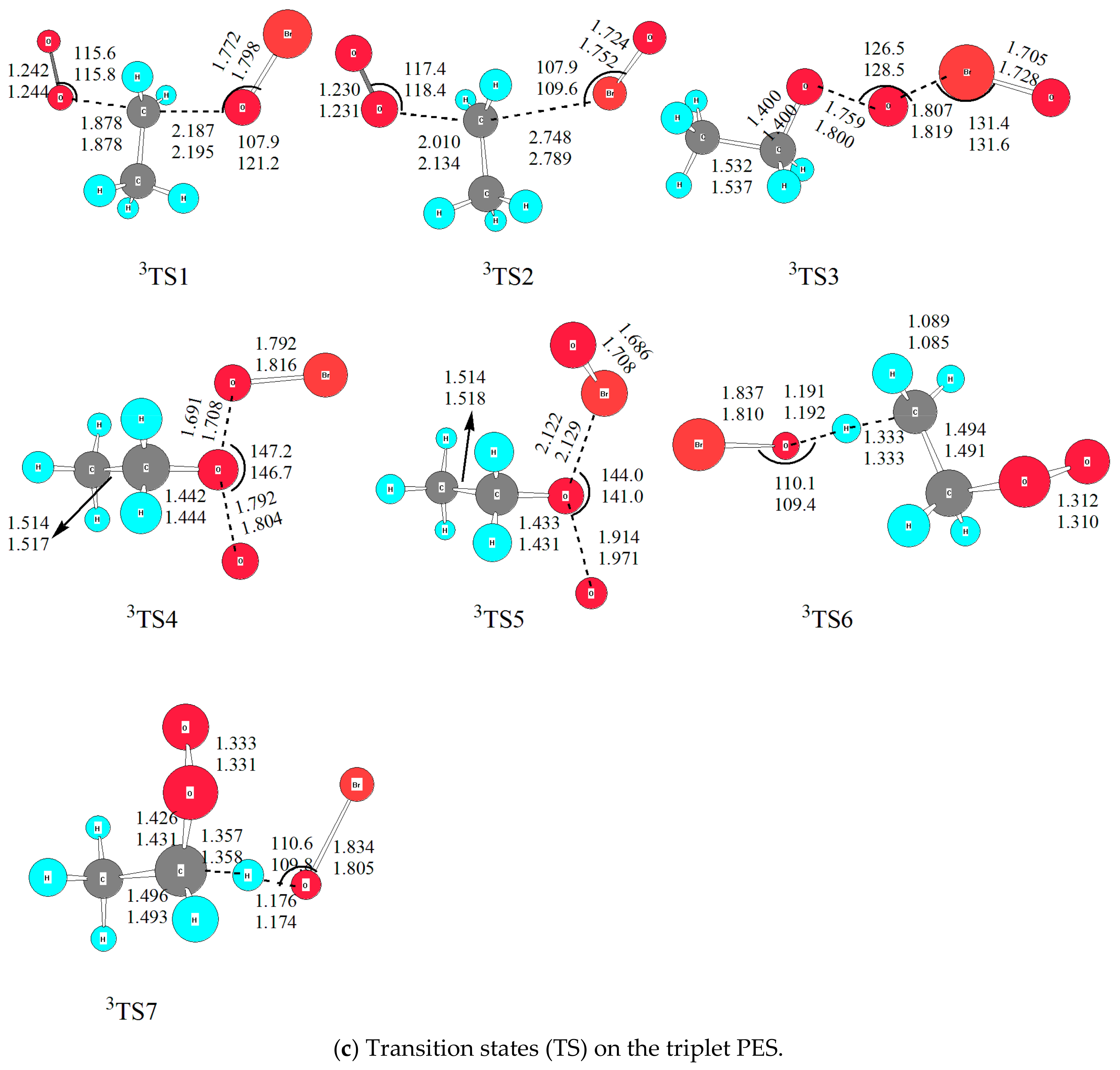
Figure 2.
Energetic profiles of the C2H5O2 + BrO reaction at the QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p) levels.
Figure 2.
Energetic profiles of the C2H5O2 + BrO reaction at the QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p) levels.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The reaction enthalpies (ΔH) (in kJ/mol) of several channels in the C2H5O2 + BrO reaction obtained form at the various levels of theory.
Table 1.
The reaction enthalpies (ΔH) (in kJ/mol) of several channels in the C2H5O2 + BrO reaction obtained form at the various levels of theory.
| C2H5O2 + BrO Reaction Channels | G4 | QCISD(T) | ||
|---|---|---|---|---|
| ΔH | ΔH a | ΔH b | ΔH c | |
| C2H5O + BrOO | −6.8 | 17.2 | 18.0 | −19.4 |
| C2H5O + OBrO | 30.0 | 39.3 | 38.8 | 36.0 |
| C2H5OBr + O2(3∑) | −190.9 | −186.3 | −189.9 | −194.1 |
| C2H5OBr + O2(1∆) | −72.8 | −62.1 | −62.8 | −67.9 |
| HOBr +CH3CHO2 | −116.7 | −109.1 | −108.9 | −110.5 |
| HOOBr + CH3CHO | −256.4 | −247.9 | −249.1 | −253.8 |
| HBr + CH3CHO + 3O2 | −310.0 | −313.3 | −312.7 | −317.1 |
a QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(d,p); b QCISD(T)/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p); c QCISD(T)/6-311++G(2df,2p)//MP2/6-311++G(2df,2p).
Table 2.
The Zero-point energy correction (ZPE), relative energies with ZPE including (ΔE) and reaction enthalpy (ΔH) (in kJ/mol) in the C2H5O2 + BrO reaction.
Table 2.
The Zero-point energy correction (ZPE), relative energies with ZPE including (ΔE) and reaction enthalpy (ΔH) (in kJ/mol) in the C2H5O2 + BrO reaction.
| Species | QCISDT | B3LYP | |
|---|---|---|---|
| ΔE | ΔH | ZPE | |
| C2H5O2 + BrO | 0 | 0 | 191.6 |
| C2H5O + BrOO | 14.9 | 18.0 | 179.4 |
| C2H5O + OBrO | 37.5 | 38.8 | 179.9 |
| C2H5Obr + O2(3∑) | −190.5 | −189.9 | 170.6 |
| C2H5Obr + O2(1∆) | −63.3 | −62.8 | 192.5 |
| HOBr + 1CH3CHO2 | −109.5 | −108.9 | 188.6 |
| HOOBr + CH3CHO | −249.8 | −249.1 | 188.9 |
| C2H5BrO + O2(3∑) | −3.3 | −1.4 | 187.3 |
| C2H5OOBr + O(3P) | 119.1 | 119.8 | 191.3 |
| C2H5ObrO + O(3P) | 150.2 | 151.8 | 188.9 |
| CH3CHO + HBrO2 | −14.8 | −15.1 | 179.3 |
| CH3CHO + HOBrO | −228.6 | −227.8 | 186.5 |
| HOBr + 3CH3CHO2 | 15.2 | 17.1 | 181.2 |
| HOBr + 3CH2CH2O2 | 19.3 | 21.3 | 181.9 |
| IM1 | −72.3 | 199.5 | |
| IM2 | −19.1 | 197.4 | |
| IM3 | −83.6 | 199.2 | |
| IM4 | 97.7 | 197.1 | |
| TS1 | 34.4 | 196.7 | |
| TS2 | 67.3 | 189.8 | |
| TS3 | 201.3 | 186.0 | |
| TS4 | 47.7 | 184.1 | |
| TS5 | 50.7 | 183.6 | |
| TS6 | 42.8 | 189.5 | |
| TS7 | −17.6 | 185.7 | |
| TS8 | 237.6 | 189.5 | |
| 3TS1 | 95.7 | 187.5 | |
| 3TS2 | 142.4 | 183.7 | |
| 3TS3 | 151.8 | 188.2 | |
| 3TS4 | 193.2 | 188.8 | |
| 3TS5 | 210.6 | 187.2 | |
| 3TS6 | 55.4 | 176.8 | |
| 3TS7 | 49.6 | 176.7 | |
Table 3.
The excitation energy TV (in eV), oscillator strength f (in atomic units) and wavelength λ (in nm) of the first five excited states of C2H5O3Br, C2H5O2BrO and C2H5OBrO2 at the TD-B3LYP/6-311++(2df,2p) level of theory.
Table 3.
The excitation energy TV (in eV), oscillator strength f (in atomic units) and wavelength λ (in nm) of the first five excited states of C2H5O3Br, C2H5O2BrO and C2H5OBrO2 at the TD-B3LYP/6-311++(2df,2p) level of theory.
| Excited States | C2H5O3Br | C2H5O2BrO | C2H5OBrO2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TV | f | λ | TV | f | λ | TV | f | λ | |
| 1 | 3.02 | 0.0002 | 410.2 | 2.23 | 0.0000 | 555.1 | 4.621 | 0.0019 | 268.3 |
| 2 | 3.77 | 0.0020 | 328.5 | 3.94 | 0.0017 | 314.4 | 4.93 | 0.0240 | 251.5 |
| 3 | 4.32 | 0.0046 | 287.0 | 4.07 | 0.1354 | 304.3 | 5.11 | 0.0001 | 242.8 |
| 4 | 4.57 | 0.1277 | 271.2 | 4.15 | 0.0019 | 299.0 | 5.131 | 0.0023 | 241.6 |
| 5 | 5.32 | 0.0142 | 233.1 | 5.74 | 0.0038 | 216.1 | 5.891 | 0.0712 | 210.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lu, C.; Tang, Y.; Zhang, W.; Qu, X.; Fu, Z. Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere. Molecules 2018, 23, 1268. https://doi.org/10.3390/molecules23061268
AMA Style
Lu C, Tang Y, Zhang W, Qu X, Fu Z. Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere. Molecules. 2018; 23(6):1268. https://doi.org/10.3390/molecules23061268
Chicago/Turabian StyleLu, Chenggang, Yizhen Tang, Wei Zhang, Xunshuai Qu, and Zhihao Fu. 2018. "Theoretical Investigations on Mechanisms and Pathways of C2H5O2 with BrO Reaction in the Atmosphere" Molecules 23, no. 6: 1268. https://doi.org/10.3390/molecules23061268