Semisynthesis, Characterization and Evaluation of New Adenosine Derivatives as Antiproliferative Agents

Abstract
:1. Introduction
2. Results and Discussion
2.1. Molecular Docking of Adenosine Derivatives
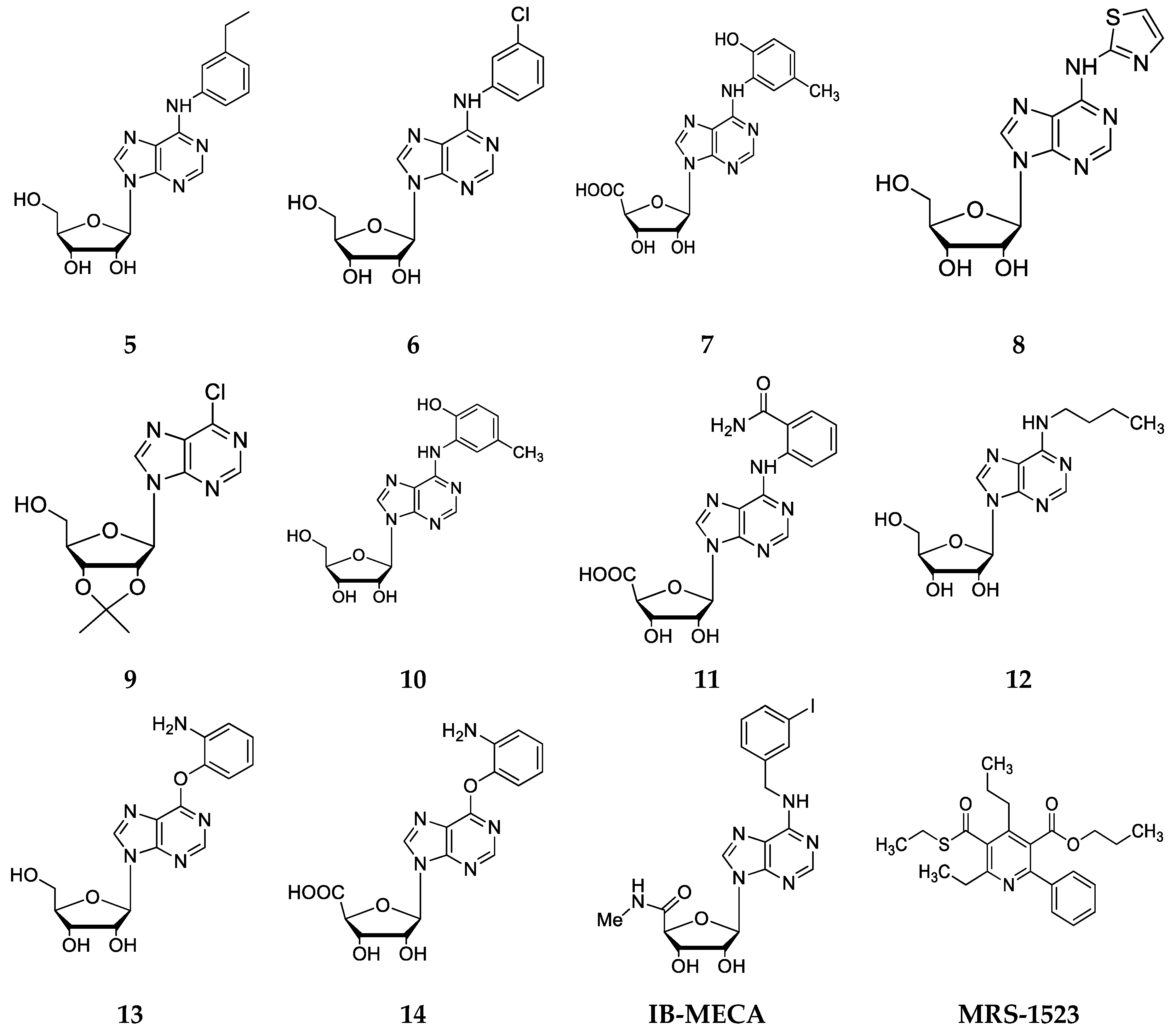
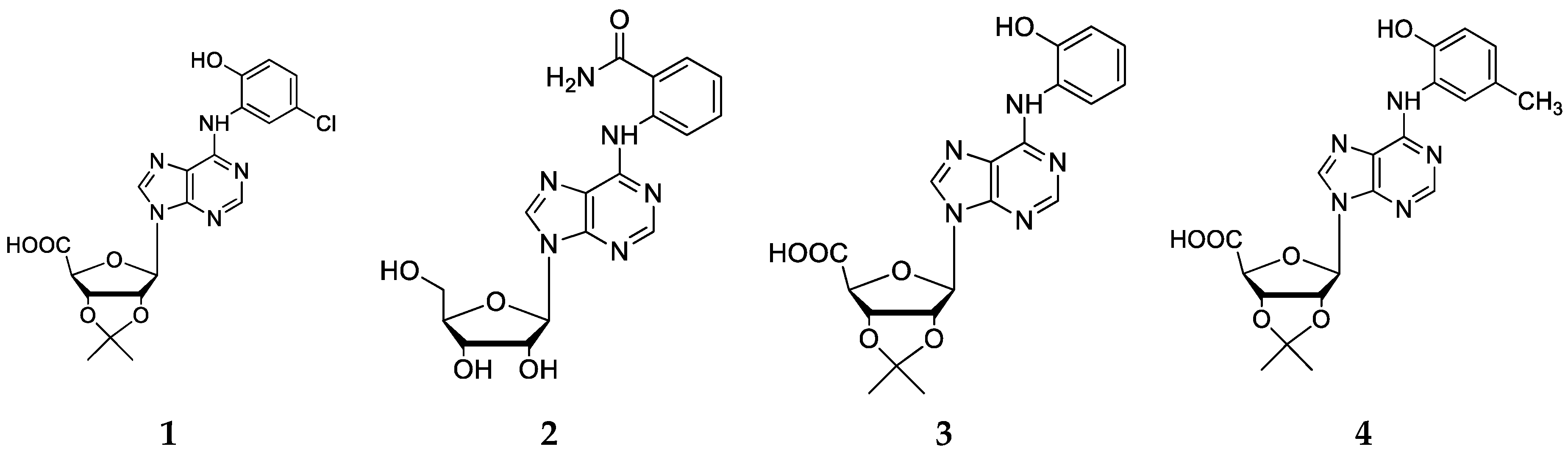
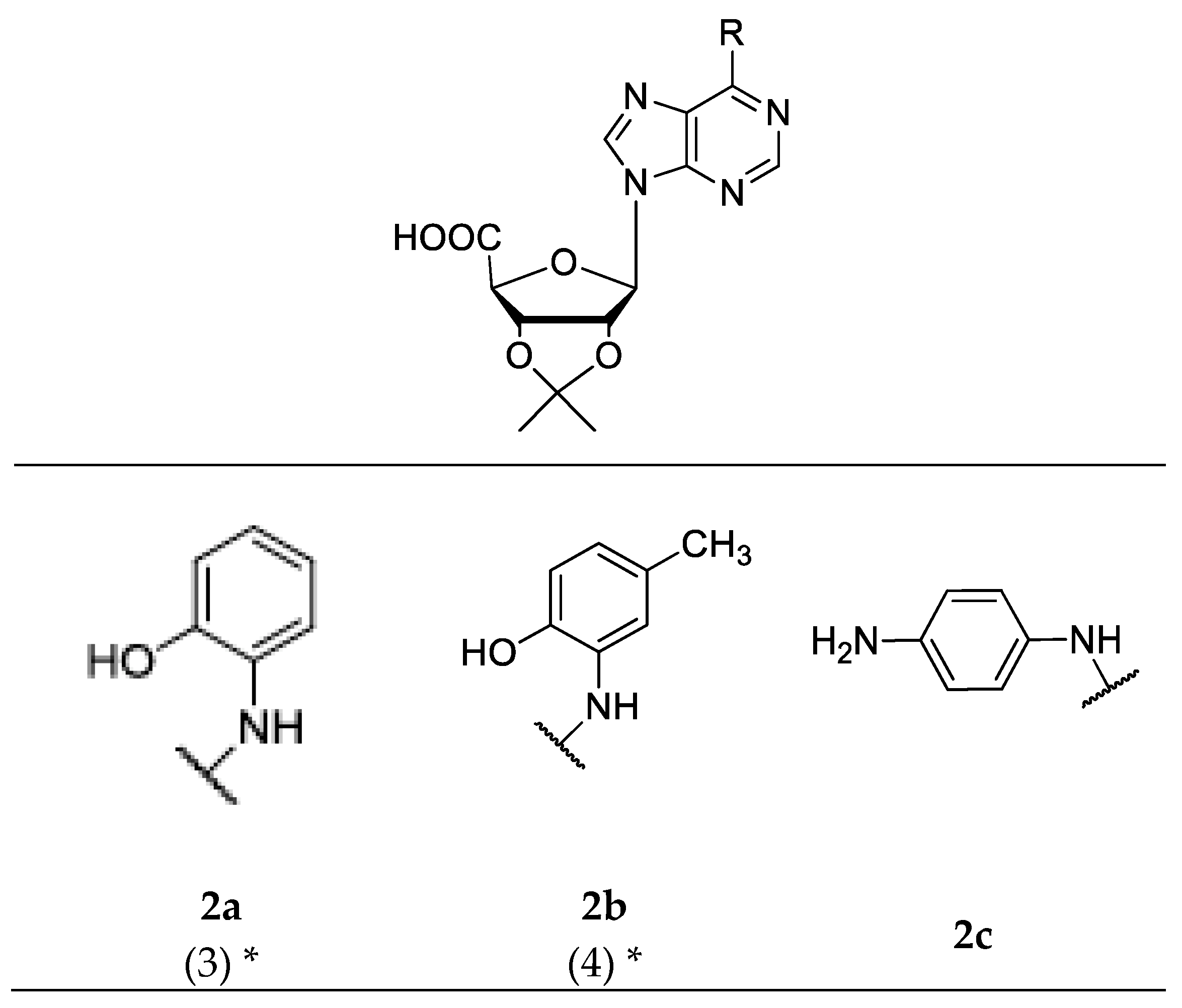
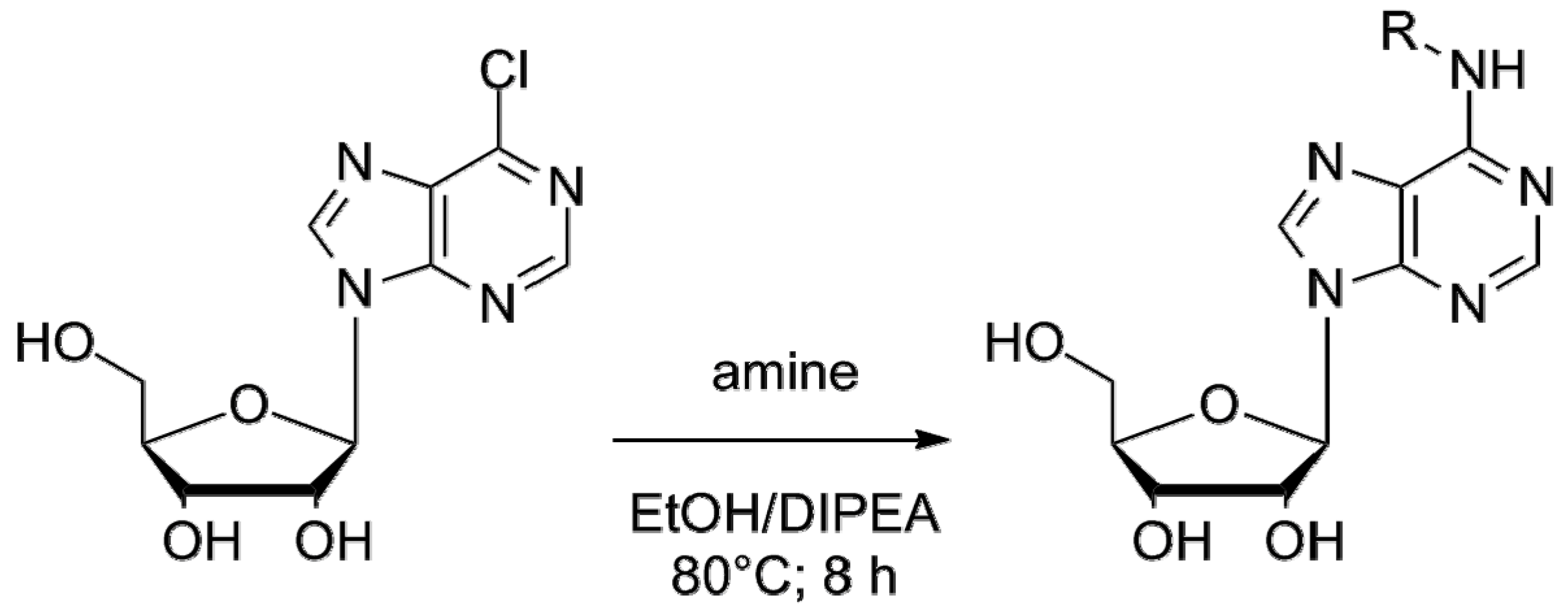
2.2. Chemical Synthesis
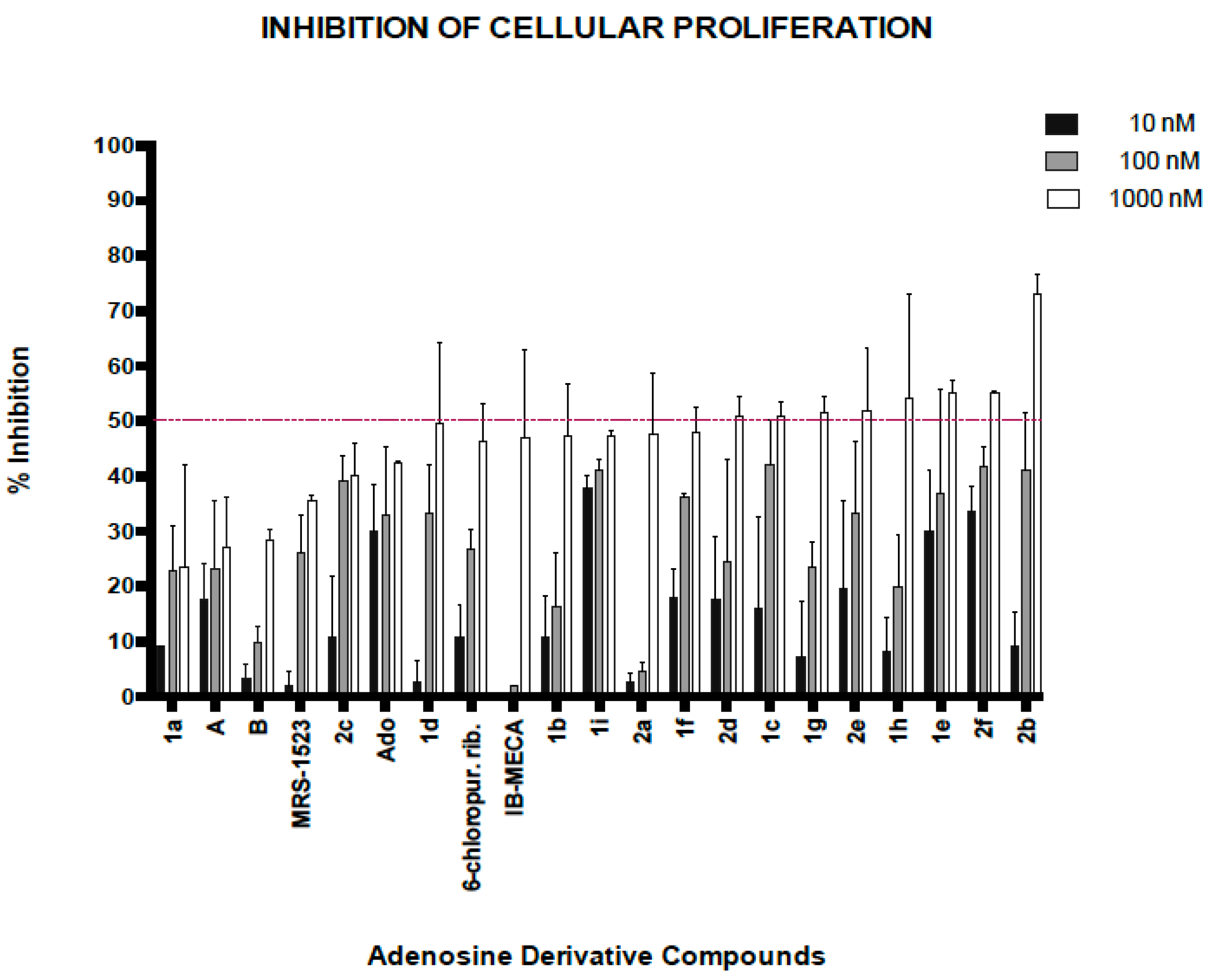
2.3. Biological Activity of Adenosine Derivatives
3. Materials and Methods
3.1. Molecular Docking’s Study of Adenosine Analogues
3.2. Selection of the Best Conformers
3.3. Chemistry
3.4. Synthesis
3.5. Biological Activity
3.5.1. Cancer Cell Line and Cell Culture Conditions
3.5.2. General Screening of Adenosine Derivatives
3.5.3. Theoretical Evaluation of ADME Properties
3.5.4. Statistical Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sanyal, N.K.; Roychoudhury, M.; Ojha, R.P. Biological activity of the nucleoside analogs: A theoretical study. Int. J. Quantum Chem. 1981, 20, 159–166. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447. [Google Scholar] [CrossRef]
- Pertusat, F.; Serpi, M.; McGuigan, C. Medicinal Chemistry of Nucleoside Phosphonate Prodrugs for Antiviral Therapy. Antivir. Chem. Chemother. 2012, 22, 181–203. [Google Scholar] [CrossRef]
- Vitali, L.A.; Petrelli, D.; Lambertucci, C.; Prenna, M.; Volpini, R.; Cristalli, G. In vitro antibacterial activity of different adenosine analogues. J. Med. Microbiol. 2012, 61, 525–528. [Google Scholar] [CrossRef]
- Oslovsky, V.E.; Drenichev, M.S.; Sun, L.; Kurochkin, N.N.; Kunetsky, V.E.; Mirabelli, C.; Neyts, J.; Leyssen, P.; Mikhailov, S.N. Fluorination of naturally occurring N6-benzyladenosine remarkably increased its antiviral activity and selectivity. Molecules 2017, 22, 1219. [Google Scholar] [CrossRef]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524. [Google Scholar] [CrossRef]
- Korelitz, B.I. Expert opinion: Experience with 6-mercaptopurine in the treatment of inflammatory bowel disease. World J. Gastroenterol. 2013, 19, 2979. [Google Scholar] [CrossRef]
- Khan, C.; Pathe, N.; Fazal, S.; Lister, J.; Rossetti, J.M. Azacitidine in the management of patients with myelodysplastic syndromes. Ther. Adv. Hematol. 2012, 3, 355–373. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Justice, R.; Pazdur, R. FDA drug approval summary: Nelarabine (Arranon®) for the treatment of T-cell lymphoblastic leukemia/lymphoma. Oncologist 2008, 13, 709–714. [Google Scholar] [CrossRef]
- Hocek, M. Syntheses of Purines Bearing Carbon Substituents in Positions 2, 6 or 8 by Metal-or Organometal-Mediated C−C Bond-Forming Reactions. Eur. J. Org. Chem. 2003, 2003, 245–254. [Google Scholar] [CrossRef]
- Ottria, R.; Casati, S.; Baldoli, E.; Maier, J.A.; Ciuffreda, P. N6-Alkyladenosines: Synthesis and evaluation of in vitro anticancer activity. Bioorg. Med. Chem. 2010, 18, 8396–8402. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; An, H.; Hatala, P.J.; Stevens, W.C., Jr.; Tao, J.; He, B. Versatile synthesis and biological evaluation of novel 3′-fluorinated purine nucleosides. Beilstein J. Org. Chem. 2015, 11, 2509. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Cacciari, B.; Dal Ben, D.; Lambertucci, C.; Moro, S.; Spalluto, G.; Volpini, R. Highlights on the development of A2A adenosine receptor agonists and antagonists. Chem. Med. Chem. 2007, 2, 260–281. [Google Scholar] [CrossRef] [PubMed]
- Ernst, D.; Schäfer, W.; Oesterhelt, D. Isolation and identification of a new, naturally occurring cytokinin (6-benzylaminopurineriboside) from an anise cell culture (Pimpinella Anisum L.). Planta 1983, 159, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Yong, J.W.H.; Goh, N.K.; Chia, L.S.; Tan, S.N.; Ong, E.S. Identification of kinetin and kinetin riboside in coconut (Cocos nucifera L.) water using a combined approach of liquid chromatography–tandem mass spectrometry, high performance liquid chromatography and capillary electrophoresis. J. Chromatogr. B 2005, 829, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Young, D.C.; Layre, E.; Pan, S.-J.; Tapley, A.; Adamson, J.; Seshadri, C.; Wu, Z.; Buter, J.; Minnaard, A.J.; Coscolla, M. In vivo biosynthesis of terpene nucleosides provides unique chemical markers of Mycobacterium tuberculosis infection. Chem. Biol. 2015, 22, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Bridges, A.J.; Bruns, R.F.; Ortwine, D.F.; Priebe, S.R.; Szotek, D.L.; Trivedi, B.K. N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)ethyl] adenosine and its uronamide derivatives. Novel adenosine agonists with both high affinity and high selectivity for the adenosine A2 receptor. J. Med. Chem. 1988, 31, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Volpini, R.; Costanzi, S.; Lambertucci, C.; Taffi, S.; Vittori, S.; Klotz, K.-N.; Cristalli, G. N6-Alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J. Med. Chem. 2002, 45, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Stemmer, S.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.; Fishman, P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011, 226, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Jung, J.-Y.; Cho, S.-D.; Hong, K.-A.; Kim, H.-J.; Shin, D.-H.; Kim, H.; Kim, H.O.; Shin, D.H.; Lee, H.W. The antitumor effect of LJ-529, a novel agonist to A3 adenosine receptor, in both estrogen receptor–positive and estrogen receptor–negative human breast cancers. Mol. Cancer Ther. 2006, 5, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Actis, A.; Croci, M.; Bergoc, R. Efectos farmacológicos de un análogo de adenosina-3′,5′-monofosfato cíclico sobre útero de ratones: Consideraciones terapéuticas. Acta Bioquim. Clin. Latinoam. 2005, 39, 197–202. [Google Scholar]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.; Klotz, K.; Gessi, S.; Borea, P. Adenosine receptors and cancer. Handb. Exp. Pharmacol. 2009, 193, 399–441. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Varani, K.; Merighi, S.; Cattabriga, E.; Iannotta, V.; Leung, E.; Baraldi, P.G.; Borea, P.A. A3 adenosine receptors in human neutrophils and promyelocytic HL60 cells: A pharmacological and biochemical study. Mol. Pharmacol. 2002, 61, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Valdés, F.; Luna, V.; Arévalo, B.; Brown, N.; Gutiérrez, M. Docking’s study of 6-substituted adenosine-5′-N-ethyluronamide analogs as selective agonists at A3 adenosine receptor and selection of compounds for to be synthesized later. Der Pharma Chem. 2015, 61, 212–225. [Google Scholar]
- Costanzi, S.; Lambertucci, C.; Vittori, S.; Volpini, R.; Cristalli, G. 2-and 8-alkynyladenosines: Conformational studies and docking to human adenosine A3 receptor can explain their different biological behavior. J. Mol. Graph. Model. 2003, 21, 253–262. [Google Scholar] [CrossRef]
- Van der Wenden, E.; Künzel, J.; Mathôt, R.; Ijzeman, A.; Danhof, M.; Soudijn, W. FC20 ribose modified adenosine analogues as potential partial agonists for the adenosine receptor. Eur. J. Pharm. Sci. 1994, 2, 105. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Kim, S.-K.; Biadatti, T.; Chen, W.; Lee, K.; Barak, D.; Kim, S.G.; Johnson, C.R.; Jacobson, K.A. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J. Med. Chem. 2002, 45, 4471–4484. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Gallicchio, E.; Friesner, R.A.; Levy, R.M. Solvent models for protein–ligand binding: Comparison of implicit solvent Poisson and surface generalized Born models with explicit solvent simulations. J. Comput. Chem. 2001, 22, 591–607. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Ohana, G.; Pathak, S.; Wasserman, L.; Barer, F.; Multani, A. Adenosine acts as an inhibitor of lymphoma cell growth: A major role for the A3 adenosine receptor. Eur. J. Cancer 2000, 36, 1452–1458. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Bratislava, Slovak Republic. Available online: http://www.molinspiration.com/cgi-bin/properties (accessed on 11 December 2017).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Release, S. 1: Desmond Molecular Dynamics System, version 3.7. Epik Version 2.2; Glide, Version 5.7; Impact, Version 5.7; LigPrep, Version 2.5; Phase, Version 3.3; Prime, Version 2.3; SiteMap, Version 2.5; Virtual Screening Workflow. DE Shaw Research: New York, NY, USA, Maestro-Desmond Interoperability Tools. Schrödinger: New York, NY, USA, 2014. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W.; Padgett, W.; Thompson, R.D.; Kusachi, S.; Bugni, W.J.; Olsson, R.A. Structure-activity relationships for N6-substituted adenosines at a brain A1-adenosine receptor with a comparison to an A2-adenosine receptor regulating coronary blood flow. Biochem. Pharmacol. 1986, 35, 2467–2481. [Google Scholar] [CrossRef]
- De Zwart, M.; De Groote, M.; Van der Klein, P.A.; Van Dun, S.; Bronsing, R.; Von Frijtag Drabbe Künzel, J.K.; Ijzerman, A.P. Phenyl-substituted N6-phenyladenosines and N6-phenyl-5′-N-ethylcarboxamidoadenosines with high activity at human adenosine A2B receptors. Drug Dev. Res. 2000, 49, 85–93. [Google Scholar] [CrossRef]
- Kwatra, M.M.; Leung, E.; Hosey, M.M.; Green, R.D. N6-Phenyladenosines: Pronounced effect of phenyl substituents on affinity for A2 adenosine receptors. J. Med. Chem. 1987, 30, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kahn, N.; Hargrave, P.A. Nucleoside inhibitors of rhodopsin kinase. Biochemistry 1990, 29, 6276–6282. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.O.; Ji, X.-D.; Siddiqi, S.M.; Olah, M.E.; Stiles, G.L.; Jacobson, K.A. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994, 37, 3614–3621. [Google Scholar] [CrossRef] [PubMed]
- Epp, J.B.; Widlanski, T.S. Facile preparation of nucleoside-5′-carboxylic acids. J. Org. Chem. 1999, 64, 293–295. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Energies | |
|---|---|---|
| Binding Energy (Kcal/mol) | ∆Gb (Kcal/mol) | |
| 1 | −44.94 | −84.71 |
| 2 | −41.69 | −79.75 |
| 3 | −38.72 | −78.28 |
| 4 | −40.73 | −77.74 |
| 5 | −44.56 | −76.52 |
| 6 | −43.68 | −76.07 |
| 7 | −38.71 | −70.76 |
| 8 | −37.88 | −69.60 |
| 9 | −30.06 | −68.64 |
| 10 | −38.95 | −68.29 |
| 11 | −46.53 | −67.00 |
| 12 | −35.32 | −66.36 |
| 13 | −37.20 | −64.83 |
| 14 | −37.41 | −57.93 |
| IB-MECA | −47.29 | −67.391 |
| MRS-1523 | −23.46 | −77.363 |
| Compound | Log P | Molecular Weight | TPSA | n-ON Acceptors | n-OHNH Donors | Volume |
|---|---|---|---|---|---|---|
| 1e | 1.09 | 358.36 | 151.58 | 10 | 6 | 302.34 |
| 1h | 1.70 | 403.39 | 144.02 | 11 | 4 | 342.14 |
| 2b | 2.11 | 426.43 | 140.87 | 11 | 3 | 358.53 |
| 2f | 2.85 | 447.83 | 140.87 | 11 | 3 | 353.32 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdés Zurita, F.; Brown Vega, N.; Gutiérrez Cabrera, M. Semisynthesis, Characterization and Evaluation of New Adenosine Derivatives as Antiproliferative Agents. Molecules 2018, 23, 1111. https://doi.org/10.3390/molecules23051111
Valdés Zurita F, Brown Vega N, Gutiérrez Cabrera M. Semisynthesis, Characterization and Evaluation of New Adenosine Derivatives as Antiproliferative Agents. Molecules. 2018; 23(5):1111. https://doi.org/10.3390/molecules23051111
Chicago/Turabian StyleValdés Zurita, Francisco, Nelson Brown Vega, and Margarita Gutiérrez Cabrera. 2018. "Semisynthesis, Characterization and Evaluation of New Adenosine Derivatives as Antiproliferative Agents" Molecules 23, no. 5: 1111. https://doi.org/10.3390/molecules23051111
APA StyleValdés Zurita, F., Brown Vega, N., & Gutiérrez Cabrera, M. (2018). Semisynthesis, Characterization and Evaluation of New Adenosine Derivatives as Antiproliferative Agents. Molecules, 23(5), 1111. https://doi.org/10.3390/molecules23051111