Synthesis of New Benzothiazole Acylhydrazones as Anticancer Agents
by
 and
and
Derya Osmaniye
1,2, 
Serkan Levent
1,2,
Abdullah Burak Karaduman
3,
Sinem Ilgın
3,
Yusuf Özkay
1,2,* and
Zafer Asım Kaplancıklı
1 1
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Anadolu University, 26470 Eskişehir, Turkey
2
Doping and Narcotic Compounds Analysis Laboratory, Faculty of Pharmacy, Anadolu University, 26470 Eskişehir, Turkey
3
Department of Pharmaceutical Toxicology, Faculty of Pharmacy, Anadolu Universty, 26470 Eskişehir, Turkey
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(5), 1054; https://doi.org/10.3390/molecules23051054
Submission received: 22 March 2018
/
Revised: 27 April 2018
/
Accepted: 27 April 2018
/
Published: 1 May 2018
(This article belongs to the Section Medicinal Chemistry)
Abstract
:During the last five decades, a large number of BT (Benzothiazole) derivatives formed one of the eligible structures in medicinal chemistry as anticancer agents. Most of the studies reveal that various substitutions at specific positions on BT scaffold modulate the antitumor property. The potential of BTs encouraged us to synthesize a number of new 2-((5-substitutedbenzothiazol-2-yl)thio)-N’-(2-(4-(substitutedphenyl)ethylidene)acetohydrazide derivatives and investigate their probable anticancer activity. 4-Substitued benzaldehyde derivatives (1a–1e) were afforded by the reaction of appropriate secondary amine and 4-fluorobenzaldehyde in DMF. Equimolar quantitates of 5-substitutedbenzothiazole-2-thiol, ethyl chloroacetate and K2CO3 were refluxed in acetone to obtain 2-((5-substitutedbenzothiazol-2-yl)thio)acetate derivatives (2a,2b), which reacted with excess of hydrazine hydrate to get 2-((5-substitutebenzothiazol-2-yl)thio)acetohydrazides (3a,3b). In the last step, 2-((5-substitutedbenzothiazol-2-yl)thio)-N’-(4-substitutedbenzylidene)acetohydrazide derivatives (4a–4j) were synthesized by the reaction of 1a–1e and 3a–3b in EtOH. The anticancer activity of target compounds was evaluated in three steps. First, an MTT test (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was performed to observe cytotoxic activity of the compounds against carcinogenic C6 (Rat brain glioma cell line), A549 (Human lung adenocarcinoma epithelial cell line), MCF-7 (Human breast adenocarcinoma cell line), and HT-29 (Human colorectal adenocarcinoma cell line) cancer cell lines. Healthy NIH3T3 (Mouse embryo fibroblast cell line) cells were also subjected to MTT assay to determine selectivity of the compounds towards carcinogenic cell lines. Secondly, inhibitory effects of selected compounds 4d, 4e, and 4h on DNA synthesis of C6 cells were investigated. Finally, flow cytometric analysis were performed to identify the death pathway of the carcinogenic cells.
1. Introduction
Cancer is still one of the most common reasons of death worldwide. In spite of the widespread research and fast changes in the cancer therapy, there is still a rising need for new treatments [1]. Discovery of new chemotherapeutics are of principal importance due to the essential capability of tumour cells to develop resistance to current agents. The progress of multiple drug resistance to antitumor drugs is a major problem in the chemotherapy. Hence, research for the recognition of novel agents for the managing of cancer is at the critical level [2].
Benzothiazole (BT) compounds display the anticancer activity by acting on various biological targets. Some important instances related to these biotargets are replication and mitosis inhibitors [3], inhibitors of thioredoxin signaling system [4], topoisomerase II inhibitor [5], cytochrome P450 (CYP) inhibitors [6,7,8], tyrosine kinase inhibitors [9], heat shock protein 90 (Hsp90) inhibitors [10], epidermal growth factor receptor (EGFR) inhibitors [11], and cathepsin D inhibitors [12].
Rational drug design is a strategy that is applied in the discovery of novel lead drugs. Drug design methodologies can be classified as ligand based and structure based. The original hit ring system, BT, was extensively modified through a structure-based drug design approach. Previous studies reveal that various groups at specific positions, particularly at the C2 and C6, on BT scaffold modulate the antitumor potency of the scaffold [13]. Most of the reported compounds with an anticancer activity possess a phenyl moiety directly linked to C2 or through a linker group [14]. A literature survey shows that the compounds include amide, urea, and semicarbazone moieties as linkers between BT and phenyl have significant anticancer activity [13,15,16,17].
N-acylarylhydrazone scaffold has been widely used as a building block in several antitumor agents owing to its flexible skeleton and the presence of both hydrogen donors and acceptors [15]. Most of the antitumor acylhydrazones have been observed through inducing apoptosis in diverse carcinogenic cells [18,19]. In addition, it has been reported that the compounds include BT and hydrazone moieties on the same structure have significant anticancer activity [20,21,22,23,24,25].
In light of the knowledge above and with a goal to discover a novel class of anticancer agents, we report herein the synthesis and antitumor effect of new compounds, including acylhydrazone moiety as a linker between BT and phenyl scaffolds.
2. Results and Discussion
2.1. Chemistry
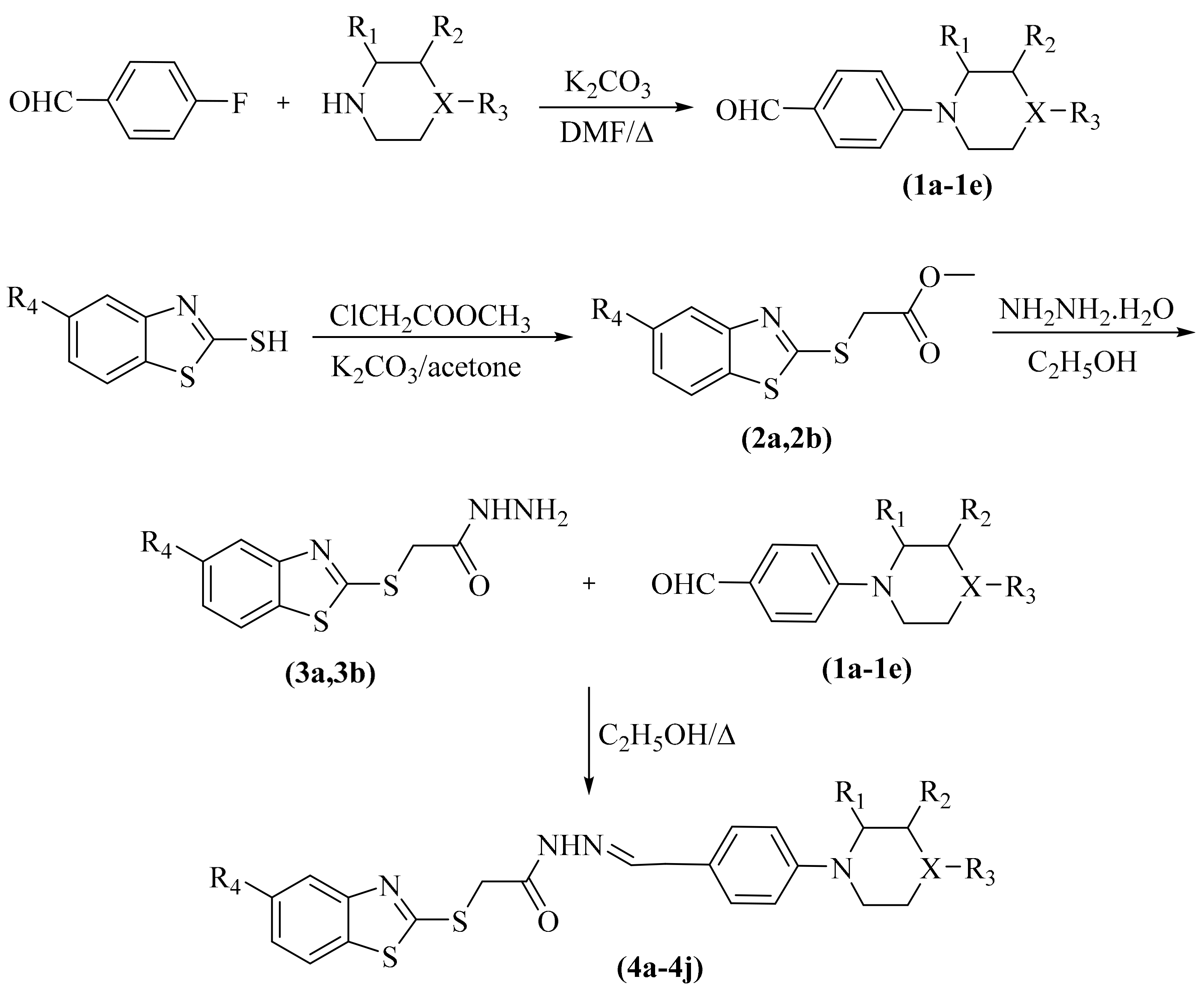
The compounds 4a–4j were synthesized as summarized in Scheme 1. Initially, 4-substituted benzaldehydes (1a–1e) were synthesized under reflux by reaction of 4-fluorobenzaldehyde and appropriate secondary amine as reported previously [26]. Secondly, 5-substitutedbenzothiazole and ethyl chloroacetate were refluxed in acetone to obtain ethyl 2-(5-substitutedbenzothiazol-2-yl-thio)acetates (2a,2b), which reacted with excess of hydrazine hydrate to afford 2-((5-substitutedbenzothiazol-2-yl)thio)acetohydrazides (3a,3b) as described previously [27]. Finally, compounds 3a and 3b were reacted with appropriate 4-seconadaryaminesubstitutedbenzaldehydes (1a–1e) to gain the target compounds 4a–4j.
Structure explanations of the final compounds were performed with IR, 1H-NMR, 13C-NMR, and HRMS spectroscopic methods (See Supplementary Materials). In the IR spectra of the compounds, stretching absorptions at 3165–3277 cm−1 proved N-H bond of the hydrazide groups. Signals belonging to carbonyl (C=O) function appeared at 1653–1664 cm−1. The stretching absorption at about 1193–1233 cm−1 were registered for C-N single bonds. The out of plane bending belonging to 1,4-disubstituted benzene was determined at 794–833 cm−1. In the 1H-NMR spectra, aliphatic protons belonging to piperidine and piperazine rings were recorded between 1.01 ppm and 4.20 ppm. Methyl groups on the piperidine ring gave doublet peaks between 0.89 ppm and 1.00 ppm. Methoxy substituents on benzothiazole and phenyl rings were observed as singlet peaks at 3.81 ppm and 3.69 ppm, respectively. Protons of methylene bridge were recorded as a singlet between 4.62 ppm and 4.66 ppm. Aromatic protons gave peaks between 6.84 ppm and 8.06 ppm. Peak splitting pattern of benzothiazole rings were recorded as two doublets with different coupling constant and one doublet of doublet, while phenyl rings gave two doublets with the same coupling constant. Protons on the azomethine moiety were observed as a singlet peak between 7.90 ppm and 7.95 ppm. N-H protons on the hydrazide moiety were recorded over 11.48 ppm. In 13C-NMR, aliphatic carbons were recorded between 13.37 ppm and 55.99 ppm. All aromatic carbons gave peaks from 105.03 to 159.17 ppm. C2 of benzothiazole was recorded around 162 ppm and carbonyl group gave peak over 168 ppm. In the HRMS spectra, all masses were matched well with the expected M + H values.
2.2. Biological Evaluation
2.2.1. Cytotoxicity Test
MTT assay, depending on the capability of metabolically active cells that change the pale yellow MTT colour to a spectrophotometrically measured blue formazan salt, is one of the most favoured cytotoxicity tests [28]. We assessed the antitumor potential of new benzothiazole-acylhydrazones on A549, C6, MCF-7, and HT-29 cell lines with various concentrations (1 mM, 0.316 mM, 0.1 mM, 0.0316 mM, 0.01 mM, 0.00316 mM, 0.001 mM, 0.000316 mM). In addition, the cytotoxic activities of compounds 4a–4j were evaluated against healthy NIH3T3 cells, owing to show the selectivity towards carcinogenic cells. The IC50 values of the compounds against cell lines are represented in Table 1.
Compound 4e was found to have the highest cytotoxic activity against A549 cell lines. Moreover, the IC50 value of (0.03 mM) this compound against A549 cells was twofold lower than that of cisplatin (0.06 mM). However, the other compounds in the series could not display a significant cytotoxic activity against A549 cell lines.
Compounds 4d, 4e, 4h showed remarkable cytotoxicity against C6 cell lines. These compounds indicated equal IC50 value (0.03 mM) to cisplatin against C6 cell lines (Figure 1).
Compounds 4c and 4d were the most active compounds against both MCF-7 and HT-29 cell lines. However, these compounds indicated lower cytotoxic activity than the reference agent cisplatin.
In general, it has been determined that 5-chloro substitution of BT enhances the cytotoxic activity against carcinogenic cell lines. In addition, comparing 2, 3, or 4-methly substitution of piperidine indicated that three or four substituents makes more contribution to the cytotoxic activity against all carcinogenic cell lines.
One of the important criteria as to being anticancer agent candidates is to display minimum or no side-effects on healthy cells. Hence, cytotoxicity of synthesized compounds 4a–4j against NIH3T3 cell line was determined. As seen in Table 1, compound 4e showed higher cytotoxicity against NIH3T3 cell lines when compared to carcinogenic cell lines. On the other hand, IC50 values of compound 4h against C6 cells were higher than its IC50 values against NIH3T3 cell lines. Moreover, compound 4d indicated lower cytotoxicity against NIH3T3 cells when compared with both carcinogenic cell lines. This finding improved the importance of compound 4d as a potential anticancer agent.
Comparing A549, C6, MCF-7, and HT-29 cell lines, the C6 cell line was found to be more susceptible to compounds. Therefore, the C6 cell line was used in further tests including DNA synthesis inhibition and flow cytometric analysis assays.
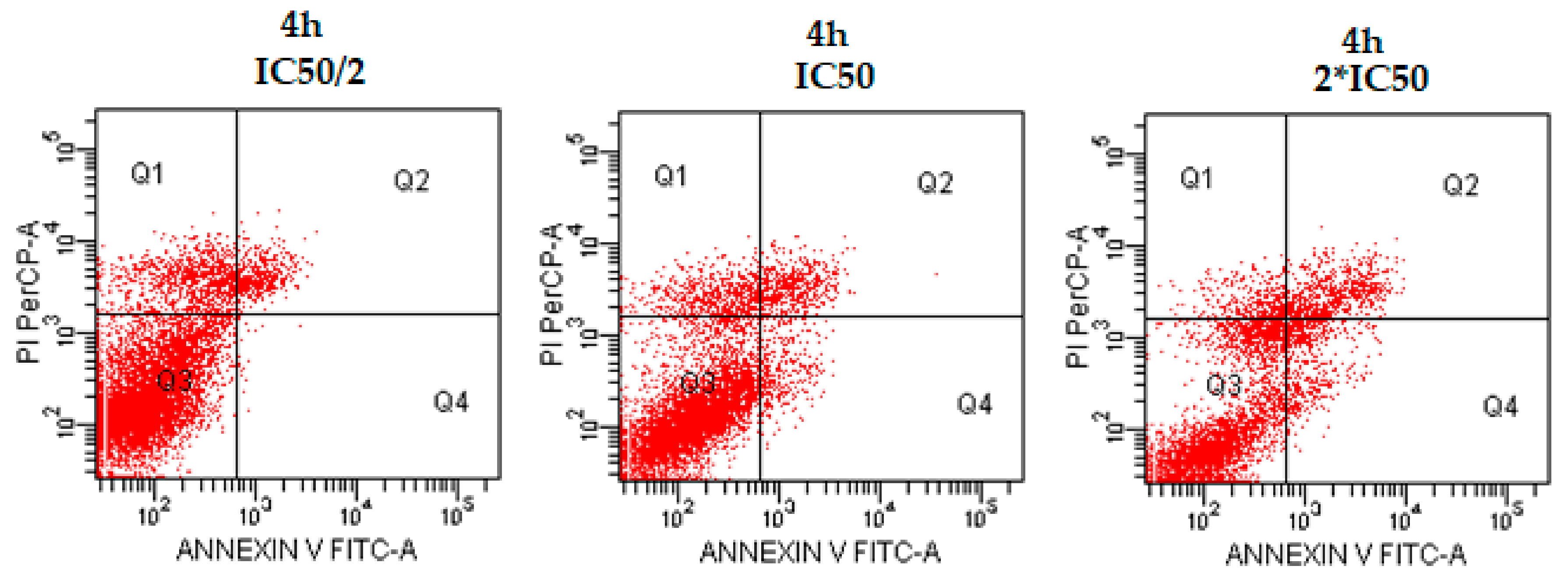
2.2.2. DNA Synthesis Inhibition Assay
This immunostaining assay is related to detecting the attendance of BrdU into nuclear DNA instead of thymidine during S-phase of the cell cycle using specific anti-BrdU antibodies [29,30]. Thus, this study was performed to evaluate inhibitory effects of compounds 4d, 4e, and 4h on proliferation of C6 cell lines. Test compounds and reference agent cisplatin were used at the concentrations of IC50/4, IC50/2, IC50, 2 × IC50, and 4 × IC50. Figure 2 represents the DNA synthesis inhibitory effect of compounds 4d, 4e, and 4h on C6 cells.
Tested compounds exhibited concentration-dependent inhibitory activity on DNA synthesis of C6 cell lines. When compared the DNA synthesis inhibitory activity of compounds, it is clear that 4d has higher inhibition potency than cisplatin at all concentrations. Compound 4e also displayed a higher antiproliferative effect than cisplatin at the concentrations of IC50/2, IC50, 2 × IC50, and 4 × IC50. However, compound 4h could not display the same ability as compounds 4d and 4e to inhibit DNA synthesis. According to these results, it can be suggested that compound 4d possesses a higher antiproliferative activity against C6 cell lines than cisplatin.
2.2.3. Flow Cytometric Analysis
In the therapy of cancer, apoptosis, or programmed cell death, is one of the distinguished mechanisms of cytotoxic agents. It is an important approach to express the cell death pathway and most of the anticancer drugs induce apoptosis in tumor cells. Annexin V-FITC is a reagent that can detect and measure quantitate of apoptotic cells by flow cytometry. Staining cells with PI and Annexin V-FITC show the differences between live, apoptotic, dead, and late apoptotic or necrotic cells [31]. Thus, this method provides evidence on the pathway of cell death.
Flow cytometry studies were performed on C6 cells for compounds 4d, 4e, and 4h and cisplatin with the aim of determining the stimulated cellular death pathway. Selected compounds and cisplatin were used at the concentrations of IC50/2, IC50, and 2 × IC50. The flow cytometric analysis results of compounds 4d, 4e, and 4h and cisplatin for C6 cell line are represented in Table 2.
Additionally, flow cytometric analysis diagrams are presented in Figure 3, Figure 4 and Figure 5. As seen in the figures, compounds 4d, 4e, and 4h exhibited comparable population of apoptotic cells to cisplatin. The percentages of apoptotic cells were determined as 5.5, 19.5, and 12.0% for compounds 4d, 4e, and 4h, respectively, at IC50 concentrations, whereas the percentage of apoptotic cells was determined as 20.9% for cisplatin at IC50 concentration. Compound 4e showed the most apoptotic activity (19.5%) when compared with other compounds at IC50 concentrations (Table 2). While compound 4d induced necrotic cell death in IC50/2 and IC50 concentrations, especially IC50/2 concentrations, compounds 4e and 4h induced necrotic cell death at IC50/2 concentration. At this point, we could emphasise that compound 4d, 4e and 4h induced apoptotic cell death, dose-dependently.
3. Materials and Methods
3.1. Chemistry
All chemicals in this study were obtained either from Sigma-Aldrich (Sigma-Aldrich Corp., St. Louis, MO, USA) or Merck (Merck KGaA, Darmstadt, Germany), and used without further chemical purifications. Melting points of the compounds were measured by using an automatic melting point determination instrument (MP90, Mettler-Toledo, Columbus, OH, USA) and were presented as uncorrected. 1H- and 13C-NMR spectra were recorded in DMSO-d6 by a Bruker digital FT-NMR spectrometer (Bruker Bioscience, Billerica, MA, USA) at 300 MHz and 75 MHz, respectively. The IR spectra of the compounds were recorded using an IRAffinity-1S Fourier transform IR (FTIR) spectrometer (Shimadzu, Tokyo, Japan). HRMS studies were performed on an LCMS-IT-TOF system (Shimadzu, Tokyo, Japan). Chemical purities of the compounds were checked by classical TLC applications performed on silica gel 60 F254 (Merck KGaA, Darmstadt, Germany).
3.1.1. General Synthesis Procedure for 4-Substitutebenzaldehyde Derivatives (1a–1e)
Appropriate secondary amine (0.007 mol), 4-fluorobenzaldehyde (0.007 mol, 0.75 mL) and K2CO3 (0.007 mol, 0.96 g) were refluxed in DMF (10 mL) for 10 h. The mixture was cooled with ice-water (50 mL), a precipitated product was filtered, washed with deionised water, dried, and recrystallized from EtOH to give compounds 1a–1e in the yields range of 63 to 72%.
3.1.2. General Procedure for Ethyl 2-(5-Substitutedbenzothiazol-2-yl-thio)acetate (2a–2b)
Corresponding 5-substitutedbenzothiazol-2-thiol (0.015 mol), ethyl 2-chloroacetate, (0.015 mol, 1.59 g), and K2CO3 (0.015 mol, 2.07 g) were refluxed in acetone for 2h. The solvent was removed under reduced pressure and the residue was recrystallized from EtOH to give compounds 2a and 2b with yields of 78% and 81%, respectively.
3.1.3. General Procedure for 2-((5-Substitutedbenzothiazol-2-yl)thio)acetohydrazide (3a–3b)
Hydrazine hydrate (5 mL) was added into EtOHic solution (20 mL) of 5-substituted ethyl 2-((benzothiazol-2-yl)thio)acetate (2a–2b) (0.012 mol) in portions with constant stirring. Then, the mixture was stirred for 3 h. The precipitated product was filtered, dried and recrystallized from EtOH to give compounds 3a and 3b with yields of 84% and 82%, respectively.
3.1.4. General Synthesis Procedure of Compounds (4a–4j)
Appropriate 2-((5-substitutedbenzothiazol-2-yl)thio) acetohydrazide (3a–3b) (0.002 mol), 4-substitutedbenzaldehyde (0.002 mol) and glacial acetic acid (0.1 mL) were refluxed in EtOH for 2 h. The mixture was cooled down in an ice-bath, precipitated product was filtered, dried and recrystallized from EtOH.
2-((5-Chlorobenzothiazol-2-yl)thio)-N’-(4-(piperidin-1-yl)benzylidene)acetohydrazide (4a). Yield: 83%, M.P. = 178–179 °C, FTIR (ATR, cm−1): 3277 (N-H), 1654 (C=O), 1232 (C-N), 1022, 813, 794. 1H-NMR (300 MHz, DMSO-d6): δ = 1.57 (6H, br.s, piperidine), 3.25 (4H, br.s, piperidine), 4.66 (2H, s, -CH2-), 6.91 (2H, d, J = 8.86 Hz, Ar-H), 7.42 (1H, dd, J1 = 2.10 Hz, J2 = 8.55 Hz, BT-H), 7.49 (2H, d, J = 8.86 Hz, Ar-H), 7.91 (1H, s, -CH=N-), 7.93 (1H, d, J = 2.10 Hz, BT-H), 8.05 (1H, d, J = 8.55 Hz, BT-H), 11.52 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 24.4, 25.4, 35.8, 48.9, 115.0, 121.1, 123.7, 124.9, 128.6, 131.7, 134.0, 144.9, 148.2, 152.8, 153.9, 162.7, 168.1. HRMS (m/z): [M + H]+ calcd for C21H21N4OS2Cl: 445.0918; found: 445.0905.
2-((5-Chlorobenzothiazol-2-yl)thio)-N’-(4-(2-methylpiperidin-1-yl)benzylidene)acetohydrazide (4b). Yield: 79%, M.P. = 138–139 °C, FTIR (ATR, cm−1): 3255 (N-H), 1654 (C=O), 1193 (C-N), 1068, 813, 794. 1H-NMR (300 MHz, DMSO-d6): δ = 1.00 (3H, d, J = 6.63 Hz, -CH3), 1.45–1.56 (4H, m, piperidine), 1.70–1.74 (2H, m, piperidine), 2.83–2.91 (1H, m, piperidine), 3.42–3.49 (1H, m, piperidine), 4.19–4.20 (1H, m, piperidine), 4.66 (2H, s, -CH2-), 6.87 (2H, d, J = 8.80 Hz, Ar-H), 7.42 (1H, dd, J1 = 2.10 Hz, J2 = 8.55 Hz, BT-H), 7.47 (2H, d, J = 8.80 Hz, Ar-H), 7.91 (1H, s, -CH=N-), 7.93 (1H, d, J = 2.10 Hz, BT-H), 8.05 (1H, d, J = 8.55 Hz, BT-H), 11.49 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 13.4, 18.7, 25.8, 30.8, 35.8, 41.8, 49.2, 114.9, 121.1, 123.7, 124.9, 128.7, 131.7, 134.0, 144.9, 148.3, 152.2, 153.9, 162.7, 168.1. HRMS (m/z): [M + H]+ calcd for C22H23N4OS2Cl: 459.1075; found: 459.1067.
2-((5-Chlorobenzothiazol-2-yl)thio)-N’-(4-(3-methylpiperidin-1-yl)benzylidene)acetohydrazide (4c). Yield: 81%, M.P. = 149–150 °C, FTIR (ATR, cm−1): 3240 (N-H), 1654 (C=O), 1230 (C-N), 1022, 792. 1H-NMR (300 MHz, DMSO-d6): δ = 0.91 (3H, d, J = 6.51 Hz, -CH3), 1.01–1.13 (1H, m, piperidine), 1.45–1.77 (4H, m, piperidine), 2.36–2.43 (1H, m, piperidine), 2.65–2.74 (1H, m, piperidine), 3.68–3.75 (2H, m, piperidine), 4.66 (2H, s, -CH2-), 6.90 (2H, d, J = 8.88 Hz, Ar-H), 7.41 (1H, dd, J1 = 2.10 Hz, J2 = 8.55 Hz, BT-H), 7.47 (2H, d, J = 8.88 Hz, Ar-H), 7.90 (1H, s, -CH=N-), 7.93 (1H, d, J = 2.10 Hz, BT-H), 8.05 (1H, d, J = 8.55 Hz, BT-H), 11.51 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 19.7, 24.8, 30.5, 32.9, 35.8, 48.3, 55.7, 114.9, 121.1, 123.7, 124.9, 128.7, 131.7, 134.0, 144.9, 148.2, 152.6, 153.9, 162.7, 168.1. HRMS (m/z): [M + H]+ calcd for C22H23N4OS2Cl: 459.1075; found: 459.1063.
2-((5-Chlorobenzothiazol-2-yl)thio)-N’-(4-(4-methylpiperidin-1-yl)benzylidene)acetohydrazide (4d). Yield: 85%, M.P. = 153–154 °C, FTIR (ATR, cm−1): 3255 (N-H), 1660 (C=O), 1226 (C-N), 1022, 817, 792. 1H-NMR (300 MHz, DMSO-d6): δ = 0.90 (3H, d, J = 6.45 Hz, -CH3), 1.09–1.22 (2H, m, piperidine), 1.48–1.55 (1H, m, piperidine), 1.63–1.67 (2H, m, piperidine), 2.68–2.75 (2H, m, piperidine), 3.74–3.79 (2H, m, piperidine), 4.66 (2H, s, -CH2-), 6.89 (2H, d, J = 8.90 Hz, Ar-H), 7.40 (1H, dd, J1 = 2.05 Hz, J2 = 8.55 Hz, BT-H), 7.48 (2H, d, J = 8.90 Hz, Ar-H), 7.89 (1H, d, J = 2.05 Hz, BT-H), 7.91 (1H, s, -CH=N-), 8.03 (1H, d, J = 8.55 Hz, BT-H), 11.53 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 22.2, 30.7, 33.7, 35.8, 48.2, 115.0, 121.1, 123.7, 124.9, 128.6, 131.7, 134.0, 144.9, 148.2, 152.6, 153.9, 162.7, 168.1. HRMS (m/z): [M + H]+ calcd for C22H23N4OS2Cl: 459.1075; found: 459.1062.
2-((5-Chlorobenzothiazol-2-yl)thio)-N’-(4-(4-(4-methoxyphenyl)piperazin-1-yl)benzylidene)acetohydrazide (4e). Yield: 78%, M.P. = 211–212 °C, FTIR (ATR, cm−1): 3201 (N-H), 1653 (C=O), 1226 (C-N), 1031, 825, 721. 1H-NMR (300 MHz, DMSO-d6): δ = 3.14 (4H, br.s, piperazine), 3.38 (4H, br.s, piperazine), 3.69 (3H, s, -OCH3), 4.68 (2H, s, -CH2-), 6.85 (2H, d, J = 9.09 Hz, Ar-H), 6.96 (2H, d, J = 9.09 Hz, Ar-H), 7.04 (2H, d, J = 8.76 Hz, Ar-H), 7.42 (1H, dd, J1 = 2.10 Hz, J2 = 8.55 Hz, BT-H), 7.55 (2H, d, J = 8.76 Hz, Ar-H), 7.94 (1H, d, J = 2.10 Hz, BT-H), 7.95 (1H, s, -CH=N-), 8.05 (1H, d, J = 8.55 Hz, BT-H), 11.57 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 35.8, 47.9, 50.1, 55.6, 114.8, 115.2, 118.2, 121.1, 123.7, 124.9, 128.6, 128.9, 131.7, 134.0, 144.8, 145.7, 148.1, 152.4, 153.7, 162.8, 168.2. HRMS (m/z): [M + H]+ calcd for C27H26N5O2S2Cl: 552.1289; found: 552.1293.
2-((5-Methoxybenzothiazol-2-yl)thio)-N’-(4-(piperidin-1-yl)benzylidene)acetohydrazide (4f). Yield: 82%, M.P. = 138–139 °C, FTIR (ATR, cm−1): 3207 (N-H), 1664 (C=O), 1230 (C-N), 1031, 810, 721. 1H-NMR (300 MHz, DMSO-d6): δ = 1.56 (6H, br.s., piperidine), 3.23 (4H, br.s., piperidine), 3.81 (3H, s, -OCH3), 4.63 (2H, s, -CH2-), 6.90 (2H, d, J = 8.95 Hz, Ar-H), 6.99 (1H, dd, J1 = 2.50 Hz, J2 = 8.80 Hz), 7.41 (1H, d, J = 2.50 Hz, BT-H), 7.48 (2H, d, J = 8.95 Hz, Ar-H), 7.87 (1H, d, J = 8.80 Hz, BT-H), 7.93 (1H, s, -CH=N-), 11.51 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 24.4, 25.4, 35.6, 48.9, 55.9, 105.0, 114.2, 115.1, 122.5, 123.5, 128.6, 144.8, 148.1, 152.8, 154.4, 159.2, 162.9, 168.2. HRMS (m/z): [M + H]+ calcd for C22H24N4O2S2: 441.1413; found: 441.1408.
2-((5-Methoxybenzothiazol-2-yl)thio)-N’-(4-(2-methylpiperidin-1-yl)benzylidene)acetohydrazide (4g). Yield: 79%, M.P. = 138–139 °C, FTIR (ATR, cm−1): 3267 (N-H), 1664 (C=O), 1230 (C-N), 1030, 824, 794. 1H-NMR (300 MHz, DMSO-d6): δ = 1.02 (3H, d, J = 6.60 Hz, -CH3), 1.47–1.58 (4H, m, piperidine), 1.73–1.78 (2H, m, piperidine), 2.83–2.91 (1H, m, piperidine), 3.42–3.49 (1H, m, piperidine), 3.81 (3H, s, -OCH3), 4.19–4.20 (1H, m, piperidine), 4.66 (2H, s, -CH2-), 6.87 (2H, d, J = 8.80 Hz, Ar-H), 6.99 (1H, dd, J1 = 2.50 Hz, J2 = 8.80 Hz), 7.41 (1H, d, J = 2.50 Hz, BT-H), 7.48 (2H, d, J = 8.95 Hz, Ar-H), 7.90 (1H, d, J = 8.80 Hz, BT-H), 7.92 (1H, s, -CH=N-), 11.50 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 13.4, 18.7, 25.8, 30.8, 35.8, 41.8, 49.8, 55.9, 105.1, 114.8, 115.5, 122.6, 123.5, 128.7, 144.4, 148.1, 152.3, 154.8, 159.2, 162.7, 168.1. HRMS (m/z): [M + H]+ calcd for C23H26N4O2S2: 455.1570; found: 455.1564.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N’-(4-(3-methylpiperidin-1-yl)benzylidene)acetohydrazide (4h). Yield: 81%, M.P. = 147–148 °C, FTIR (ATR, cm−1): 3165 (N-H), 1660 (C=O), 1228 (C-N), 1029, 808, 783. 1H-NMR (300 MHz, DMSO-d6): δ = 0.89 (3H, d, J = 6.54 Hz, -CH3), 1.03–1.08 (1H, m, piperidine), 1.48–1.76 (4H, m, piperidine), 2.34–2.42 (1H, m, piperidine), 2.64–2.72 (1H, m, piperidine), 3.66–3.73 (2H, m, piperidine), 3.81 (3H, s, -OCH3), 4.63 (2H, s, -CH2-), 6.89 (2H, d, J = 8.77 Hz, Ar-H), 6.99 (1H, dd, J1 = 2.46 Hz, J2 = 8.80 Hz BT-H), 7.41 (1H, d, J = 2.46 Hz), 7.47 (2H, d, J = 8.77 Hz, Ar-H), 7.85 (1H, d, J = 8.80 Hz, BT-H), 7.93 (1H, s, -CH=N-), 11.52 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 19.6, 24.8, 30.5, 32.9, 35.6, 48.3, 55.7, 105.0, 114.2, 114.9, 122.5, 123.4, 128.6, 144.8, 148.1, 152.5, 154.4, 159.2, 162.9, 168.2. HRMS (m/z): [M + H]+ calcd for C23H26N4O2S2: 455.1570; found: 455.1569.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N’-(4-(4-methylpiperidin-1-yl)benzylidene)acetohydrazide (4i). Yield: 79%, M.P. = 149–150 °C, FTIR (ATR, cm−1): 3215 (N-H), 1664 (C=O), 1228 (C-N), 1031, 833, 804. 1H-NMR (300 MHz, DMSO-d6): δ = 0.92 (3H, d, J = 6.39 Hz, -CH3), 1.03–1.08 (1H, m, piperidine), 1.11–1.23 (2H, m, piperidine), 1.52–1.57 (1H, m, piperidine), 1.64–1.69 (2H, m, piperidine), 2.69–2.77 (2H, m, piperidine), 3.75 (1H, br.s., piperidine), 3.81 (3H, s, -OCH3), 4.62 (2H, s, -CH2-), 6.91 (2H, d, J = 8.70 Hz, Ar-H), 6.99 (1H, dd, J1 = 2.30 Hz, J2 = 8.80 Hz, BT-H), 7.40 (1H, d, J = 2.30 Hz), 7.48 (2H, d, J = 8.70 Hz, Ar-H), 7.85 (1H, d, J = 8.80 Hz, BT-H), 7.92 (1H, s, -CH=N-), 11.50 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 22.2, 30.7, 33.7, 35.6, 48.2, 55.9, 105.0, 114.2, 115.1, 122.5, 123.5, 128.6, 144.8, 148.1, 152.5, 154.4, 159.2, 162.9, 168.2. HRMS (m/z): [M + H]+ calcd for C23H26N4O2S2: 455.1570; found: 455.1566.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N’-(4-(4-(4-methoxyphenyl)piperazin-1-yl)benzylidene)acetohydrazide (4j). Yield: 80%, M.P. = 199–200 °C, FTIR (ATR, cm−1): 3226 (N-H), 1656 (C=O), 1224 (C-N), 1029, 812, 715. 1H-NMR (300 MHz, DMSO-d6): δ = 3.14 (4H, br.s., piperazine), 3.39 (4H, br.s., piperazine), 3.69 (3H, s, -OCH3), 3.81 (3H, s, -OCH3), 4.64 (2H, s, -CH2-), 6.84 (2H, d, J = 9.15 Hz, Ar-H), 6.96 (2H, d, J = 9.15 Hz), 6.97–7.05 (3H, m, Ar-H, BT-H), 7.41 (1H, d, J = 2.46 Hz, BT-H), 7.55 (2H, d, J = 8.85 Hz, Ar-H), 7.88 (1H, d, J = 8.79 Hz, BT-H), 7.95 (1H, s, -CH=N-), 11.54 (1H, s, -NH). 13C-NMR (75 MHz, DMSO-d6): δ = 35.6, 47.9, 50.1, 55.7, 55.9, 105.0, 114.2, 114.8, 115.2, 118.2, 122.5, 124.5, 128.6, 144.7, 145.7, 147.9, 152.4, 153.7, 154.4, 159.2, 162.9, 168.3. HRMS (m/z): [M + H]+ calcd for C28H29N5O3S2: 548.1785; found: 548.1783.
3.2. Biological Evaluation
3.2.1. Cytotoxicity Test
Metabolic activity of viable cells was measured by MTT assay based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium salt to a formazan product, which can be quantified spectrophotometrically to determine the percent of viable cells [32].
Anticancer activities of new benzothiazole acylhydrazones derivatives were evaluated against A549 C6, MCF-7 and HT-29 cell lines. The selectivity of their cytotoxic effects was evaluated on NIH/3T3 mouse embryonic fibroblast cells. NIH/3T3 cells were incubated in DMEM supplemented with fetal calf serum, penicillin (100 IU/mL), streptomycin (100 mg/mL) and 7.5% NaHCO3 at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Carcinogenic cells were incubated in RPMI medium supplemented with fetal calf serum, penicillin (100 IU/mL), streptomycin (100 mg/mL) and 7.5% NaHCO3 at 37 °C in a humidified atmosphere of 95% air and 5% CO2. All cell lines were seeded at a density of 1 × 104 cells into the 96-well plates. After 24 h of incubating period, the culture mediums were removed and test compounds were added at concentrations of 0.000316–1 mM. After a 24 h incubation period, OD of samples were measured by a microplate reader (Biotek, Winooski, VT, USA) at 540 nm. Inhibition % at concentrations was determined using the formula below and IC50 values were calculated by nonlinear regression analysis using the SigmaPlot v.10 package program (Manufacturer, City, US State abbrev. if applicable, Country) [33,34,35,36]. Cisplatin was used as a positive control:
% inhibition = 100 − (mean sample × 100/mean solvent)
3.2.2. DNA Synthesis Inhibition Assay
The BrdU cell proliferation method was performed to analyse the effects of compounds 4d, 4e and 4h on proliferation of C6 cells. C6 cells were seeded into the 96-well plates at a density of 1 × 104 cells. Compounds were added into each well at five different concentrations (IC50/4, IC50/2, IC50, 2 × IC50, and 4 × IC50) and the plates were incubated for 24 h. At the end of the incubation period, BrdU solution was added and cells were reincubated for 2 h at 37 °C. Anti-BrdU-POD (100 mL) was added and the mixture was incubated for 90 min. Microplates were washed with PBS for three times. After adding substrate solution, the mixture was incubated for 15 min. OD of the wells were determined at 492 nm. Proliferation of control cells was assessed as 100% and growth inhibition % of cells, treated with test compounds and cisplatin were calculated [37].
3.2.3. Flow Cytometric Analysis
Death pathway of the carcinogenic cell lines was detected by Annexin V-FITC Apoptosis Detection Kit (BD, Pharmingen, City, US State abbrev. if applicable, Country) as reported in the manufacturer’s instructions. Cisplatin and compounds 4d, 4e, and 4h, which possess the highest cytotoxic activity, were used at their IC50/2, IC50, and 2 × IC50 concentrations. After 24 h incubation period, cells were harvested by centrifugation at 1200 rpm for 5 min. at room temperature, rinsed with cold water twice and then suspended at 1 × 106 cells/mL concentration in Annexin V-FITC binding buffer. Propidium iodide (5 mL) and annexin V-FITC (5 mL) were added for staining the cells and the fluorescence measurements was performed using a flow cytometer. FCSExpress software (version, Manufacturer, City, US State abbrev. if applicable, Country) was used to display the percent of normal and apoptotic cells at different stages [37]. In the diagrams, Q1, Q2, Q3, and Q4 demonstrate the necrotic cells (positive for PI and negative for annexin/FITC), late apoptotic or necrotic cells (positive for annexin and PI), live cells (negative for annexin and PI), and apoptotic cells (negative for PI and positive for annexin), respectively. The experiments were carried out in triplicate.
4. Conclusions
A major challenge to medicinal chemists is determination of new structures that may be beneficial in the design of novel, selective and less-toxic anticancer agents. For this purpose, we synthesized new 2-((5-substituebenzothiazol-2-yl)thio)-N’-(2-(4-substitutedphenylethylidene)acetohydrazide (4a–4j) and evaluated their anticancer activity against A549, C6, MCF-7, and HT-29 cells. Cytotoxic activities of the synthesized compounds against healthy NIH3T3 cell lines were also investigated. It was determined that compounds 4d, 4e, and 4h showed higher cytotoxicity than other compounds against C6 cell lines. Moreover, these compounds did not kill healthy cells at concentrations with cytotoxic effects against cancer cells, so it could be said that compounds 4d, 4e, and 4h displayed selective inhibition towards cancer cells. Especially, the IC50 value of compound 4d against NIH3T3 cell lines was calculated as 1 mM < similar to the IC50 value of cisplatin. Structures of the test compounds vary from each other in terms of substituents on BT and phenyl substructures. Evaluating the substituent effect on cytotoxic activity, compounds 4a–4e bearing 5-chlorobenzothiazole have been noted with greater effects. In addition, 4-(4-methylpiperidin-1-yl)phenyl moiety in compound 4d caused a selective cytotoxic effect.
Consequently, anticancer activity screening of novel benzothiazole-acylhydrazones indicated that compound 4d, which is the lead compound of the series, possesses significant selective cytotoxic effects on cancer cells. Furthermore, compound 4d shows higher antiproliferative activity than cisplatin. However, it has induced apoptosis in cancer cells, dose-dependently. On the other hand, compound 4e (SI = 0.33), which has lower selectivity index (SI) compared to 4d, induced apoptotic cell death in cancer cells. At this point, we could be emphasized that findings of this preliminary study will not only direct us to synthesize similar compounds with greater anticancer activity, but also may have an influence on medicinal chemists to synthesize more active compounds.
Supplementary Materials
The following are available online, Spectra 1-12: IR, HRMS, 1H-NMR, and 13C-NMR spectra of compounds 4d, 4e and 4h.
Author Contributions
Y.O. and Z.A.K. conceived and designed the experiments; D.O. performed the synthesis; S.L. performed analysis studies; A.B.K. and S.I. performed activity tests; D.O., S.L., A.B.K., S.I., Y.O. and Z.A.K. wrote the paper.
Acknowledgments
This study was financially supported by Anadolu University Scientific Projects Fund, Project No.: 1601S019.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M. Synthesis and in vitro antitumor activity of new series of benzothiazole and pyrimido[2,1-b]benzothiazole derivatives. Eur. J. Med. Chem. 2014, 85, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Tuylu, B.A.; Zeytinoglu, H.S.; Isikdag, I. Synthesis and mutagenicity of 2-aryl-substitute (o-hydroxy-, m-bromo-, o-methoxy-, o-nitro-phenyl or 4-pyridyl) benzothiazole derivatives on Salmonella typhimurium and human lymphocytes exposed in vitro. Biologia 2007, 62, 626–632. [Google Scholar]
- Lion, C.J.; Matthews, C.S.; Wells, G.; Bradshaw, T.D.; Stevens, M.F.; Westwell, A.D. Antitumour properties of fluorinated benzothiazole substituted hydroxycyclohexa-2,5-dienones (‘quinols’). Bioorg. Med. Chem. Lett. 2006, 16, 5005–5008. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Park, H.J.; Lee, S.K.; Kim, S.W.; Han, G.; Choo, H.Y. Solid phase combinatorial synthesis of benzothiazoles and evaluation of topoisomerase II inhibitory activity. Bioorg. Med. Chem. 2006, 14, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, I.; Chua, M.S.; Browne, H.L.; Trapani, V.; Bradshaw, T.D.; Westwell, A.D. Antitumor benzothiazoles. 14. Synthesis and in vitro biological properties of fluorinated 2-(4-aminophenyl) benzothiazoles. J. Med. Chem. 2001, 44, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, I.; Jennings, S.A.; Vishnuvajjala, B.R.; Westwell, A.D.; Stevens, M.F. Antitumor benzothiazoles. 16. Synthesis and pharmaceutical properties of antitumor 2-(4-aminophenyl)benzothiazole amino acid prodrugs. J. Med. Chem. 2002, 45, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, I.; Bradshaw, T.D.; Matthews, C.S.; Stevens, M.F.; Westwell, A.D. Antitumour benzothiazoles. Part 20: 3′-cyano and 3′-alkynylsubstituted 2-(4′-aminophenyl)benzothiazoles as new potent and selective analogues. Bioorg. Med. Chem. Lett. 2003, 13, 471–474. [Google Scholar] [CrossRef]
- Bhuva, H.A.; Kini, S.G. Synthesis, anticancer activity and docking of some substituted benzothiazoles as tyrosine kinase inhibitors. J. Mol. Graph. Model. 2010, 29, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fan, J.; Vu, K.; Hong, K.; Le Brazidec, J.Y.; Shi, J. 7′-substituted benzothiazolothio- and pyridinothiazolothio-purines as potent heat shock protein 90 inhibitors. J. Med. Chem. 2006, 49, 5352–5362. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Singh, S.K.; Thakur, B.; Ray, P.; Singh, S.K. Design and Synthesis of Novel Schiff Base-Benzothiazole Hybrids as Potential Epidermal Growth Factor Receptor (EGFR) Inhibitors. Anti-Cancer Agents Med. Chem. 2016, 16, 722–739. [Google Scholar] [CrossRef]
- Dumas, J.; Brittelli, D.; Chen, J.; Dixon, B.; Hatoum-Mokdad, H.; König, G.; Sibley, R.; Witowsky, J.; Wong, S. Synthesis and structure activity relationships of novel small molecule cathepsin D inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 2531–2536. [Google Scholar] [CrossRef]
- Singh, M.; Singh, S.K. Benzothiazoles: How Relevant in Cancer Drug Design Strategy? Anti-Cancer Agents Med. Chem. 2014, 14, 127–146. [Google Scholar] [CrossRef]
- Caputo, R.; Calabro, M.R.; Micale, N.; Schimmer, A.D.; Ali, M.; Zappala, M.; Grasso, S. Synthesis of benzothiazole derivatives and their biological evaluation as anticancer agents. Med. Chem. Res. 2012, 21, 2644–2651. [Google Scholar] [CrossRef]
- Bao, G.; Du, B.; Ma, Y.; Zhao, M.; Gong, P.; Zhai, X. Design, Synthesis and Antiproliferative Activity of Novel Benzothiazole Derivatives Conjugated with Semicarbazone Scaffold. Med. Chem. 2016, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.H.; Wang, Z.; Xia, Y.; Ye, T.H.; Deng, M.; Xu, Y.Z.; Wei, Y.Q.; Yu, L.T. Synthesis and Biological Evaluation of Novel Benzothiazole-2-thiol Derivatives as Potential Anticancer Agents. Molecules 2012, 17, 3933–3944. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Sinhmar, A.; Sharma, A.; Rajak, H.; Pal Pathak, D. Medicinal significance of benzothiazole scaffold: An insight view. J. Enzym. Inhib. Med. Chem. 2013, 28, 240–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Drewe, J.; Tseng, B.; Kasibhatla, S.; Cai, S.X. Discovery and SAR of indole-2-carboxylic acid benzylidene-hydrazides as a new series of potent apoptosis inducers using a cell-based HTS assay. Bioorg. Med. Chem. 2004, 12, 3649–3655. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Q.P.; Hsu, D.C.; Goode, D.R.; Novotny, C.J.; Totten, R.K.; Hergenrother, P.J. Procaspase-3 activation as an anti-cancer strategy: Structure-activity relationship of procaspase-activating compound 1 (PAC-1) and its cellular co-localization with caspase-3. J. Med. Chem. 2009, 52, 5721–5731. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, E.B.; de Brito, M.A.; Vasconcelos, T.R.; de Moraes, M.O.; Montenegro, R.C.; Yoneda, J.D.; Leal, K.Z. Synthesis and anticancer activity of (E)-2-benzothiazole hydrazones. Eur. J. Med. Chem. 2014, 30, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.F.; Azevedo, E.C.; Ferreira, V.F.; Araújo, A.J.; dos Santos, E.A.; Pessoa, C.; Costa-Lotufo, L.V.; Montenegro, R.C.; de Moraes, M.O.; Vasconcelos, T.R.A. Synthesis and Antitumor Evaluation of (E)-2-Benzothiazole Hydrazones. Lett. Drug Des. Discov. 2010, 7, 551–555. [Google Scholar] [CrossRef]
- Ma, J.; Chen, D.; Lu, K.; Wang, L.; Han, X.; Zhao, Y.; Gong, P. Design, Synthesis, And Structure-Activity Relationships of Novel Benzothiazole Derivatives Bearing the Ortho-Hydroxy N-Carbamoylhydrazone Moiety as Potent Antitumor Agents. Eur. J. Med. Chem. 2014, 17, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Prangya, S.; Pany, P.; Bommisetti, P.; Diveshkumar, K.V.; Pradeepkumar, P.I. Benzothiazole hydrazones of furylbenzamides preferentially stabilize c-MYC and c-KIT1 promoter G-quadruplex DNAs. Org. Biomol. Chem. 2016, 14, 5779–5793. [Google Scholar]
- Ma, J.; Zhang, G.; Han, X.; Bao, G.; Wang, L.; Zhai, X.; Gong, P. Synthesis and Biological Evaluation of Benzothiazole Derivatives Bearing the ortho-Hydroxy-N-acylhydrazone Moiety as Potent Antitumor Agents. Arch. Pharm. Chem. Life Sci. 2014, 347, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Sleebs, B.E.; Kersten, W.J.A.; Kulasegaram, S.; Nikolakopoulos, G.; Hatzis, E.; Moss, R.M.; Parisot, J.P.; Yang, H.; Czabotar, P.E.; Fairlie, W.D.; et al. Discovery of Potent and Selective Benzothiazole Hydrazone Inhibitors of Bcl-XL. J. Med. Chem. 2013, 56, 5514–5540. [Google Scholar] [CrossRef] [PubMed]
- Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; İnci, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules 2017, 22, 1381. [Google Scholar] [CrossRef] [PubMed]
- Ilgın, S.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; Acar Çevik, U.; Kaya Çavuşoğlu, B.; Özkay, Y.; Kaplancıklı, Z.A. Design and Synthesis of New Benzothiazole Compounds as Selective hMAO-B Inhibitors. Molecules 2017, 22, 2187. [Google Scholar] [CrossRef] [PubMed]
- Varache-Lembege, M.; Moreau, S.; Larrouture, S.; Montaudon, D.; Robert, J.; Nuhrich, A. Synthesis and antiproliferative activity of aryl- and heteroaryl-hydrazones derived from xanthone carbaldehydes. Eur. J. Med. Chem. 2008, 43, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Malikova, J.; Swaczynova, J.; Kolar, Z.; Strnad, M. Anticancer and antiproliferative activity of natural brassinosteroids. Phytochemistry 2008, 69, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Maghni, K.; Nicolescu, O.M.; Martin, J.G. Suitability of cell metabolic colorimetric assays for assessment of CD4+ T cell proliferation: Comparison to 5-bromo-2-deoxyuridine (BrdU) ELISA. J. Immunol. Methods 1999, 223, 185–194. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Sağlık, B.N.; Ilgın, S.; Özkay, Y. Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur. J. Med. Chem. 2016, 124, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgın, S.; Atlı, Ö.; Demirel, R.; Kaplancıklı, Z.A. A novel series of thiazolyl-pyrazoline derivatives: Synthesis and evaluation of antifungal activity, cytotoxicity and genotoxicity. Eur. J. Med. Chem. 2015, 92, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgın, S.; Atlı, Ö.; Demirci, F.; Kaplancıklı, Z.A. Synthesis and in vitro evaluation of new nitro-substituted thiazolyl hydrazone derivatives as anticandidal and anticancer agents. Molecules 2014, 19, 14809–14820. [Google Scholar] [CrossRef] [PubMed]
- Yurttaş, L.; Demirayak, S.; Ilgın, S.; Atlı, Ö. In vitro antitumor activity evaluation of some 1,2,4-triazine derivatives bearing piperazine amide moiety against breast cancer cells. Bioorg. Med. Chem. 2014, 22, 6313–6323. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4a–4j are available from the authors. |
Scheme 1.
Synthesis way of the target compounds (4a–4j).

| Compounds | R1 | R2 | R3 | R4 | X |
|---|---|---|---|---|---|
| 4a | -H | -H | -H | -Cl | -CH |
| 4b | -CH3 | -H | -H | -Cl | -CH |
| 4c | -H | -CH3 | -H | -Cl | -CH |
| 4d | -H | -H | -CH3 | -Cl | -CH |
| 4e | -H | -H | 4-methoxyphenyl | -Cl | -N |
| 4f | -H | -H | -H | -OCH3 | -CH |
| 4g | -CH3 | -H | -H | -OCH3 | -CH |
| 4h | -H | -CH3 | -H | -OCH3 | -CH |
| 4i | -H | -H | -CH3 | -OCH3 | -CH |
| 4j | -H | -H | 4-methoxyphenyl | -OCH3 | -N |
Figure 1.
Inhibition values (%) of the compounds 4d, 4e, and 4h and cisplatin for each concentration against C6 cell line following a 24 h incubation period in MTT assay. All data were expressed as mean ± standard deviation.
Figure 1.
Inhibition values (%) of the compounds 4d, 4e, and 4h and cisplatin for each concentration against C6 cell line following a 24 h incubation period in MTT assay. All data were expressed as mean ± standard deviation.

Figure 2.
DNA synthesis inhibitory activity % of the compounds 4d, 4e, and 4h and cisplatin against C6 cell line for 24 h incubation period.
Figure 2.
DNA synthesis inhibitory activity % of the compounds 4d, 4e, and 4h and cisplatin against C6 cell line for 24 h incubation period.

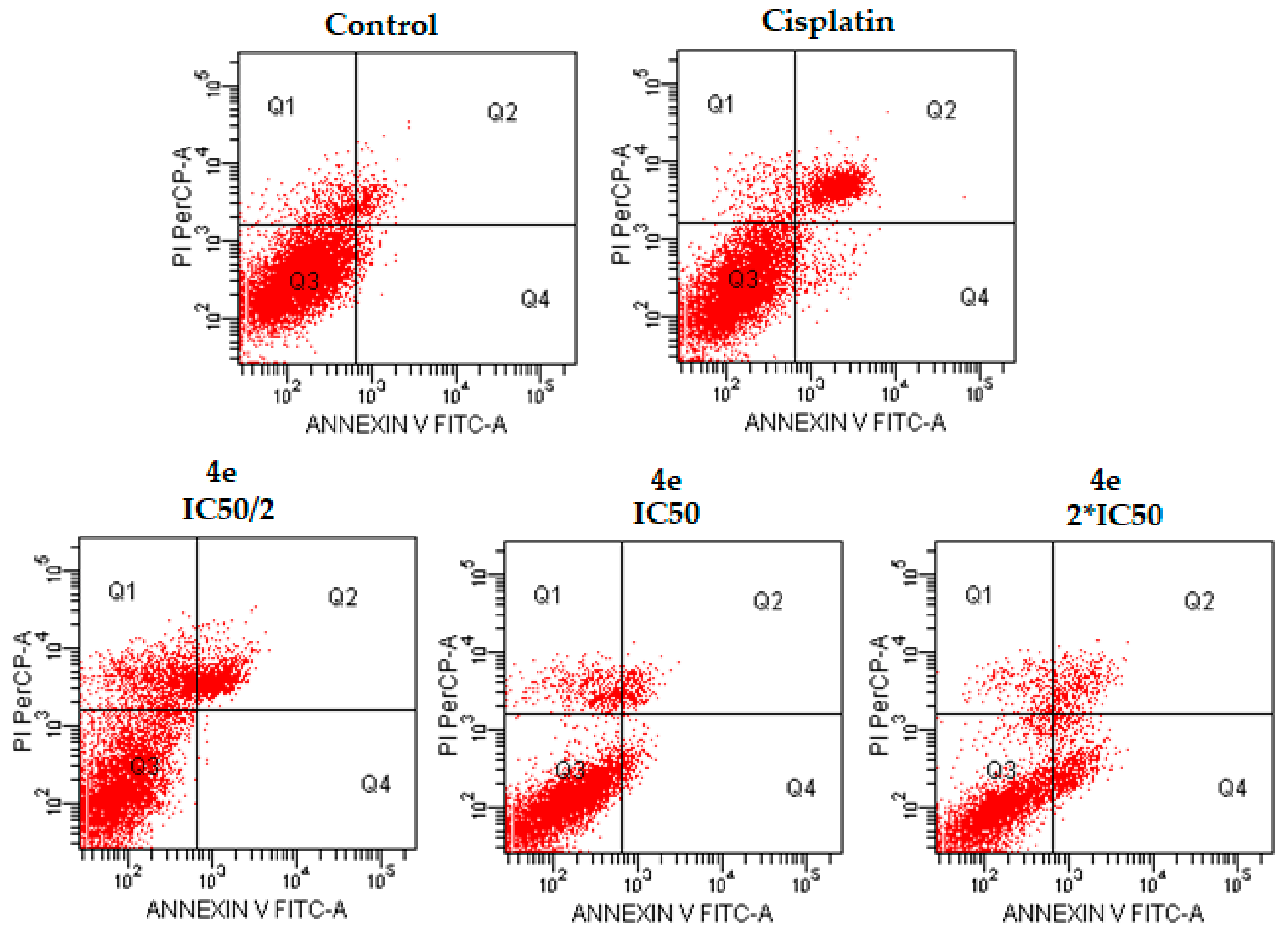
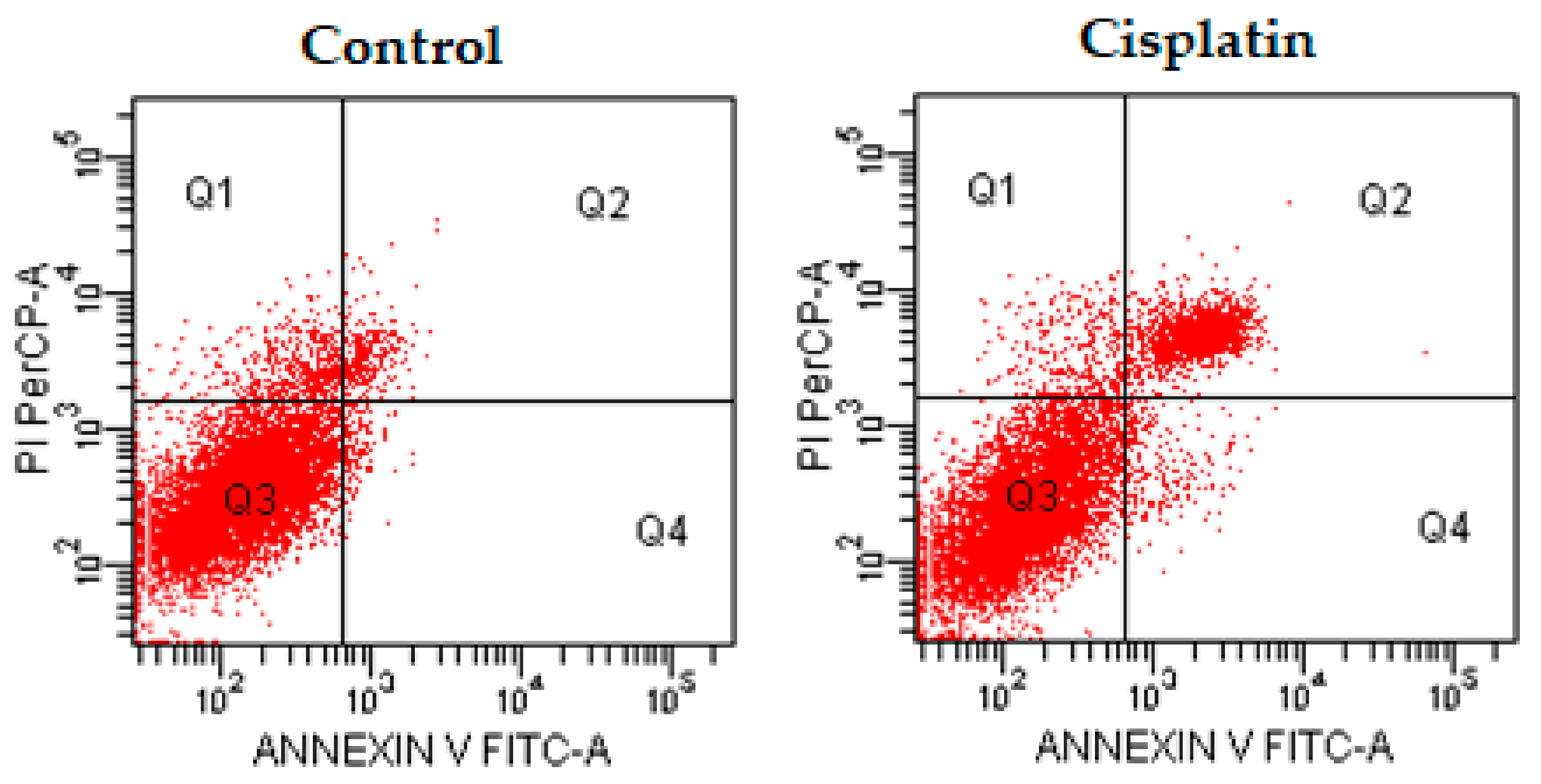
Figure 3.
The flow cytometric analysis diagram of compound 4d and cisplatin for C6 cell line. Annexin V-PI analysis in C6 cell line, following 24 h incubation period of compounds 4d at IC50/2, IC50, 2 × IC50 concentrations, which are 0.015, 0.03 and 0.06 mM, respectively, following 24 h incubation period. FITC Annexin V method in C6 cell line, following a 24 h incubation period of cisplatin at IC50 (0.03 mM). A diagram can be divided into four regions that are defined as follows: the percentage of necrotic cells (PI/FITC. +/−; Q1); the percentage of late apoptotic cells (Q2; PI/FITC +/+); the percentage of viable cells (Q3; PI/FITC −/−) and the percentage of early apoptotic cells (Q4; PI/FITC −/+). Percentages of Q1, Q2, Q3, and Q4 of compound 4d and cisplatin were presented in Table 2.
Figure 3.
The flow cytometric analysis diagram of compound 4d and cisplatin for C6 cell line. Annexin V-PI analysis in C6 cell line, following 24 h incubation period of compounds 4d at IC50/2, IC50, 2 × IC50 concentrations, which are 0.015, 0.03 and 0.06 mM, respectively, following 24 h incubation period. FITC Annexin V method in C6 cell line, following a 24 h incubation period of cisplatin at IC50 (0.03 mM). A diagram can be divided into four regions that are defined as follows: the percentage of necrotic cells (PI/FITC. +/−; Q1); the percentage of late apoptotic cells (Q2; PI/FITC +/+); the percentage of viable cells (Q3; PI/FITC −/−) and the percentage of early apoptotic cells (Q4; PI/FITC −/+). Percentages of Q1, Q2, Q3, and Q4 of compound 4d and cisplatin were presented in Table 2.

Figure 4.
The flow cytometric analysis diagram of compound 4e and cisplatin for C6 cell line. Annexin V-PI analysis in C6 cell line, following a 24 h incubation period of compounds 4e at IC50/2, IC50, 2 × IC50 concentrations, which are 0.015, 0.03 and 0.06 mM, respectively, following a 24 h incubation period. An FITC Annexin V method in C6 cell line, following a 24 h incubation period of cisplatin at IC50 (0.03 mM). A diagram can be divided into four regions that are defined as follows: the percentage of necrotic cells (PI/FITC. +/−; Q1); the percentage of late apoptotic cells (Q2; PI/FITC +/+); the percentage of viable cells (Q3; PI/FITC −/−) and the percentage of early apoptotic cells (Q4; PI/FITC −/+). Percentages of Q1, Q2, Q3, and Q4 of compound 4e and cisplatin were presented in Table 2.
Figure 4.
The flow cytometric analysis diagram of compound 4e and cisplatin for C6 cell line. Annexin V-PI analysis in C6 cell line, following a 24 h incubation period of compounds 4e at IC50/2, IC50, 2 × IC50 concentrations, which are 0.015, 0.03 and 0.06 mM, respectively, following a 24 h incubation period. An FITC Annexin V method in C6 cell line, following a 24 h incubation period of cisplatin at IC50 (0.03 mM). A diagram can be divided into four regions that are defined as follows: the percentage of necrotic cells (PI/FITC. +/−; Q1); the percentage of late apoptotic cells (Q2; PI/FITC +/+); the percentage of viable cells (Q3; PI/FITC −/−) and the percentage of early apoptotic cells (Q4; PI/FITC −/+). Percentages of Q1, Q2, Q3, and Q4 of compound 4e and cisplatin were presented in Table 2.

Figure 5.
The flow cytometric analysis diagram of compound 4h and cisplatin for C6 cell line. Annexin V-PI analysis in C6 cell line, following 24 h incubation period of compounds 4h at IC50/2, IC50, 2 × IC50 concentrations, which are 0.015, 0.03 and 0.06 mM, respectively, following a 24 h incubation period. FITC Annexin V method in C6 cell line, following 24 h incubation period of cisplatin at IC50 (0.03 mM). Diagram can be divided into four regions that are defined as follows: the percentage of necrotic cells (PI/FITC. +/−; Q1); the percentage of late apoptotic cells (Q2; PI/FITC +/+); the percentage of viable cells (Q3; PI/FITC −/−) and the percentage of early apoptotic cells (Q4; PI/FITC −/+). Percentages of Q1, Q2, Q3, and Q4 of compound 4h and cisplatin were presented in Table 2.
Figure 5.
The flow cytometric analysis diagram of compound 4h and cisplatin for C6 cell line. Annexin V-PI analysis in C6 cell line, following 24 h incubation period of compounds 4h at IC50/2, IC50, 2 × IC50 concentrations, which are 0.015, 0.03 and 0.06 mM, respectively, following a 24 h incubation period. FITC Annexin V method in C6 cell line, following 24 h incubation period of cisplatin at IC50 (0.03 mM). Diagram can be divided into four regions that are defined as follows: the percentage of necrotic cells (PI/FITC. +/−; Q1); the percentage of late apoptotic cells (Q2; PI/FITC +/+); the percentage of viable cells (Q3; PI/FITC −/−) and the percentage of early apoptotic cells (Q4; PI/FITC −/+). Percentages of Q1, Q2, Q3, and Q4 of compound 4h and cisplatin were presented in Table 2.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cytotoxic activity of the compounds against A549, C6, MCF-7, HT-29 and NIH3T3 cell lines.
| Compounds | IC50 (mM) | ||||
|---|---|---|---|---|---|
| A549 | C6 | MCF-7 | HT-29 | NIH3T3 | |
| 4a | 1< | 0.10 | 0.49 | 0.52 | 0.10 |
| 4b | 1 | 0.10 | 1< | 1 | 0.10 |
| 4c | 0.52 | 0.10 | 0.10 | 0.30 | 0.03 |
| 4d | 1< | 0.03 | 0.10 | 0.30 | 1< |
| 4e | 0.03 | 0.03 | 0.30 | 1< | 0.01 |
| 4f | 1< | 1< | 1< | 1< | 0.03 |
| 4g | 1< | 1< | 1< | 1< | 0.10 |
| 4h | 1< | 0.03 | 1< | 1< | 0.10 |
| 4i | 0.49 | 0.10 | 0.30 | 1 | 0.03 |
| 4j | 1< | 1< | 1< | 1< | 1< |
| Cisplatin | 0.06 | 0.03 | 0.05 | 0.06 | 1< |
Table 2.
The flow cytometric analysis results of compounds 4d, 4e, and 4h and cisplatin for C6 cell line.
Table 2.
The flow cytometric analysis results of compounds 4d, 4e, and 4h and cisplatin for C6 cell line.
| Compound | IC50/2 | IC50 | 2 × IC50 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | Q1 | Q2 | Q3 | Q4 | Q1 | Q2 | Q3 | Q4 | |
| 4d | 16.4 | 12.3 | 71.2 | 0.1 | 6.9 | 3.1 | 87.6 | 2.4 | 4.5 | 7.1 | 74.5 | 13.9 |
| 4e | 13.5 | 3.8 | 82.6 | 0 | 9.8 | 16.0 | 70.7 | 3.5 | 10.1 | 14.0 | 73.3 | 2.5 |
| 4h | 10.8 | 4.9 | 84.1 | 0.3 | 8.6 | 7.4 | 79.4 | 4.6 | 5.4 | 10.5 | 75.2 | 8.9 |
| Cisplatin | N.D. | N.D. | N.D. | N.D. | 3.5 | 18.3 | 75.6 | 2.6 | N.D. | N.D. | N.D. | N.D. |
N.D.: Not Determined.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Osmaniye, D.; Levent, S.; Karaduman, A.B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Benzothiazole Acylhydrazones as Anticancer Agents. Molecules 2018, 23, 1054. https://doi.org/10.3390/molecules23051054
AMA Style
Osmaniye D, Levent S, Karaduman AB, Ilgın S, Özkay Y, Kaplancıklı ZA. Synthesis of New Benzothiazole Acylhydrazones as Anticancer Agents. Molecules. 2018; 23(5):1054. https://doi.org/10.3390/molecules23051054
Chicago/Turabian StyleOsmaniye, Derya, Serkan Levent, Abdullah Burak Karaduman, Sinem Ilgın, Yusuf Özkay, and Zafer Asım Kaplancıklı. 2018. "Synthesis of New Benzothiazole Acylhydrazones as Anticancer Agents" Molecules 23, no. 5: 1054. https://doi.org/10.3390/molecules23051054