
Confinement Effects on Chemical Equilibria: Pentacyano(Pyrazine)Ferrate(II) Stability Changes within Nanosized Droplets of Water


 and
and
Abstract
:
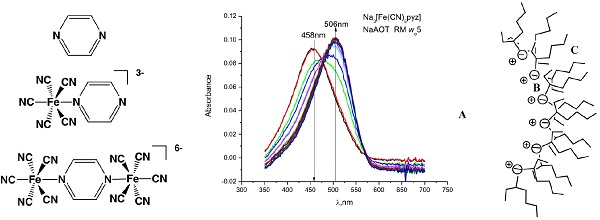{kind=link}
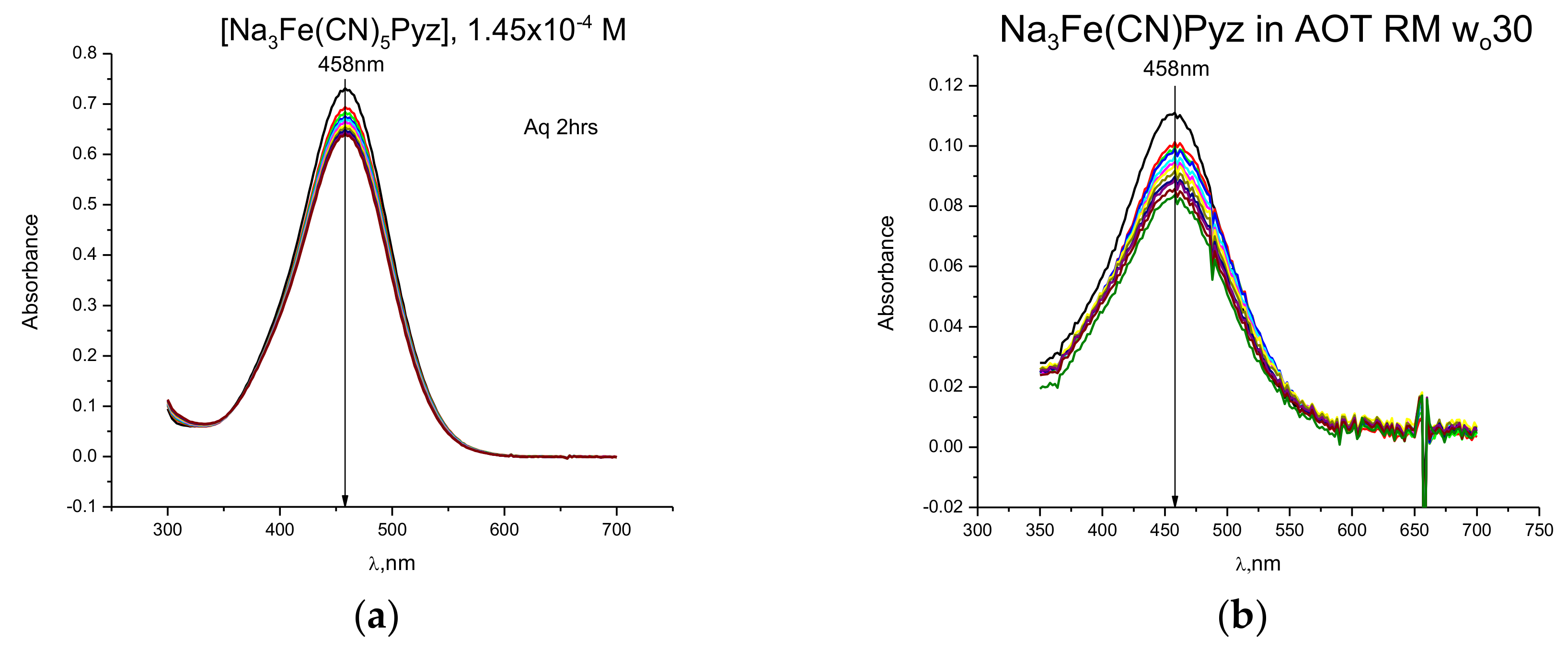{kind=link}
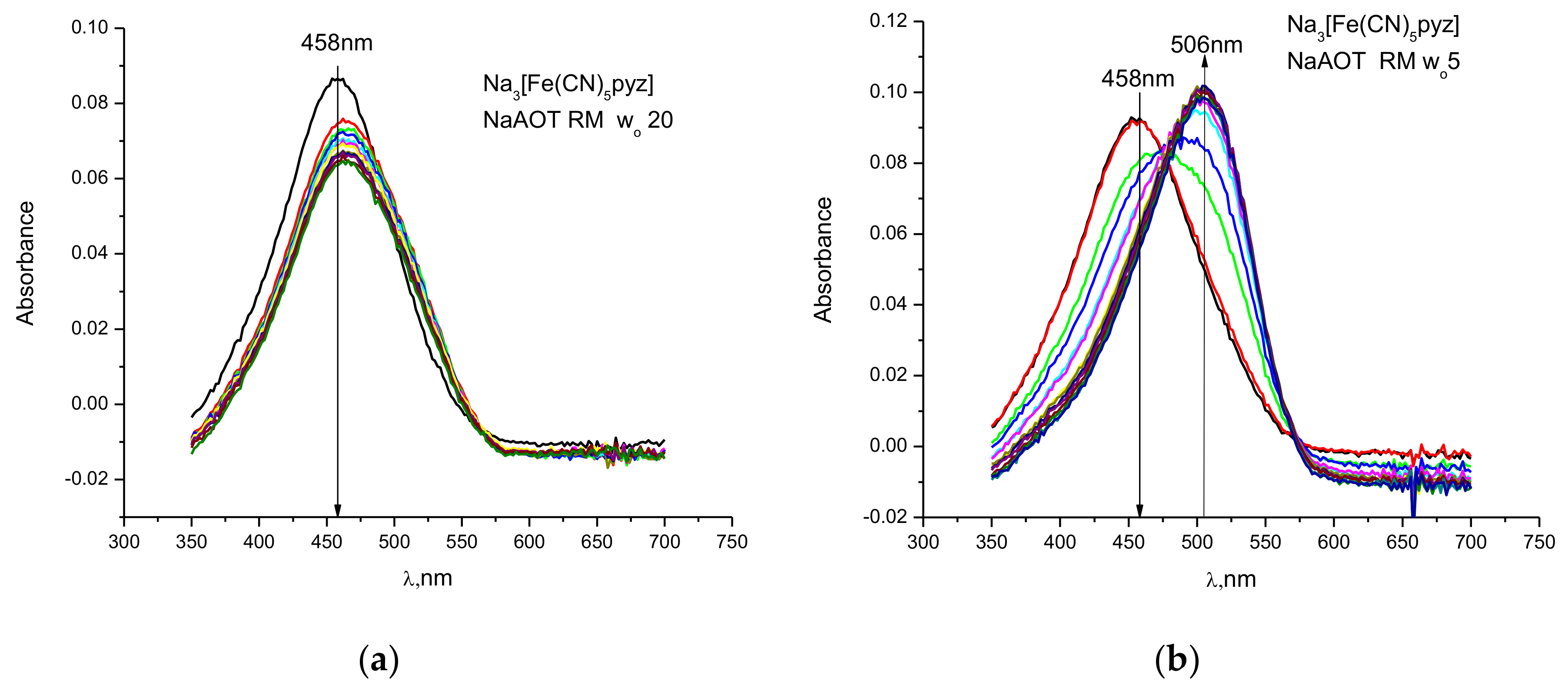{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
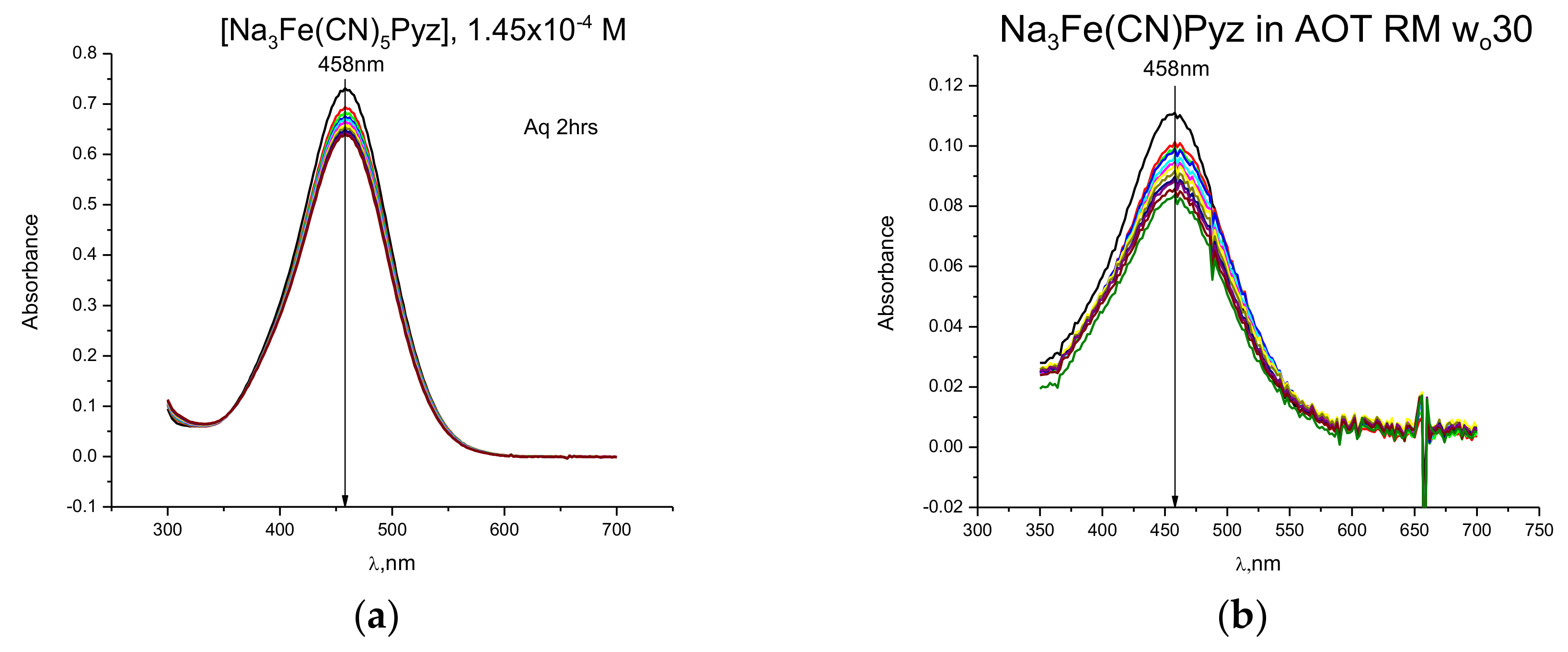
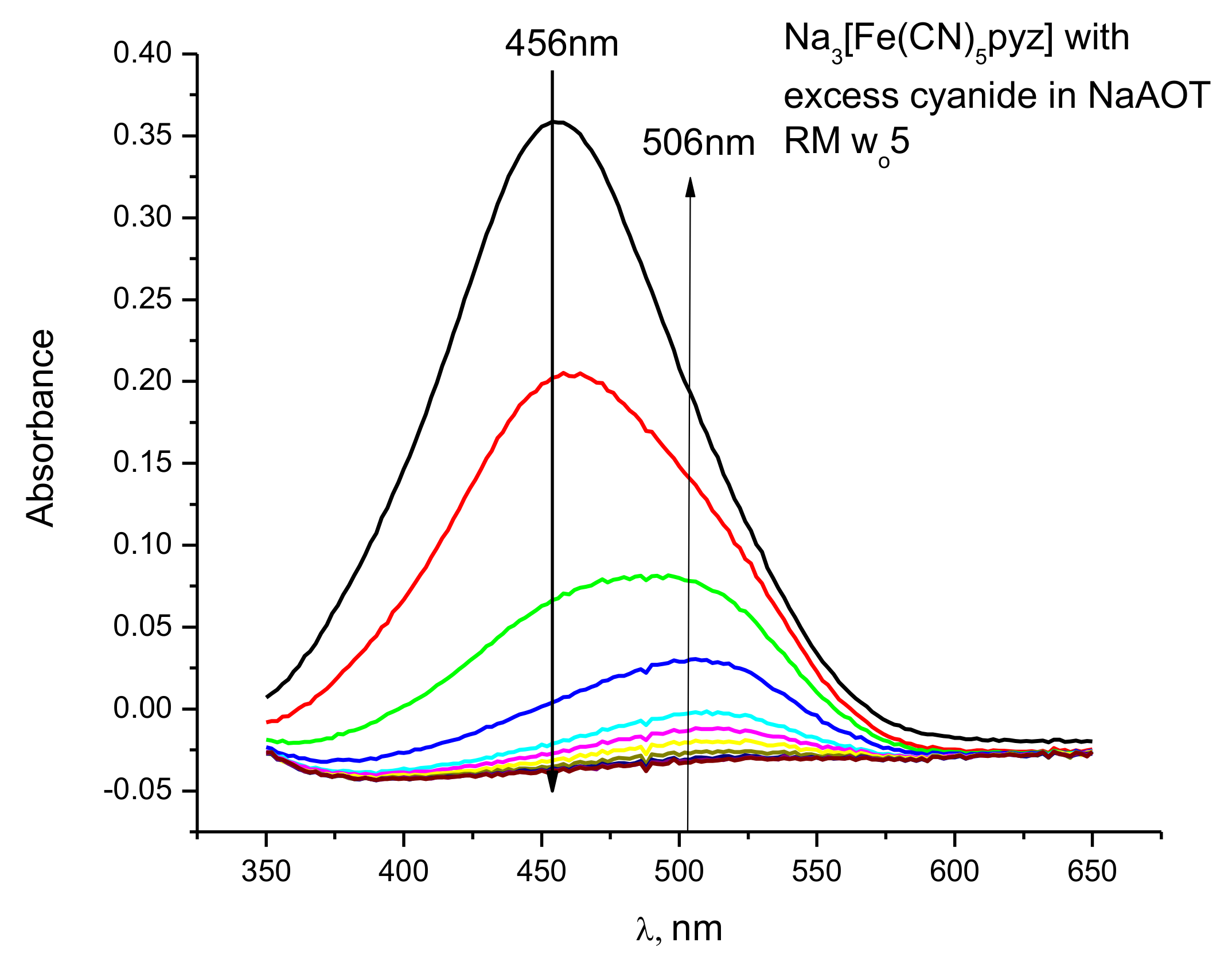
2.1. Kinetic Measurements Using UV-Vis Spectroscopy
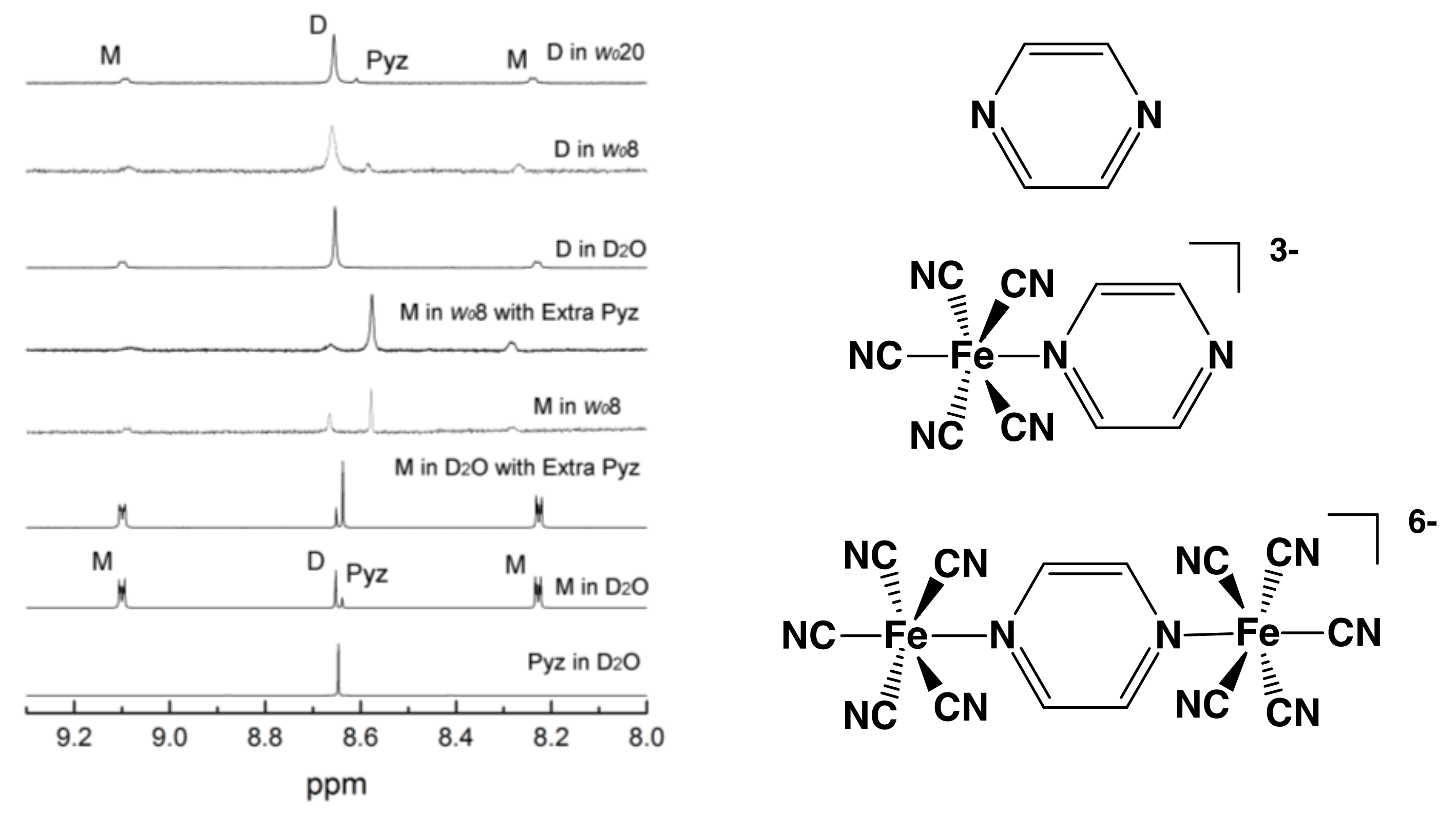
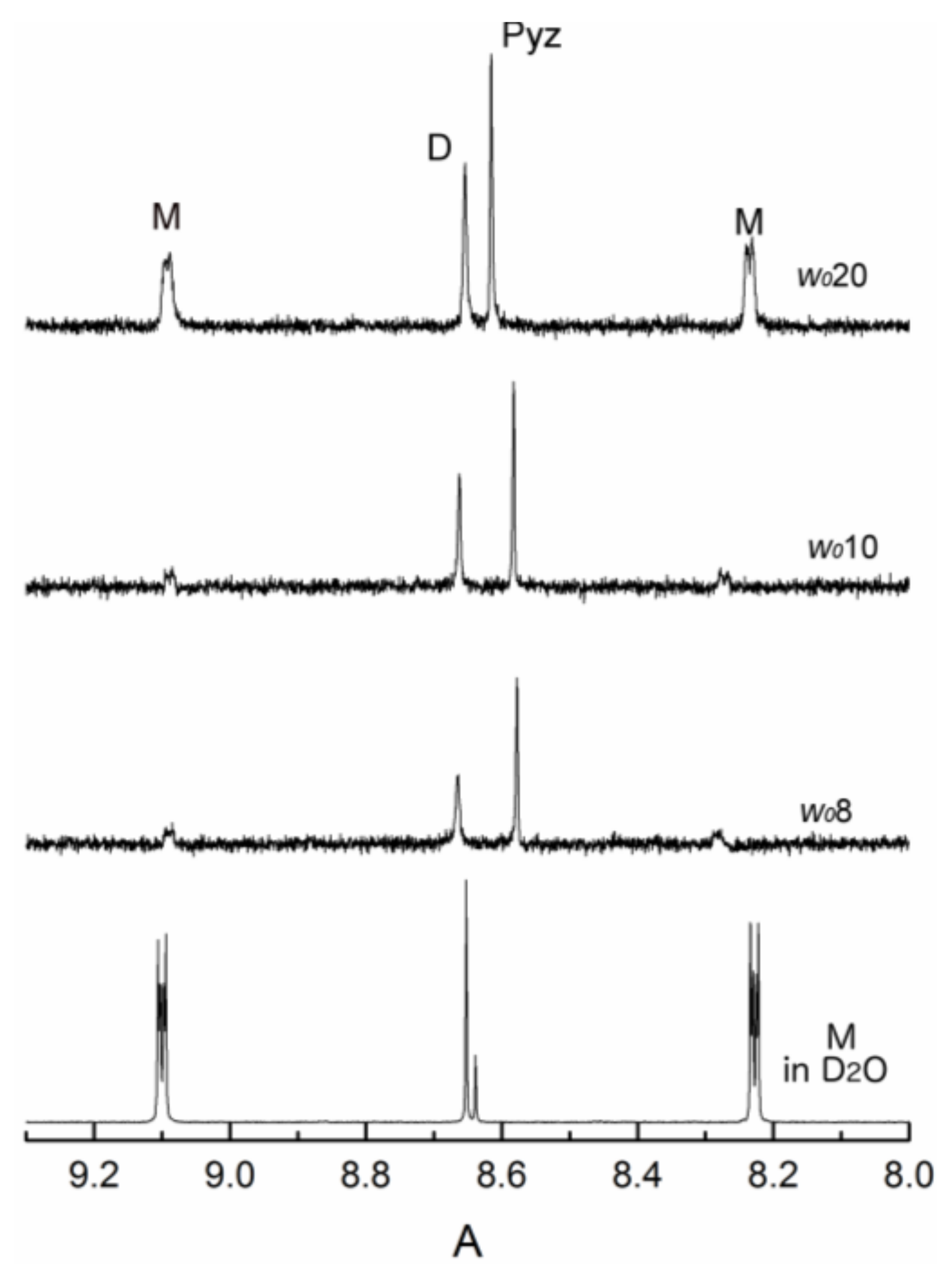
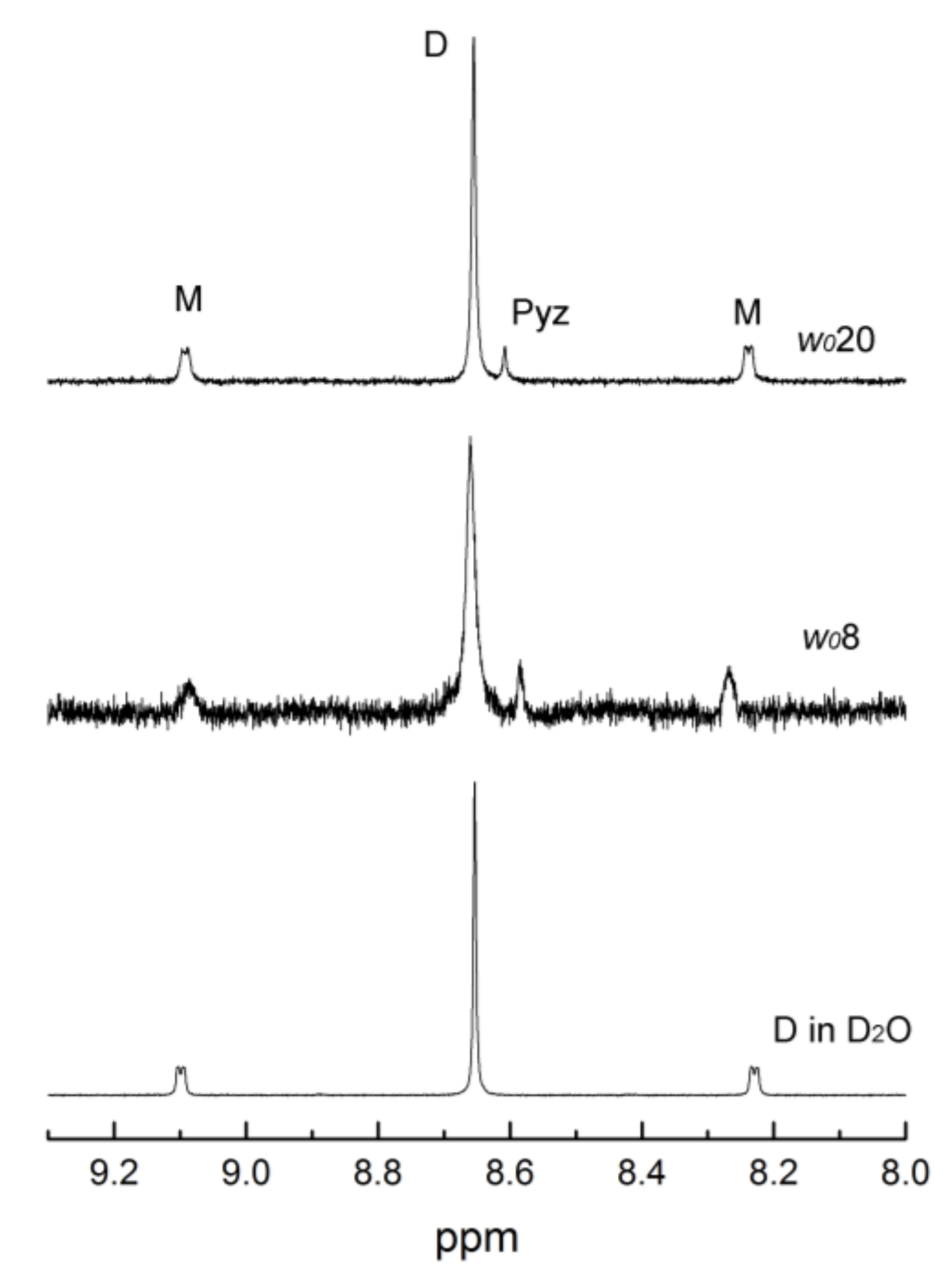
2.2. Thermodynamic Measurements using 1H-NMR Spectroscopy
3. Materials and Methods
3.1. General Materials and Methods
3.2. Preparation of Solutions
3.2.1. Metal Complex Solutions
3.2.2. Reverse Micelle (RM) Solutions for Spectroscopic Measurements
3.3. Methods
3.3.1. UV-Visible Spectroscopy
3.3.2. 1H-NMR Spectroscopy
3.3.3. Dynamic Light Scattering (DLS)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferreira, P.; Xue, W.-M.; Bencze, E.; Herdtweck, E.; Kuehn, F.E. Bidentate Lewis Base Adducts of Methyltrioxorhenium(VII) and Their Application in Catalytic Epoxidation. Inorg. Chem. 2001, 40, 5834–5841. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Sonnenberg, J.L.; Hay, P.J.; Martin, R.L.; Bursten, B.E.; Schlegel, H.B. Theoretical Investigation of Uranyl Dihydroxide: Oxo Ligand Exchange, Water Catalysis, and Vibrational Spectra. J. Phys. Chem. A 2005, 109, 8579–8586. [Google Scholar] [CrossRef] [PubMed]
- Moloney, M.G.; Paul, D.R.; Thompson, R.M.; Wright, E. Ligand exchange and catalysis in phenylation reactions mediated by lead tetracarboxylates. J. Organomet. Chem. 1998, 560, 77–88. [Google Scholar] [CrossRef]
- Ortiz-Maldonado, M.; Ballou, D.P.; Massey, V. A Rate-Limiting Conformational Change of the Flavin in p-Hydroxybenzoate Hydroxylase Is Necessary for Ligand Exchange and Catalysis: Studies with 8-Mercapto- and 8-Hydroxy-Flavins. Biochemistry 2001, 40, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Basolo, F. Base hydrolysis of cobalt (III) ammines. Bull. Hist. Chem. 1996, 19, 66–71. [Google Scholar]
- Wilkins, R.G. Factors influencing the rates of dissociation of metal complexes. IV. A comparison of nickel(II)- and copper(II)diamine complexes. J. Chem. Soc. 1962, 4475–4478. [Google Scholar] [CrossRef]
- Hubbard, C.D.; Van Eldik, R. Mechanistic studies of reactions of coordination compounds. Some recent highlights. J. Coord. Chem. 2007, 60, 1–51. [Google Scholar] [CrossRef]
- Swaddle, T.W. Ligand Substitution Dynamics in Metal Complexes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 339–393. [Google Scholar] [CrossRef]
- Lincoln, S.E. Mechanistic studies of metal aqua ions: A semi-historical perspective. Helv. Chim. Acta 2005, 88, 523–545. [Google Scholar] [CrossRef]
- Richens, D.T. Ligand substitution reactions at inorganic centers. Chem. Rev. 2005, 105, 1961–2002. [Google Scholar] [CrossRef] [PubMed]
- Macartney, D.H. Properties and reactions of substituted pentacyanoferrate(II) complexes. Rev. Inorg. Chem. 1988, 9, 101–151. [Google Scholar] [CrossRef]
- Wilkins, R.G. The Study of Kinetics and Mechanism of Reactions of Transition Metal Complexes; Allyn and Bacon: Boston, MA, USA, 1974; p. 403. [Google Scholar]
- Bachmann, P.A.; Walde, P.; Luisi, P.L.; Lang, J. Self-replicating micelles: Aqueous micelles and enzymatically driven reactions in reverse micelles. J. Am. Chem. Soc. 1991, 113, 8204–8209. [Google Scholar] [CrossRef]
- Gupte, A.; Nagarajan, R.; Kilara, A. Enzyme reactions in reverse micelles. Dev. Food Sci. 1995, 37A, 1–74. [Google Scholar] [CrossRef]
- Li, Y.; Li, G.; Ma, C. Enzymology in surfactant association systems. J. Dispers. Sci. Technol. 2000, 21, 409–432. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Ru, X.; Jiang, Y.; Hu, M.; Li, S.; Zhai, Q. Catalytic performance and thermostability of chloroperoxidase in reverse micelle: Achievement of a catalytically favorable enzyme conformation. J. Ind. Microbiol. Biotechnol. 2011, 38, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Trujillo, A.M.; Pharazyn, P.S.; Cohen, M.D. How environment affects drug activity: Localization, compartmentalization and reactions of a vanadium insulin-enhancing compound, dipicolinatooxovanadium(V). Coord. Chem. Rev. 2011, 255, 2178–2192. [Google Scholar] [CrossRef]
- O’Connor, C.J.; Fendler, E.J.; Fendler, J.H. Catalysis by reversed micelles in nonplanar solvents. Aquation and electron-transfer reactions of chromium(III) and cobalt(III) complexes in benzene. J. Chem. Soc. Dalton Trans. 1974, 625–631. [Google Scholar] [CrossRef]
- O’Connor, C.J.; Fendler, E.J.; Fendler, J.H. Catalysis by reversed micelles in nonpolar solvents. Trans-cis isomerization of bis(oxalato)diaquochromate(III). J. Am. Chem. Soc. 1974, 96, 370–375. [Google Scholar] [CrossRef]
- Burgess, J.; Patel, M.S. Pentacyanoferrates(II): Solvatochromism and reactivity in micelles and in reversed micelles. J. Chem. Soc. Faraday Trans. 1993, 89, 783–787. [Google Scholar] [CrossRef]
- Yamagishi, A.; Masui, T.; Watanabe, F. Temperature-jump studies of the ligand substitution reactions of tetrahedral cobalt(II) complexes solubilized in reversed micelles. Inorg. Chem. 1981, 20, 513–518. [Google Scholar] [CrossRef]
- Venkateswarlu, G.; Syamala, P.; Rao, P.V.S.; Ramakrishna, K. Kinetics of dissociation of tris(1,10-phenanthroline)iron(II) in aqueous and reverse micellar media—Catalysis of CuII, NiII and CoII ions. J. Indian Chem. Soc. 2003, 80, 86–90. [Google Scholar]
- Baruah, B.; Roden, J.M.; Sedgwick, M.; Correa, N.M.; Crans, D.C.; Levinger, N.E. When is water not water? Exploring water confined in large reverse micelles using a highly charged inorganic molecular probe. J. Am. Chem. Soc. 2006, 128, 12758–12765. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.; Rithner, C.D.; Inafuku, R.A.; Crans, D.C.; Levinger, N.E. Interaction of dipicolinatodioxovanadium(V) with polyatomic cations and surfaces in reverse micelles. Langmuir 2005, 21, 6250–6258. [Google Scholar] [CrossRef] [PubMed]
- Chatkon, A.; Barres, A.; Samart, N.; Boyle, S.; Haller, K.J.; Crans, D.C. Guanylurea metformium double salt of decavanadate, (HGU+)4(HMet+)2(V10O286−)·2H2O. Inorg. Chem. Acta 2014, 420, 85–91. [Google Scholar] [CrossRef]
- Crans, D.C.; Baruah, B.; Gaidamauskas, E.; Lemons, B.G.; Lorenz, B.B.; Johnson, M.D. Impairment of ascorbic acid’s anti-oxidant properties in confined media: Inter and intramolecular reactions with air and vanadate at acidic pH. J. Inorg. Biochem. 2008, 102, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Lorenz, B.B.; Wilkins, P.C.; Lemons, B.G.; Baruah, B.; Lamborn, N.; Stahla, M.; Chatterjee, P.B.; Richens, D.T.; Crans, D.C. Switching off Electron Transfer Reactions in Confined Media: Reduction of [Co.(dipic)2]- and [Co.(edta)]- by Hexacyanoferrate(II). Inorg. Chem. 2012, 51, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Lemons, B.G.; Richens, D.T.; Anderson, A.; Sedgwick, M.; Crans, D.C.; Johnson, M.D. Stabilization of a vanadium(V)-catechol complex by compartmentalization and reduced solvation inside reverse micelles. New J. Chem. 2013, 37, 75–81. [Google Scholar] [CrossRef]
- Lemons, B.G.; Crans, D.C.; Johnson, M.D. Electron transfer rate enhancements in nanosized waterpools. Eur. J. Inorg. Chem. 2014, 27, 4537–4540. [Google Scholar] [CrossRef]
- Molina, P.G.; Silber, J.J.; Correa, N.M.; Sereno, L. Electrochemistry in AOT Reverse Micelles. A Powerful Technique to Characterize Organized Media. J. Phys. Chem. C 2007, 111, 4269–4276. [Google Scholar] [CrossRef]
- De, T.; Maitra, A. Solution behavior of Aerosol OT in non-polar solvents. Adv. Colloid Interface Sci. 1995, 59, 95–193. [Google Scholar] [CrossRef]
- Maitra, A. Determination of size parameters of water aerosol OT oil reverse micelles from their nuclear magnetic resonance data. J. Phys. Chem. 1984, 88, 5122–5125. [Google Scholar] [CrossRef]
- Gaidamauskas, E.; Cleaver, D.P.; Chatterjee, P.B.; Crans, D.C. Effect of Micellar and Reverse Micellar Interface on Solute Location: 2,6-Pyridinedicarboxylate in CTAB Micelles and CTAB and AOT Reverse Micelles. Langmuir 2010, 26, 13153–13161. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Moilanen, D.E.; Fayer, M.D. Water dynamics—The effects of ions and nanoconfinement. J. Phys. Chem. B 2008, 112, 5279–5290. [Google Scholar] [CrossRef] [PubMed]
- Belafi-Bako, K.; Nagy, E. Characteristics of AOT/isooctane/water system. Hung. J. Ind. Chem. 1994, 22, 51–56. [Google Scholar]
- Boyer, B.; Lamaty, G.; Mary, F.; Mouanga, J.; Roque, J.P. Reactivity in reverse micelles. Effects of micelle size and the role of the cosurfactant. New J. Chem. 1992, 16, 375–379. [Google Scholar]
- Fayer, M.D.; Levinger, N.E. Analysis of Water in Confined Geometries and at Interfaces. Ann. Rev. Anal. Chem. 2010, 3, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Piletic, I.R.; Moilanen, D.E.; Spry, D.B.; Levinger, N.E.; Fayer, M.D. Testing the core/shell model of nanoconfined water in reverse micelles using linear and nonlinear IR spectroscopy. J. Phys. Chem. A 2006, 110, 4985–4999. [Google Scholar] [CrossRef] [PubMed]
- Day, R.A.; Robinson, B.H.; Julian, H.R.; Clarke, J.H.R.; Doherty, J.V. Characterisation of water-containing reversed micelles by viscosity and dynamic light scattering methods. J. Chem. Soc. Faraday Trans. 1 1979, 75, 132–139. [Google Scholar] [CrossRef]
- Crans, D.C.; Baruah, B.; Ross, A.; Levinger, N.E. Impact of confinement and interfaces on coordination chemistry: Using oxovanadate reactions and proton transfer reactions as probes in reverse micelles. Coord. Chem. Rev. 2009, 253, 2178–2185. [Google Scholar] [CrossRef]
- Ooms, K.J.; Bolte, S.E.; Baruah, B.; Choudhary, M.A.; Crans, D.C.; Polenova, T. V-51 solid-state NMR and density functional theory studies of eight-coordinate non-oxo vanadium complexes: Oxidized amavadin. Dalton Trans. 2009, 3262–3269. [Google Scholar] [CrossRef]
- Durantini, A.M.; Falcone, R.D.; Silber, J.J.; Correa, N.M. A New Organized Media: Glycerol:N,N-Dimethylformamide Mixtures/AOT/n-Heptane Reversed Micelles. The Effect of Confinement on Preferential Solvation. J. Phys. Chem. B 2011, 115, 5894–5902. [Google Scholar] [CrossRef] [PubMed]
- Malin, J.M.; Schmidt, C.F.; Toma, H.E. Carbon-13 and proton nuclear magnetic resonance spectra of some pentacyanoferrate(II) complexes. Inorg. Chem. 1975, 14, 2924–2928. [Google Scholar] [CrossRef]
- Felix, F.; Hauser, U.; Siegenthaler, H.; Wenk, F.; Ludi, A. Mixed valence properties of μ-pyrazine-decacyanodiiron(II, III). Inorg. Chim. Acta 1975, 15, L7–L8. [Google Scholar] [CrossRef]
- Toma, H.E.; Malin, J.M. Kinetics of formation and stability constants of some pentacyanoferrate(II) complexes of aromatic nitrogen heterocycles. Inorg. Chem. 1973, 12, 2080–2083. [Google Scholar] [CrossRef]
- Willard, D.M.; Riter, R.E.; Levinger, N.E. Dynamics of polar solvation in lecithin/water/cyclohexane reverse micelles. J. Am. Chem. Soc. 1998, 120, 4151–4160. [Google Scholar] [CrossRef]
- Raju, B.B.; Costa, S.M.B. Excited-state behavior of 7-diethylaminocoumarin dyes in AOT reversed micelles: Size effects. J. Phys. Chem B 1999, 103, 4309–4317. [Google Scholar] [CrossRef]
- Crans, D.C.; Trujillo, A.M.; Bonetti, S.; Rithner, C.D.; Baruah, B.; Levinger, N.E. Penetration of Negatively Charged Lipid Interfaces by the Doubly Deprotonated Dipicolinate. J. Org. Chem. 2008, 73, 9633–9640. [Google Scholar] [CrossRef] [PubMed]
- Zingaretti, L.; Correa, N.M.; Boscatto, L.; Chiacchiera, S.M.; Durantini, E.N.; Bertolotti, S.G.; Rivarola, C.R.; Silber, J.J. Distribution of amines in water/AOT/n-hexane reverse micelles: Influence of the amine chemical structure. J. Colloid Interface Sci. 2005, 286, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Rithner, C.D.; Baruah, B.; Gourley, B.L.; Levinger, N.E. Molecule probe location in reverse micelles determined by NMR dipolar interactions. J. Am. Chem. Soc. 2006, 128, 4437–4445. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lombardo, I.; Baruah, B.; Alvarez, S.; Werst, K.R.; Segaline, N.A.; Levinger, N.E.; Crans, D.C. Size and shape trump charge in interactions of oxovanadates with self-assembled interfaces: Application of Continuous Shape Measure analysis to the decavanadate anion. New J. Chem. 2016, 40, 962–975. [Google Scholar] [CrossRef]
- Samart, N.; Beuning, C.; Haller, K.; Rithner, C.D.; Crans, D.C. Interaction of a biguanide compound with membrane model interface systems: Probing properties of antimalaria and antidiabetic compounds. Langmuir 2014, 30, 8697–8706. [Google Scholar] [CrossRef] [PubMed]
- Lepori, C.M.O.; Correa, N.M.; Silber, J.J.; Falcone, R.D. How the cation 1-butyl-3-methylimidazolium impacts the interaction between the entrapped water and the reverse micelles interface created with an ionic liquid-like surfactant. Soft Matter 2016, 12, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.O.; Fernandez, M.A.; Silber, J.J.; de Rossi, R.H.; Correa, N.M. Inhibited phenol ionization in reverse micelles: Confinement effect at the nanometer scale. Chemphyschem 2012, 13, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Agazzi, F.; Correa, N.M.; Rodriquez, J. Molecular dynamic simulation of water/BHDC cationic reverse micelles. Structural characterization, dynamical properties, and influence of solvent on intermicellar interactions. Langmuir 2014, 30, 9643–9653. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Levinger, N.E. The conundrum of pH in water nanodroplets: Sensing pH in reverse micelle water pools. Acc. Chem. Res. 2012, 45, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, M.; Cole, R.L.; Rithner, C.D.; Crans, D.C.; Levinger, N.E. Correlating Proton Transfer Dynamics to Probe Location in Confined Environments. J. Am. Chem. Soc. 2012, 134, 11904–11907. [Google Scholar] [CrossRef] [PubMed]
- Sripradite, J.; Miller, S.A.; Johnson, M.D.; Tongraar, A.; Crans, D.C. How Interfaces Affect the Acidity of the Anilinium Ion. Chem. Eur. J. 2016, 22, 3873–3880. [Google Scholar] [CrossRef] [PubMed]
- Levinger, N.E.; Rubenstrunk, L.C.; Baruah, B.; Crans, D.C. Acidification of reverse micellar nanodroplets by CO2. J. Am. Chem. Soc. 2011, 133, 7205–7213. [Google Scholar] [CrossRef] [PubMed]
- Blach, D.; Pessego, M.; Silber, J.J.; Correa, N.M.; García-Río, L.; Falcone, R.D. Ionic liquids entrapped in reverse micelles as nanoreactors for bimolecular nucleophilic substitution reaction. Effect of the confinement on the chloride ion availability. Langmuir 2014, 30, 12130–12137. [Google Scholar] [CrossRef] [PubMed]
- Crosio, M.A.; Correa, N.M.; Silber, J.J.; Falcone, R.D. Aprotic ionic liquid, when entrapped in cationic reverse micelles, can be used as a suitable solvent for a bimolecular nucleophilic substitution reaction. Org. Biomol. Chem. 2016, 14, 3170–3177. [Google Scholar] [CrossRef] [PubMed]
- Correa, N.M.; Durantini, E.N.; Silber, J.J. Influence of anionic and cationic reverse micelles on nucleophilic aromatic substitution reactions between 1-fluoro-2,4-dinitrobenzene and piperidine. J. Org. Chem. 2000, 65, 6427–6433. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Schoeberl, S.; Gaidamauskas, E.; Baruah, B.; Roess, D.A. Antidiabetic vanadium compound and membrane interfaces: Interface facilitated metal complex hydrolysis. J. Biol. Inorg. Chem. 2011, 16, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Chatkon, A.; Chatterjee, P.B.; Sedgwick, M.A.; Haller, K.J.; Crans, D.C. Counterion affects interaction with interfaces: The antidiabetic drugs metformin and decavanadate. Eur. J. Inorg. Chem. 2013, 2013, 1859–1868. [Google Scholar] [CrossRef]
- Moyano, F.; Setjen, E.; Silber, J.J.; Correa, N.M. Enzymatic hydrolysis of N-benzoyl-l-tyrosine p-nitroanilide by α-chymotrypsin in DMSO-water/AOT/n-heptane reverse micelles.A unique interfacial effect on the enzymatic activity. Langmuir 2013, 29, 8245–8254. [Google Scholar] [CrossRef] [PubMed]
- Biasutti, M.A.; Abuin, E.B.; Silber, J.J.; Correa, N.M.; Lissi, E.A. Kinetics of reactions catalyzed by enzymes in solutions of surfactant. Adv. Colloid Interface Sci. 2008, 136, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Solis, A.K.C.; Correa, N.M.; Molina, P.G. Determination of benzyl-hexadecyldimethylammonium 1,4-bis(2-ethylhexyll)sulfosuccinate vesicle permeability by using square wave voltammetry and an enzymatic reaction. Langmuir 2017, 33, 12080–12086. [Google Scholar] [CrossRef] [PubMed]
- Stahla, M.L.; Baruah, B.; James, D.M.; Johnson, M.D.; Levinger, N.E.; Crans, D.C. 1H NMR studies of aerosol-OT reverse micelles with alkali and magnesium counterions: Preparation and analysis of MAOTs. Langmuir 2008, 24, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Zulauf, M.; Eicke, H.-F. Inverted Micelles and Microemulsions in the Ternary-System H2O-Aerosol-OT-Isooctane as Studied by Photon Correlation Spectroscopy. J. Phys. Chem. 1979, 83, 480–486. [Google Scholar] [CrossRef]
- Winter, P.W.; Al-Qatati, A.; Wolf-Ringwall, A.; Schoeberl, S.; Van Orden, A.K.; Barisas, B.G.; Roess, D.A.; Crans, D.C. The anti-diabetic bis(maltolato)oxovanadium(IV) (BMOV) reduce insulin receptor lateral diffusion and increase receptor confinement in membrane microdomains. Dalton Trans. 2012, 41, 6419–6430. [Google Scholar] [CrossRef] [PubMed]
- Bakas, L.; Verza, G.; Cortizo, A. Effect of vanadium compounds on the lipid organization of liposomes and cell membranes. Biol. Trace Elem. Res. 2001, 80, 269–279. [Google Scholar] [CrossRef]
- Sarkar, Y.; Majumder, R.; Das, S.; Ray, A.; Parui, P.P. Detection of curvature-radius-dependent interfacial pH/polarity for amphiphilic self-assemblies:positive versus negative curvature. Langmuir 2018. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.A.; Salazar, J.V.; Salinas, M.; Gaitani, C.M.; Nurkiewicz, T.; Negrete, G.R.; Garcia, C.D. Self-assembled nanospheres for encapsulation and aerosolization of rifampicin. RSC Adv. 2016, 6, 12959–12963. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, G.M.; Wunderliallenspach, H. Partition-Coefficients in-Vitro—Artificial Membranes as a Standardized Distribution Model. Eur. J. Pharm. Sci. 1994, 1, 273–282. [Google Scholar] [CrossRef]
- Ataf, A.A.; Lal, B.; Badshah, A.; Usman, M.; Chatterjee, P.B.; Huq, F.; Ullah, S.; Crans, D.C. Synthesis, structural characterization, modal membrane interaction and anti-tumor cell line studies of nitrophenyl ferrocenes. J. Mol. Structure. 2016, 1113, 162–170. [Google Scholar] [CrossRef]
- Jain, M.K. Introduction to Biological Membranes, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1988. [Google Scholar]
Sample Availability: Compounds are readily available by synthesis from commercially available material according to literature procedures. |


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borunda, T.; Myers, A.J.; Mary Fisher, J.; Crans, D.C.; Johnson, M.D. Confinement Effects on Chemical Equilibria: Pentacyano(Pyrazine)Ferrate(II) Stability Changes within Nanosized Droplets of Water. Molecules 2018, 23, 858. https://doi.org/10.3390/molecules23040858
Borunda T, Myers AJ, Mary Fisher J, Crans DC, Johnson MD. Confinement Effects on Chemical Equilibria: Pentacyano(Pyrazine)Ferrate(II) Stability Changes within Nanosized Droplets of Water. Molecules. 2018; 23(4):858. https://doi.org/10.3390/molecules23040858
Chicago/Turabian StyleBorunda, Teofilo, Alexander J. Myers, J. Mary Fisher, Debbie C. Crans, and Michael D. Johnson. 2018. "Confinement Effects on Chemical Equilibria: Pentacyano(Pyrazine)Ferrate(II) Stability Changes within Nanosized Droplets of Water" Molecules 23, no. 4: 858. https://doi.org/10.3390/molecules23040858
APA StyleBorunda, T., Myers, A. J., Mary Fisher, J., Crans, D. C., & Johnson, M. D. (2018). Confinement Effects on Chemical Equilibria: Pentacyano(Pyrazine)Ferrate(II) Stability Changes within Nanosized Droplets of Water. Molecules, 23(4), 858. https://doi.org/10.3390/molecules23040858