Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis

 , , , , , ,
, , , , , ,  , ,
, ,  and
and 
Abstract
: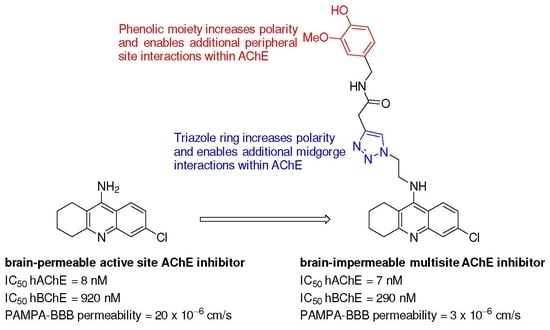
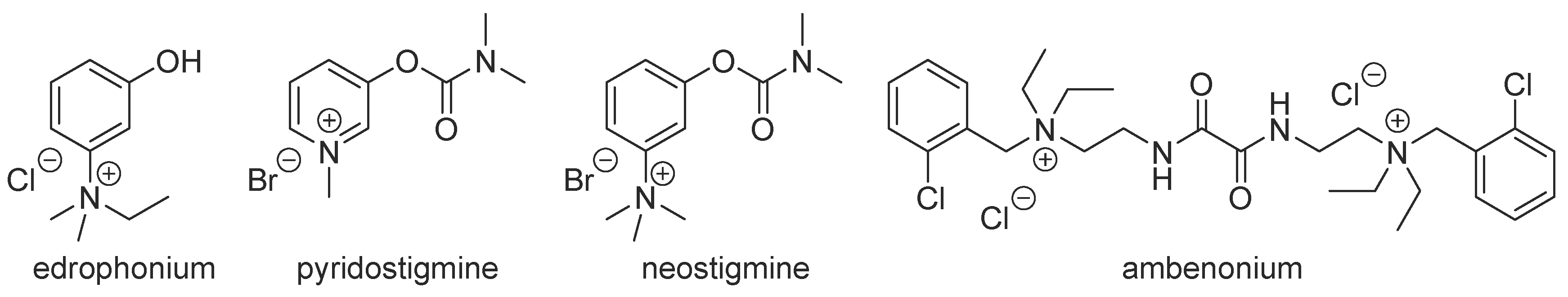
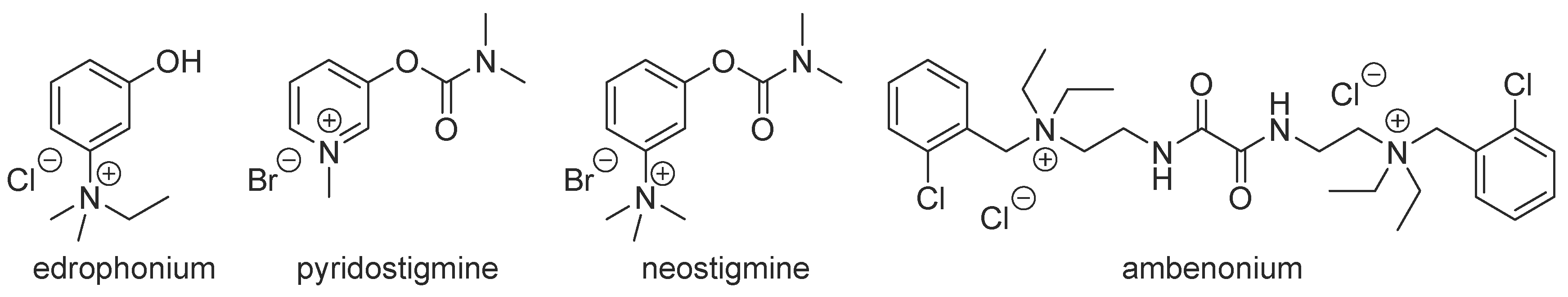
1. Introduction
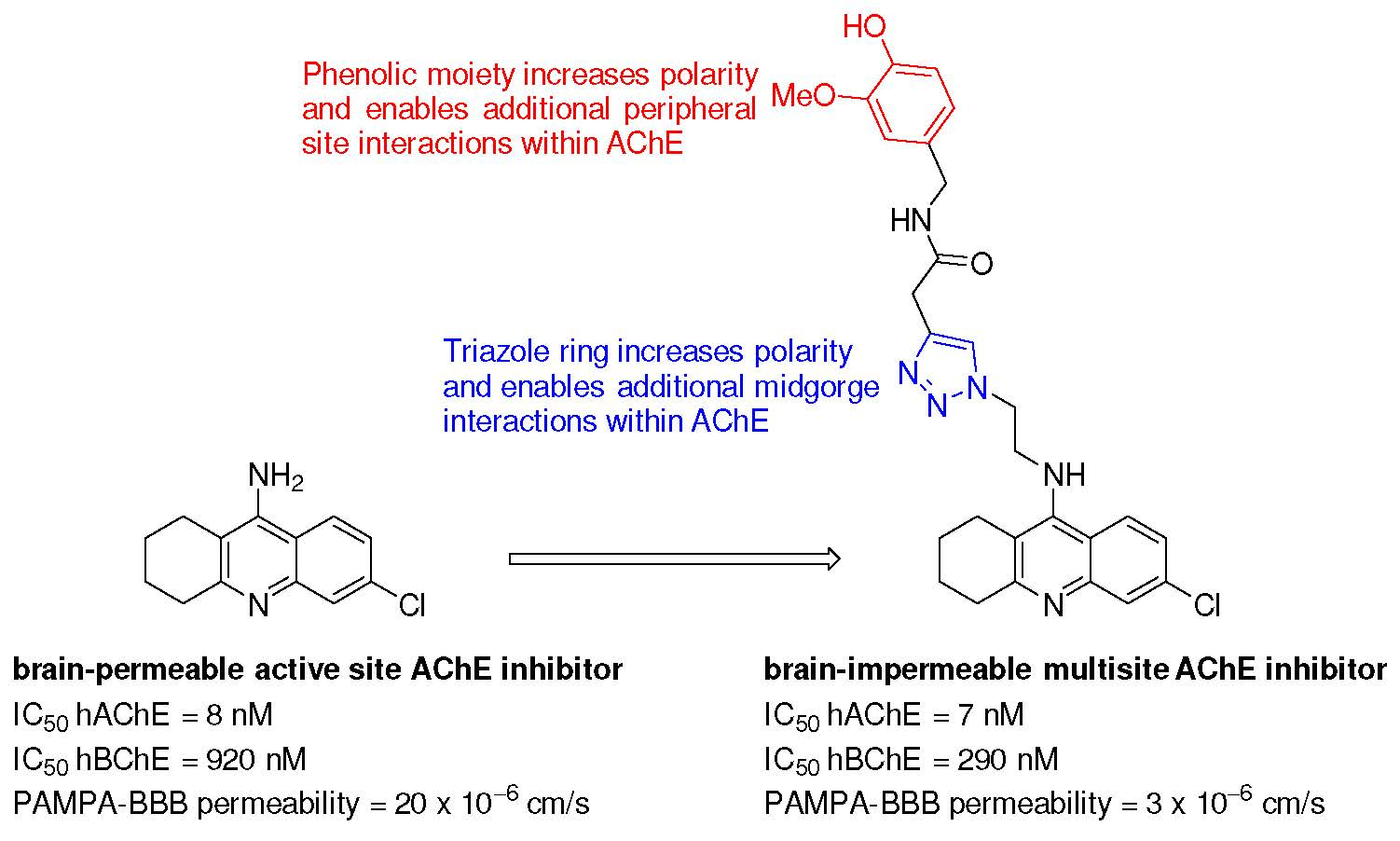
2. Results and Discussion
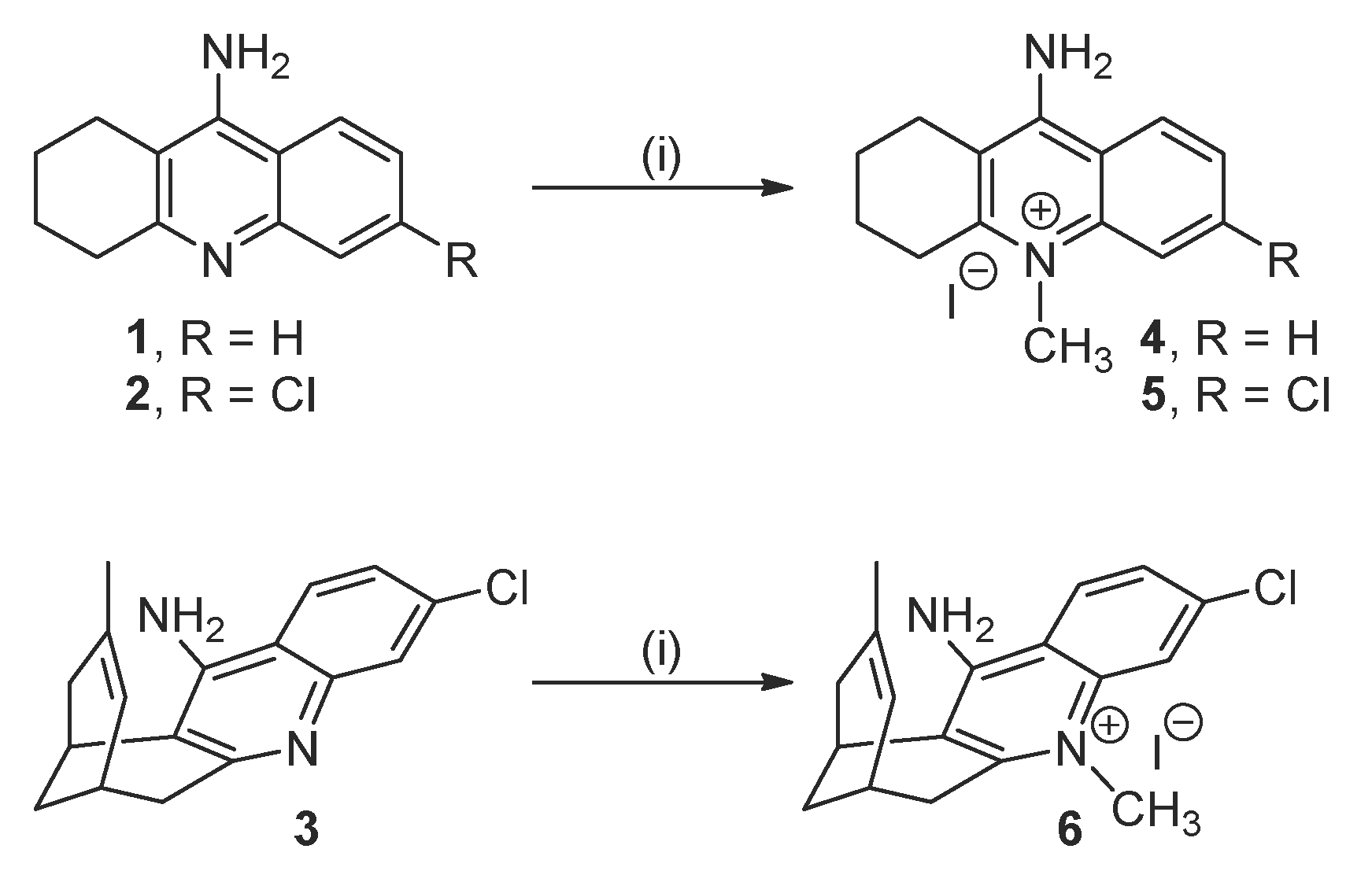
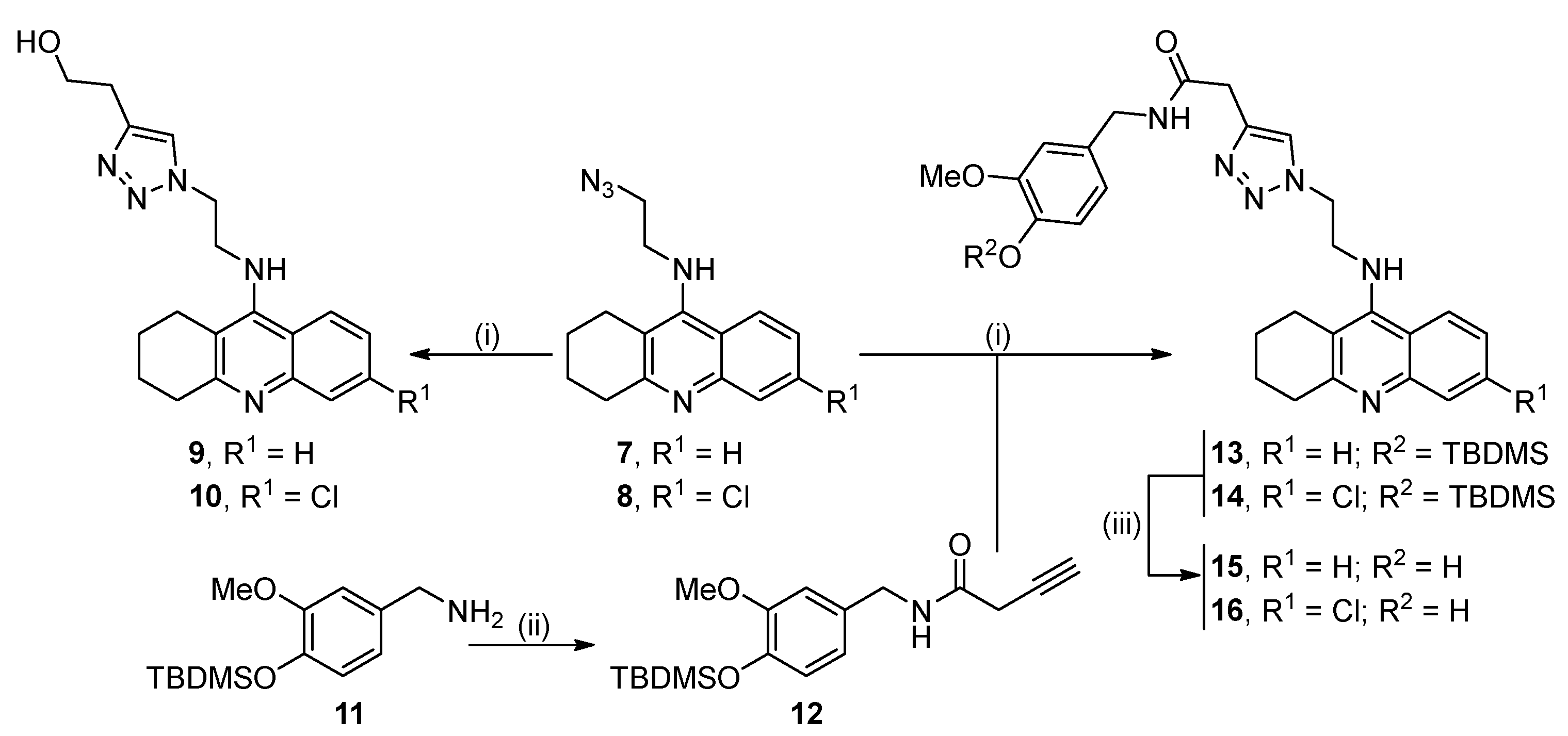
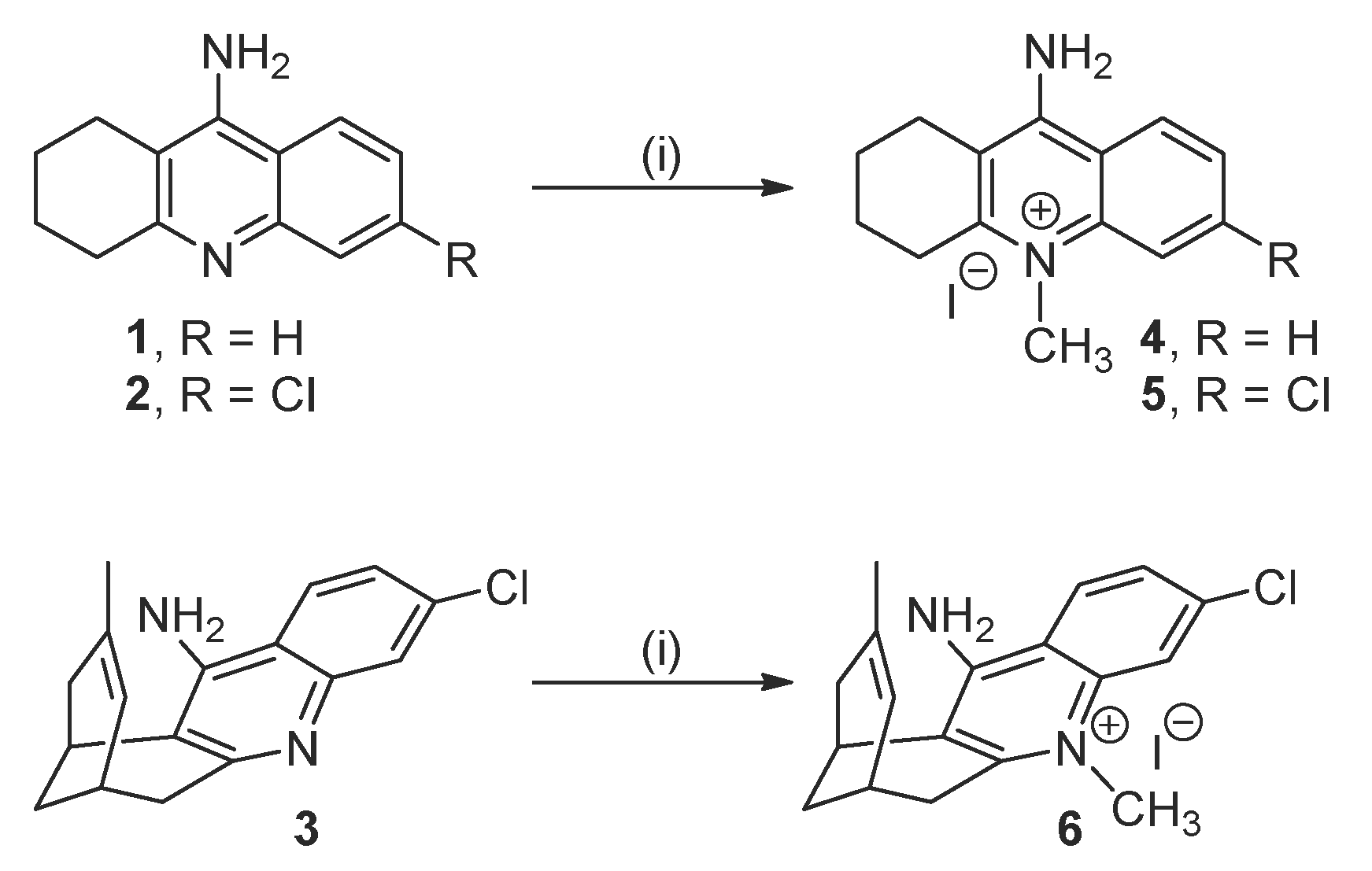
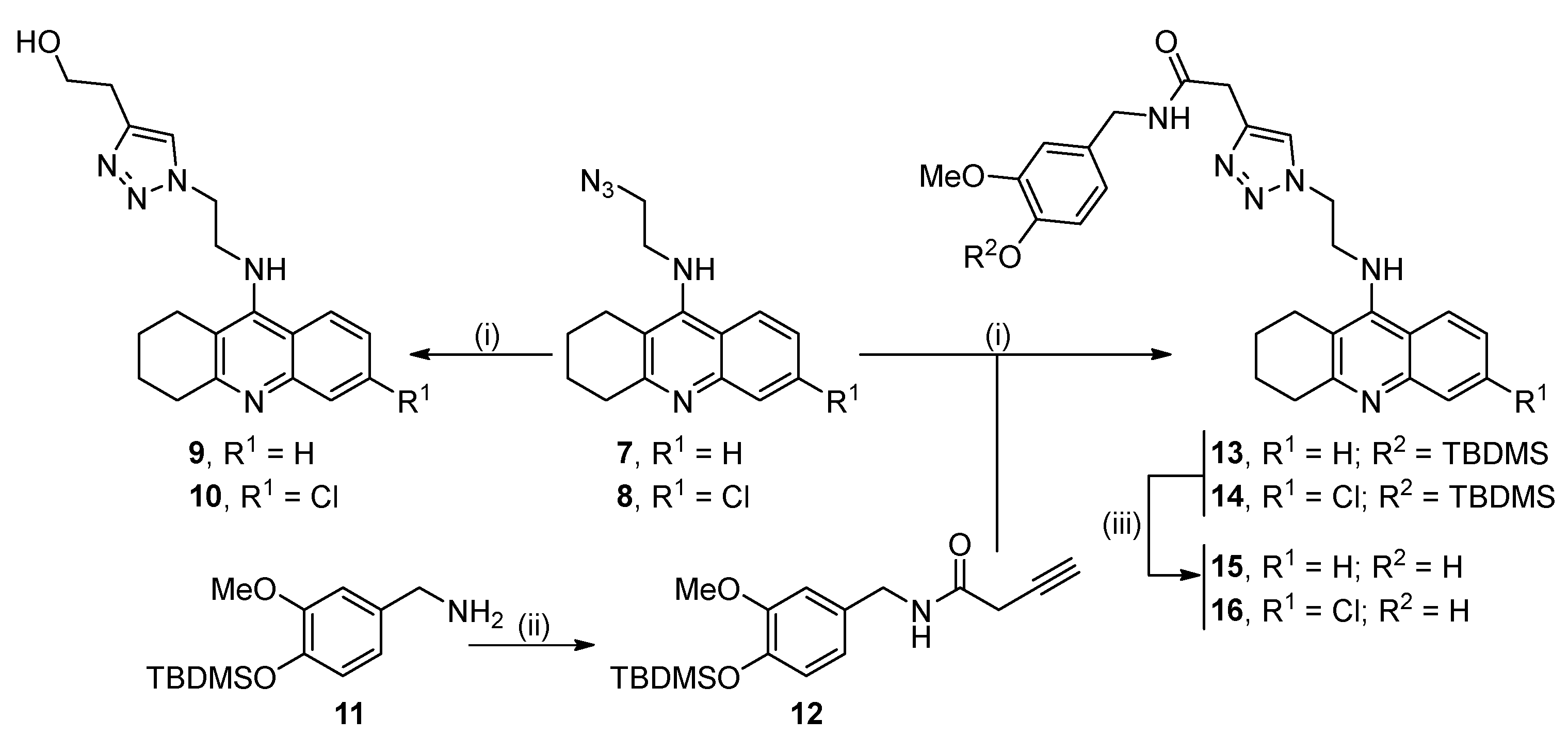
2.1. Synthesis of the Target Compounds
2.2. Biological Profiling of the Target Compounds
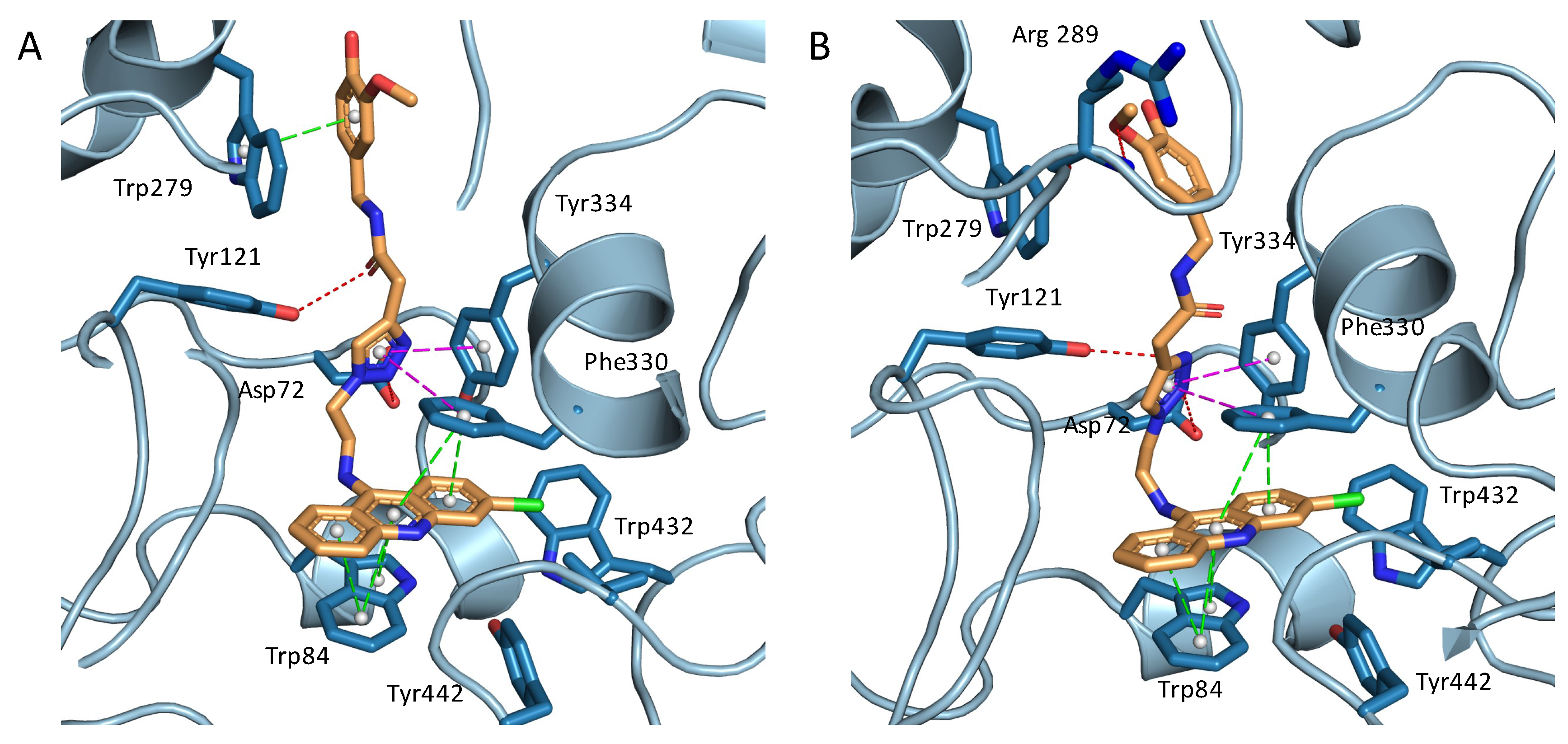
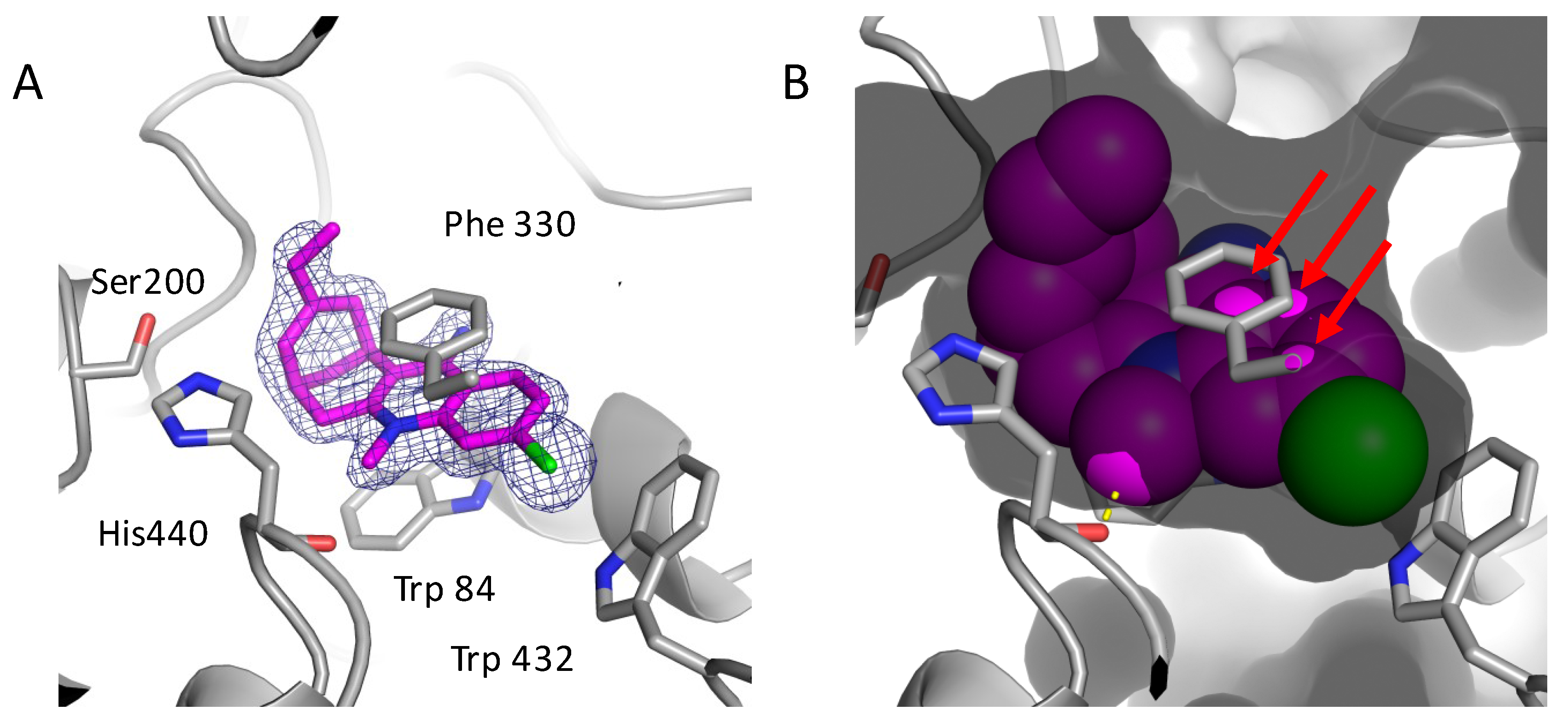
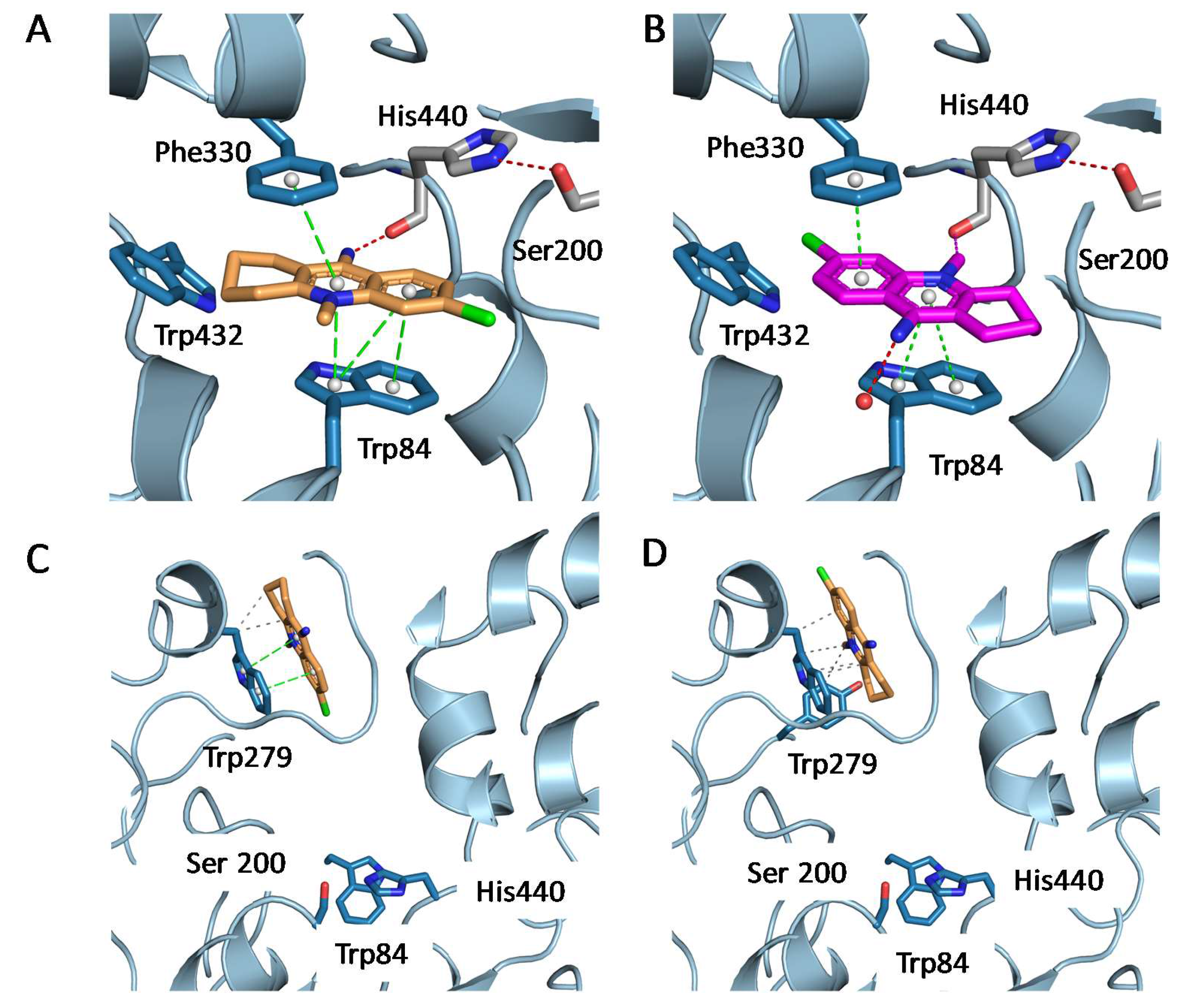
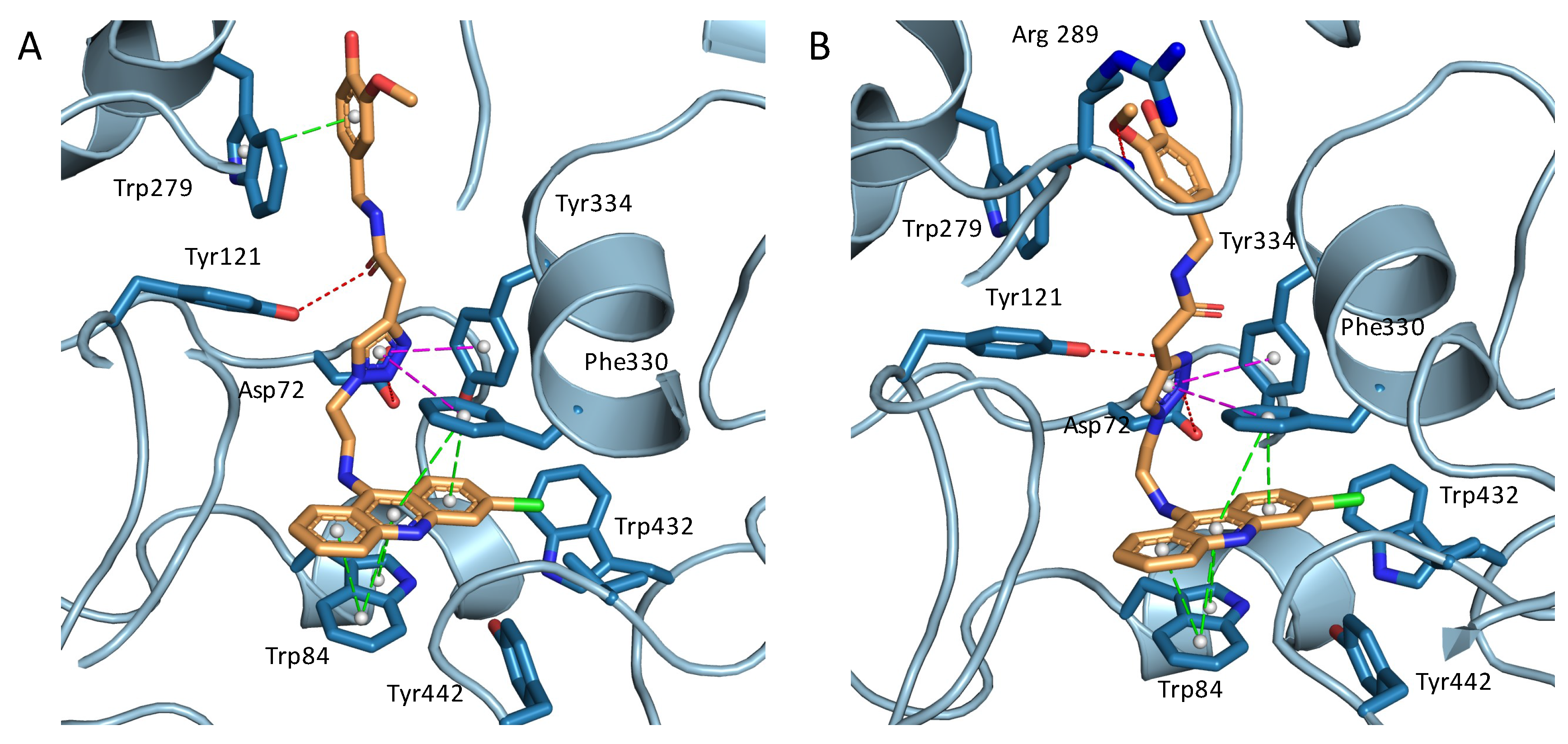
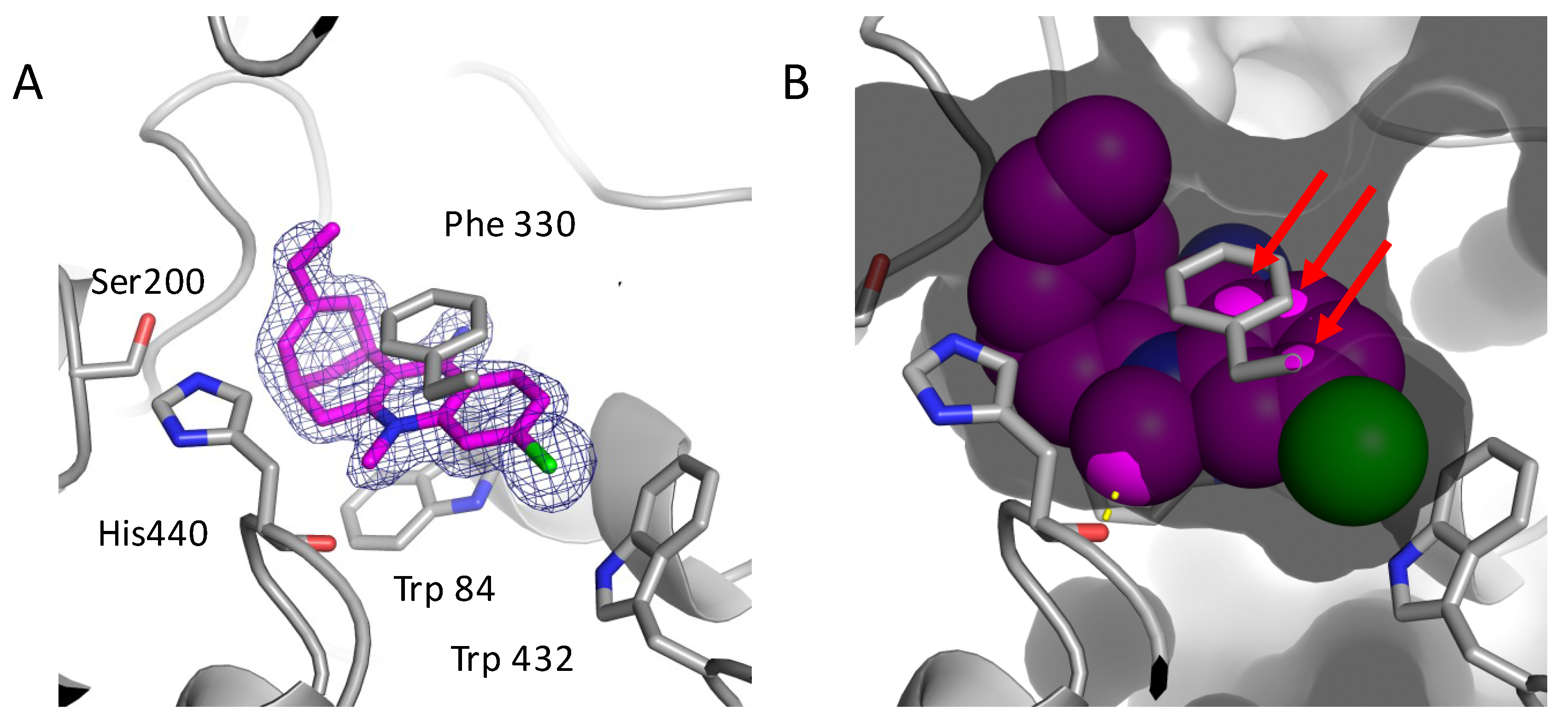
2.3. Structural Characterization of Complexes of Compounds 5, 6, and 16 with TcAChE
3. Materials and Methods
3.1. Chemistry
General Methods
3.2. Biological Profiling
3.2.1. Evaluation of hAChE and hBChE Inhibitory Activity
3.2.2. PAMPA-BBB Assay
3.3. Structural Biology
3.3.1. Crystallization and Data Collection
3.3.2. Data Processing and Refinement
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Melzer, N.; Ruck, T.; Fuhr, P.; Gold, R.; Hohlfeld, R.; Marx, A.; Melms, A.; Tackenberg, B.; Schalke, B.; Schneider-Gold, C.; et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: A supplement to the guidelines of the German Neurological Society. J. Neurol. 2016, 263, 1473–1494. [Google Scholar] [CrossRef] [PubMed]
- Conti-Fine, B.M.; Milani, M.; Kaminski, H.J. Myasthenia gravis: Past, present, and future. J. Clin. Investig. 2006, 116, 2843–2854. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.S.; Cardwell, C.R.; McCarron, P.O.; McConville, J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Binks, S.; Vincent, A.; Palace, J. Myasthenia gravis: A clinical-immunological update. J. Neurol. 2016, 263, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Brenner, T.; Nizri, E.; Irony-Tur-Sinai, M.; Hamra-Amitay, Y.; Wirguin, I. Acetylcholinesterase inhibitors and cholinergic modulation in Myasthenia Gravis and neuroinflammation. J. Neuroimmunol. 2008, 201–202, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Punga, A.R.; Stålberg, E. Acetylcholinesterase inhibitors in myasthenia gravis: To be or not to be? Muscle Nerve 2009, 39, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Alkhawajah, N.M.; Oger, J. Treatment of myasthenia gravis in the aged. Drugs Aging 2015, 32, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Komloova, M.; Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Structure-activity relationship of quaternary acetylcholinesterase inhibitors—Outlook for early myasthenia gravis treatment. Curr. Med. Chem. 2010, 17, 1810–1824. [Google Scholar] [CrossRef] [PubMed]
- Lindovský, J.; Petrov, K.; KrůšeK, J.; Reznik, V.S.; Nikolsky, E.E.; Vyskočil, F. Effect of tissue-specific acetylcholinesterase inhibitor C-547 on α3β4 and αβεδ acetylcholine receptors in COS cells. Eur. J. Pharmacol. 2012, 688, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.K.; Cowley, P.; Muir, A.W.; Palin, R.; Pow, E.; Prosser, A.B.; Taylor, R.; Zhang, M.Q. Quaternary salts of E2020 analogues as acetylcholinesterase inhibitors for the reversal of neuromuscular block. Bioorg. Med. Chem. Lett. 2002, 12, 2565–2568. [Google Scholar] [CrossRef]
- Palin, R.; Clark, J.K.; Cowley, P.; Muir, A.W.; Pow, E.; Prosser, A.B.; Taylor, R.; Zhang, M.Q. Novel piperidinium and pyridinium agents as water-soluble acetylcholinesterase inhibitors for the reversal of neuromuscular blockade. Bioorg. Med. Chem. Lett. 2002, 12, 2569–2572. [Google Scholar] [CrossRef]
- Musilek, K.; Komloova, M.; Zavadova, V.; Holas, O.; Hrabinova, M.; Pohanka, M.; Dohnal, V.; Nachon, F.; Dolezal, M.; Kuca, K.; et al. Preparation and in vitro screening of symmetrical bispyridinium cholinesterase inhibitors bearing different connecting linkage—Initial study for Myasthenia gravis implications. Bioorg. Med. Chem. Lett. 2010, 20, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Komloova, M.; Musilek, K.; Horova, A.; Holas, O.; Dohnal, V.; Gunn-Moore, F.; Kuca, K. Preparation, in vitro screening and molecular modelling of symmetrical bis-quinolinium cholinesterase inhibitors—Implications for early Myasthenia gravis treatment. Bioorg. Med. Chem. Lett. 2011, 21, 2505–2509. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Komloova, M.; Holas, O.; Hrabinova, M.; Pohanka, M.; Dohnal, V.; Nachon, F.; Dolezal, M.; Kuca, K. Preparation and in vitro screening of symmetrical bis-isoquinolinium cholinesterase inhibitors bearing various connecting linkage—Implications for early Myasthenia gravis treatment. Eur. J. Med. Chem. 2011, 46, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Mary, A.; Renko, D.Z.; Guillou, C.; Thal, C. Potent acetylcholinesterase inhibitors: Design, synthesis, and structure-activity relationships of bis-interacting ligands in the galanthamine series. Bioorg. Med. Chem. 1998, 6, 1835–1850. [Google Scholar] [CrossRef]
- Freeman, S.E.; Dawson, R.M. Tacrine: A pharmacological review. Prog. Neurobiol. 1991, 36, 257–277. [Google Scholar] [CrossRef]
- Gregor, V.E.; Emmerling, M.R.; Lee, C.; Moore, C.J. The synthesis and in vitro acetylcholinesterase and butyrylcholinesterase inhibitory activity of tacrine (Cognex®) derivatives. Bioorg. Med. Chem. Lett. 1992, 2, 861–864. [Google Scholar] [CrossRef]
- Camps, P.; El Achab, R.; Görbig, D.M.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; et al. Synthesis, in vitro pharmacology, and molecular modeling of very potent tacrine-huperzine A hybrids as acetylcholinesterase inhibitors of potential interest for the treatment of Alzheimer’s disease. J. Med. Chem. 1999, 42, 3227–3242. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; El Achab, R.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; Luque, F.J. New tacrine-huperzine A hybrids (huprines): Highly potent tight-binding acetylcholinesterase inhibitors of interest for the treatment of Alzheimer’s disease. J. Med. Chem. 2000, 43, 4657–4666. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; Cusack, B.; Mallender, W.D.; El Achab, R.; Morral, J.; Muñoz-Torrero, D.; Rosenberry, T.L. Huprine X is a novel high-affinity inhibitor of acetylcholinesterase that is of interest for treatment of Alzheimer’s disease. Mol. Pharmacol. 2000, 57, 409–417. [Google Scholar] [PubMed]
- Huisgen, R. 1,3-Dipolar cycloaddition—Introduction, survey, mechanism. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; Wiley: New York, NY, USA, 1984; Volume 1, pp. 1–176. [Google Scholar]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Bourne, Y.; Kolb, H.C.; Radić, Z.; Sharpless, K.B.; Taylor, P.; Marchot, P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.G.; Green, L.G.; Grynszpan, F.; Radić, Z.; Carlier, P.R.; Taylor, P.; Finn, M.G.; Sharpless, K.B. Click chemistry in situ: Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew. Chem. Int. Ed. 2002, 41, 1053–1057. [Google Scholar] [CrossRef]
- Oueis, E.; Santoni, G.; Ronco, C.; Syzgantseva, O.; Tognetti, V.; Joubert, L.; Romieu, A.; Weik, M.; Jean, L.; Sabot, C.; et al. Reaction site-driven regioselective synthesis of AChE inhibitors. Org. Biomol. Chem. 2014, 12, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Oueis, E.; Sabot, C.; Renard, P.Y. New insights into the kinetic target-guided synthesis of protein ligands. Chem. Commun. 2015, 51, 12158–12169. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, D.; Davis, C.A.; Herrero, B.D.R.; Ganellin, C.R.; Dunn, P.M.; Jenkinson, D.H. Synthesis and structure-activity relationships of dequalinium analogues as K+ channel blockers. Investigations on the role of the charged heterocycle. J. Med. Chem. 1995, 38, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Manetsch, R.; Krasiński, A.; Radić, Z.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In situ click chemistry: Enzyme inhibitors made to their own specifications. J. Am. Chem. Soc. 2004, 126, 12809–12818. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, H.; Xu, M. Synthesis and bioactivity of novel tacrine-indole hybrids by microwave-assisted Huisgen [3 + 2] cycloaddition reaction. Chin. J. Org. Chem. 2012, 32, 413–419. [Google Scholar] [CrossRef]
- Camps, P.; Formosa, X.; Galdeano, C.; Gómez, T.; Muñoz-Torrero, D.; Scarpellini, M.; Viayna, E.; Badia, A.; Clos, M.V.; Camins, A.; et al. Novel donepezil-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation. J. Med. Chem. 2008, 51, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.G.; Metzger, A.; Brescia, M.-R.; Qin, L.-Y.; Appell, K.C.; Brain, C.T.; Hallett, A.; Ganju, P.; Denholm, A.A.; Wareing, J.R.; et al. Sulfonamido-aryl ethers as bradykinin B1 receptor antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Dvir, H.; Wong, D.M.; Harel, M.; Barril, X.; Orozco, M.; Luque, F.J.; Muñoz-Torrero, D.; Camps, P.; Rosenberry, T.L.; Silman, I.; et al. 3D Structure of Torpedo californica acetylcholinesterase complexed with huprine X at 2.1 Å resolution: Kinetic and molecular dynamics correlates. Biochemistry 2002, 41, 2970–2981. [Google Scholar] [CrossRef] [PubMed]
- Savini, L.; Gaeta, A.; Fattorusso, C.; Catalanotti, B.; Campiani, G.; Chiasserini, L.; Pellerano, C.; Novellino, E.; McKissic, D.; Saxena, A. Specific targeting of acetylcholinesterase and butyrylcholinesterase recognition sites. Rational design of novel, selective, and highly potent cholinesterase inhibitors. J. Med. Chem. 2003, 46, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Elsinghorst, P.W.; Cieslik, J.S.; Mohr, K.; Tränkle, C.; Gütschow, M. First gallamine-tacrine hybrid: Design and characterization at cholinesterases and the M2 muscarinic receptor. J. Med. Chem. 2007, 50, 5685–5695. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, A.D.; Lushchekina, S.V.; Petrov, K.A.; Kots, E.D.; Nachon, F.; Villard-Wandhammer, M.; Zueva, I.V.; Krejci, E.; Reznik, V.S.; Zobov, V.V.; et al. Slow-binding inhibition of acetylcholinesterase by an alkylammonium derivative of 6-methyluracil: Mechanism and possible advantages for myasthenia gravis treatment. Biochem. J. 2016, 473, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Petrov, K.A.; Girard, E.; Nikitashina, A.D.; Colasante, C.; Bernard, V.; Nurullin, L.; Leroy, J.; Samigullin, D.; Colak, O.; Nikolsky, E.; et al. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by α7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 2014, 34, 11870–11883. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Beeson, D.; Palace, J. Therapeutic strategies for congenital myasthenic syndromes. Ann. N. Y. Acad. Sci. 2018, 1412, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget drug design strategy: Quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Cryst. 2017, D73, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Carletti, E.; Colletier, J.-P.; Weik, M.; Nachon, F.; Jean, L.; Renard, P.-Y. Huprine derivatives as sub-nanomolar human acetylcholinesterase inhibitors: From rational design to validation by X-ray crystallography. ChemMedChem 2012, 7, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Frolow, F.; Varon, L.; Toker, L.; Futerman, A.H.; Silman, I. Purification and crystallization of a dimeric form of acetylcholinesterase from Torpedo californica subsequent to solubilization with phosphatidylinositol-specific phospholipase. J. Mol. Biol. 1988, 203, 821–823. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4–6, 9, 10, and 13 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | hAChE IC50 (μM) 1 | hBChE IC50 (μM) 1 | PAMPA-BBB Pe (10−6 cm/s) (Prediction) 2 |
|---|---|---|---|
| 4 | 12.0 ± 1.00 | 2.85 ± 0.19 | 1.70 ± 1.50 (CNS−) |
| 5 | 2.45 ± 0.28 | 22.3 ± 1.50 | 1.20 ± 0.80 (CNS−) |
| 6 | 0.06 ± 0.01 3 | 3.25 ± 0.15 | 10.90 ± 1.50 (CNS+) |
| 9 | 1.73 ± 0.12 | 0.17 ± 0.01 | 0.03 ± 0.001 (CNS−) |
| 10 | 0.20 ± 0.01 | 3.25 ± 0.17 | 0.02 ± 0.001 (CNS−) |
| 15 | 0.25 ± 0.01 | 0.05 ± 0.00 4 | 0.03 ± 0.001 (CNS−) |
| 16 | 0.01 ± 0.00 5 | 0.29 ± 0.01 | 2.97 ± 0.61 (CNS±) |
| pyridostigmine | 1.38 ± 0.07 | 2.13 ± 0.20 | - |
| tacrine | 0.32 ± 0.01 | 0.05 ± 0.00 6 | - |
| 6-chlorotacrine | 0.01 ± 0.00 7 | 0.50 ± 0.03 | 19.8 ± 0.4 (CNS+) |
| huprine Y | 8 | 0.18 ± 0.01 | 23.8 ± 2.7 (CNS+) |
| Compound | Reported Pe 1 | Experimental Pe 2 |
|---|---|---|
| Cimetidine | 0.0 | 0.7 ± 0.1 |
| Lomefloxacin | 1.1 | 0.8 ± 0.1 |
| Norfloxazin | 0.1 | 0.9 ± 0.1 |
| Ofloxazin | 0.8 | 1.0 ± 0.1 |
| Hydrocortisone | 1.9 | 1.4 ± 0.1 |
| Piroxicam | 2.5 | 1.8 ± 0.1 |
| Clonidine | 5.3 | 6.5 ± 0.1 |
| Corticosterone | 5.1 | 6.7 ± 0.1 |
| Imipramine | 13.0 | 12.3 ± 0.1 |
| Promazine | 8.8 | 13.8 ± 0.3 |
| Progesterone | 9.3 | 16.8 ± 0.3 |
| Desipramine | 12.0 | 17.8 ± 0.1 |
| Testosterone | 17.0 | 24.7 ± 1.4 |
| Verapamil | 16.0 | 25.4 ± 1.6 |
| 5 | 6 | 16 | |
|---|---|---|---|
| Resolution range (Å) | 44.04–1.79 (1.854–1.79) | 46.03–1.82 (1.82–1.89) | 45.7–1.9(1.968–1.9) |
| Space group | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| Unit cell (Å) | 91.44 106.61 150.53 | 91.74 106.44 150.68 | 91.82 106.81 151.67 |
| Total reflections | 634,220 (58,489) | 642,785 (63,116) | 1,072,187 (87,007) |
| Unique reflections | 138,453 (13,737) | 130,469 (12,879) | 117,742 (11,618) |
| Multiplicity | 4.6 (4.3) | 4.9 (4.9) | 7.7 (7.5) |
| Completeness (%) | 99.66 (99.75) | 98.4 (91.8) | 99.71 (99.38) |
| Mean I/sigma(I) | 12.9 (1.5) | 12.6 (1.6) | 8.8 (1.0) |
| Wilson B-factor (Å2) | 24.63 | 24.09 | 29.21 |
| R-merge | 0.067 (0.943) | 0.075 (1.054) | 0.128 (1.286) |
| CC1/2 | 0.998 (0.686) | 0.999 (0.719) | 0.998 (0.588) |
| Reflections used in refinement | 138,303 (13,721) | 159,670 (14,805) | 117,502 (11,589) |
| Reflections used for R-free | 6854 (681) | 7990 (773) | 5876 (580) |
| R-work | 0.1797 (0.3422) | 0.1896 (0.3100) | 0.1880 (0.3357) |
| R-free | 0.2125 (0.3788) | 0.2193 (0.3114) | 0.2177 (0.3411) |
| Number of non-hydrogen atoms | 9962 | 9719 | 9640 |
| macromolecules | 8591 | 8583 | 8564 |
| ligands | 321 | 153 | 163 |
| solvent | 1040 | 983 | 916 |
| Protein residues | 1067 | 1064 | 1068 |
| RMS (bonds) (Å) | 0.006 | 0.007 | 0.007 |
| RMS (angles) (°) | 0.88 | 0.86 | 0.88 |
| Ramachandran favored (%) | 96.29 | 96.30 | 96.03 |
| Ramachandran allowed (%) | 3.71 | 3.70 | 3.88 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.09 |
| Rotamer outliers (%) | 0.74 | 1.40 | 2.40 |
| Clashscore | 4.19 | 3.67 | 3.45 |
| Average B-factor (Å2) | 31.03 | 31.94 | 35.77 |
| macromolecules | 29.49 | 30.88 | 34.72 |
| ligands | 45.26 | 45.93 | 50.66 |
| solvent | 39.32 | 38.98 | 42.98 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galdeano, C.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Bartolini, M.; Pérez, B.; Clos, M.V.; Silman, I.; Jean, L.; Colletier, J.-P.; Renard, P.-Y.; et al. Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis. Molecules 2018, 23, 634. https://doi.org/10.3390/molecules23030634
Galdeano C, Coquelle N, Cieslikiewicz-Bouet M, Bartolini M, Pérez B, Clos MV, Silman I, Jean L, Colletier J-P, Renard P-Y, et al. Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis. Molecules. 2018; 23(3):634. https://doi.org/10.3390/molecules23030634
Chicago/Turabian StyleGaldeano, Carles, Nicolas Coquelle, Monika Cieslikiewicz-Bouet, Manuela Bartolini, Belén Pérez, M. Victòria Clos, Israel Silman, Ludovic Jean, Jacques-Philippe Colletier, Pierre-Yves Renard, and et al. 2018. "Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis" Molecules 23, no. 3: 634. https://doi.org/10.3390/molecules23030634
APA StyleGaldeano, C., Coquelle, N., Cieslikiewicz-Bouet, M., Bartolini, M., Pérez, B., Clos, M. V., Silman, I., Jean, L., Colletier, J.-P., Renard, P.-Y., & Muñoz-Torrero, D. (2018). Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis. Molecules, 23(3), 634. https://doi.org/10.3390/molecules23030634