Antioxidant and Anti-Osteoporosis Activities of Chemical Constituents of the Stems of Zanthoxylum piperitum

Abstract
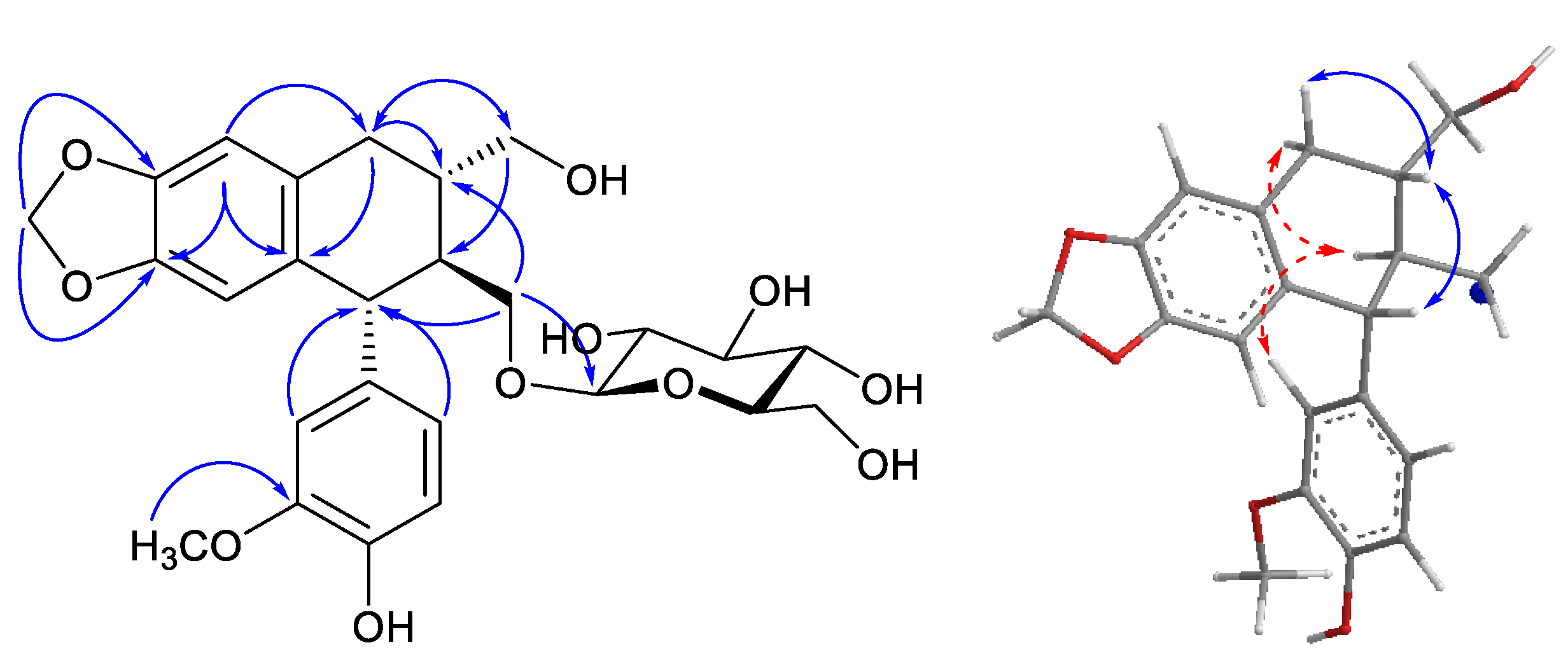
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis and Sugar Identification
3.5. Product Characterization
3.6. Oxygen Radical Absorbance Capacity (ORAC) Assay
3.7. Reducing Capacity (CUPRAC) Assay
3.8. Tartrate-Resistant Acid Phosphatase (TRAP) Assay
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harris, P.E.; Bouloux, P.-M.G. Metabolic Bone Disease. In Endocrinology in Practice, 2nd ed.; Harris, P.E., Bouloux, P.-M.G., Eds.; CRC Press: London, UK, 2014; pp. 243–261. ISBN 978-1841849515. [Google Scholar]
- Rosen, C.J.; Bouxsein, M.L. Mechanisms of Disease: Is Osteoporosis the Obesity of Bone? Nat. Clin. Pract. Rheumatol. 2006, 2, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.; Mathews, C.; Villanueva, A.; Kleerekoper, M.; Frame, B.; Rao, D. Relationships between Surface, Volume, and Thickness of Iliac Trabecular Bone in Aging and in Osteoporosis. Implications for the Microanatomic and Cellular Mechanisms of Bone Loss. J. Clin. Investig. 1983, 72, 1396–1409. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Deal, C. Osteoporosis in Elderly: Prevention and Treatment. Clin. Geriatr. Med. 2002, 18, 529–555. [Google Scholar] [CrossRef]
- Basu, S.; Michaëlsson, K.; Olofsson, H.; Johansson, S.; Melhus, H. Association Between Oxidative Stress and Bone Mineral Density. Biochem. Biophys. Ress Communs. 2001, 288, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Maggio, D.; Barabani, M.; Pierandrei, M.; Polidori, M.C.; Catani, M.; Mecocci, P.; Senin, U.; Pacifici, R.; Cherubini, A. Marked Decrease in Plasma Antioxidants in Aged Osteoporotic women: Results of a Cross-Sectional Study. J. Clin. Endocrinol. Metab. 2003, 88, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.J.; Barrett-Connor, E.L.; Schneider, D.L. Vitamin c Supplement Use and Bone Mineral Density in Postmenopausal Women. J. Bone. Miner. Res. 2001, 16, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Inada, K.; Ogawa, T.-o.; Ito, H.; Yoshida, T. Aliphatic Acid Amides of the Fruits of Zanthoxylum piperitum. Phytochemistry. 2004, 65, 2599–2604. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kubota, K. Formation by Mechanical Stimulus of the Flavor Compounds in Young Leaves of Japanese Pepper (Xanthoxylum piperitum DC.). J. Agric. Food Chem. 2001, 49, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Yoshihara, K.; Hirose, Y. Constituents of Fruit Oil from Japanese Pepper. Bull. Chem. Soc. Jap. 1968, 41, 1945–1950. [Google Scholar] [CrossRef]
- Machmudah, S.; Izumi, T.; Sasaki, M.; Goto, M. Extraction of Pungent Components from Japanese Pepper (Xanthoxylum piperitum DC.) Using Supercritical CO2. Sep. Purif. Technol. 2009, 68, 159–164. [Google Scholar] [CrossRef]
- Kikuchi, M.; Kikuchi, M. Studies on the Constituents of Swertia japonica Makino II. On the Structures of New Glycosides. Chem. Pharm. Bull. 2005, 53, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.F.; Lee, I.S.; Dat, N.T.; Shen, G.; Kang, J.S.; Kim, D.H.; Kim, Y.H. Inhibitory Lignans Against NFAT Transcription Factor from Acanthopanax koreanum. Arch. Pharm. Res. 2004, 27, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Lin, H.; Qian, Z. Two New Furofuran Lignans from Kandelia obovata. Heterocycl. Int. J. Rev. Commun. Heterocycl. Chem. 2013, 87, 1093–1098. [Google Scholar] [CrossRef]
- Choi, J.S.; Young, H.S.; Park, J.C.; Choi, J.-H.; Woo, W.S. Flavonoids from the Leaves of Rhododendron brachycarpum. Arch. Pharm. Res. 1986, 9, 233–236. [Google Scholar] [CrossRef]
- Pyo, M.-K.; Koo, Y.-K.; YunChoi, H.-S. Anti-platelet Effect of the Phenolic Constituents Isolated from the Leaves of Magnolia obovata. Nat. Prod. Sci. 2002, 8, 147–151. [Google Scholar]
- Fukunaga, T.; Nishiya, K.; Kajikawa, I.; Watanabe, Y.; Suzuki, N.; Takeya, K.; Itokawa, H. Chemical Studies on the Constituents of Hyphear tanakae Hosokawa from Different Host Trees. Chem. Pharm. Bull. 1988, 36, 1180–1184. [Google Scholar] [CrossRef]
- Flamini, G.; Antognoli, E.; Morelli, I. Two Flavonoids and Other Compounds from the Aerial Parts of Centaurea bracteata from Italy. Phytochemistry 2001, 57, 559–564. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Khamthong, N.; Sukpondma, Y.; Phongpaichit, S.; Hutadilok-Towatana, N.; Graidist, P.; Sakayaroj, J.; Kirtikara, K. Cyclohexene, Diketopiperazine, Lactone and Phenol Derivatives from the Sea Fan-derived Fungi Nigrospora sp. PSU-F11 and PSU-F12. Arch. Pharm. Res. 2010, 33, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.-J.; Chiang, H.-C. Phenylpropanoids from Aralia bipinnata. Phytochemistry. 1995, 39, 825–827. [Google Scholar] [CrossRef]
- Zhao, C.; Nagatsu, A.; Hatano, K.; Shirai, N.; Kato, S.; Ogihara, Y. New Lignan Glycosides from Chinese Medicinal Plant, Sinopodophillum emodi. Chem. Pharm. Bull. 2003, 51, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Maryanoff, B.E.; McComsey, D.F.; Craig, J.C. Chiroptical Properties and Absolute Configuration of Pyrroloisoquinoline Antidepressants. Chirality 1998, 10, 169–172. [Google Scholar] [CrossRef]
- Wen, Q.; Lin, X.; Liu, Y.; Xu, X.; Liang, T.; Zheng, N.; Kintoko; Huang, R. Phenolic and Lignan Glycosides from the Butanol Extract of Averrhoa carambola L. root. Molecules 2012, 17, 12330–12340. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.W.; Chai, H.B.; Keller, W.J.; Kinghorn, A.D. Lignans and Other Constituents of the Fruits of Euterpe oleracea (Acai) with Antioxidant and Cytoprotective Activities. J. Agric. Food Chem. 2008, 56, 7759–7764. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and Antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, E.A.; Gillespie, K.M. Estimation of Total Phenolic Content and Other Oxidation Substrates in Plant Tissues Using Folin–Ciocalteu Reagent. Nat. Prot. 2007, 2, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-T.; Lee, S.-H.; Li, W.; Sun, Y.-N.; Yang, S.-Y.; Jang, H.-D.; Kim, Y.-H. Evaluation of the Antioxidant and Anti-osteoporosis Activities of Chemical Constituents of the Fruits of Prunus mume. Food Chem. 2014, 156, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Kapoor, H.C. Antioxidants in fruits and vegetables–the millennium’s health. Int. J. Food Sci. Technol. 2001, 36, 703–725. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Deiana, M.; Jenner, A.; Halliwell, B.; Kaur, H.; Banni, S.; Corongiu, F.P.; Dessí, M.A.; Aeschbach, R. Effect of hydroxytyrosol found in extra virgin olive oil on oxidative DNA damage and on low-density lipoprotein oxidation. J. Agric. Food Chem. 1998, 46, 5181–5187. [Google Scholar] [CrossRef]
- Lee, S.-H.; Ding, Y.; Yan, X.T.; Kim, Y.-H.; Jang, H.-D. Scopoletin and scopolin isolated from artemisia iwayomogi suppress differentiation of osteoclastic macrophage RAW 264.7 cells by scavenging reactive oxygen species. J. Nat. Prod. 2013, 76, 615–620. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC a | δH b (mult., J in Hz) | δC a | δH b (mult., J in Hz) | |
| 1 | 138.6 | - | 135.6 | - |
| 2 | 114.0 | 6.60 (d, 1.8) | 110.7 | 6.83 (d, 2.1) |
| 3 | 149.2 | - | 148.2 | - |
| 4 | 146.2 | - | 147.1 | - |
| 5 | 116.2 | 6.65 (d, 7.8) | 116.0 | 6.66 (d, 8.2) |
| 6 | 123.5 | 6.55 (dd, 7.8, 1.8) | 119.9 | 6.71 (dd, 8.2, 2.1) |
| 7 | 49.7 | 3.68 * | 84.3 | 4.73 (d, 6.9) |
| 8 | 45.5 | 1.87 * | 51.8 | 2.41 (q, 6.8) |
| 9 | 70.8 | 3.54 (dd. 6.2, 10.3) | 68.5 | 3.50 (dd, 6.2, 10.3) |
| 3.69 (dd, 6.2, 10.3) | 4.11 (dd, 6.2, 10.3) | |||
| 1′ | 131.4 | - | 136.1 | - |
| 2′ | 134.5 | - | 122.7 | 6.59 (dd, 8.2, 1.3) |
| 3′ | 110.5 | 6.04 (s) | 109.1 | 6.63 (d, 8.2) |
| 4′ | 147.3 | - | 147.3 | - |
| 5′ | 147.2 | - | 149.2 | - |
| 6′ | 108.9 | 6.48 (s) | 110.1 | 6.64 (dd, 8.2, 1.3) |
| 7′ | 34.2 | 2.76 (dd, 10.1, 16.1) | 33.9 | 2.91 (dd, 3.1, 12.4) |
| 2.63 (dd, 2.8, 16.1) | 2.45 (dd, 3.1, 12.4) | |||
| 8′ | 41.0 | 1.87 * | 44.2 | 2.63 (m) |
| 9′ | 65.5 | 3.69 * | 73.7 | 3.87 (dd, 6.2, 8.2) |
| 3.59 * | 3.63 (dd, 6.2, 8.2) | |||
| 3-OCH3 | 56.6 | 3.70 * | 56.4 | 3.76 (s) |
| -OCH2O- | 101.9 | 5.70 (s) | 102.2 | 5.80 (s) |
| Glc-1 | 104.1 | 3.96 (d, 7.3) | 104.7 | 4.20 (d, 7.6) |
| 2 | 75.2 | 3.07 (m) | 75.2 | 3.15 (dd, 9.1, 7.8) |
| 3 | 78.3 | 3.19 (m) | 78.3 | 3.21 (m) |
| 4 | 71.5 | 3.20 (m) | 71.7 | 3.28 (m) |
| 5 | 78.0 | 2.99 (m) | 78.0 | 3.34 (m) |
| 6 | 62.6 | 3.56 (m) | 62.9 | 3.61 (m) |
| 3.70 (m) | 3.84 (m) | |||
| Compound (10 μM) | Peroxyl Radical-scavenging Capacity (TE, μM) a | Reducing Capacity (Copper(I) Ions, μM) |
|---|---|---|
| 1 | 13.26 ± 0.33 | 9.60 ± 0.26 |
| 2 | 15.79 ± 0.64 | 10.69 ± 0.11 |
| 3 | 14.47 ± 0.98 | 14.51 ± 0.17 |
| 4 | 12.98 ± 0.30 | 20.42 ± 0.55 |
| 5 | 15.81 ± 0.33 | 16.39 ± 0.36 |
| 6 | 19.09 ± 0.09 | 25.66 ± 0.32 |
| 7 | 22.12 ± 0.76 | 27.97 ± 0.34 |
| 8 | 22.89 ± 0.87 | 10.19 ± 0.11 |
| 9 | 26.91 ± 1.05 | 33.04 ± 0.17 |
| 10 | 20.53 ± 0.89 | 0.04 ± 0.06 |
| 11 | 5.91 ± 0.11 | 0.08 ± 0.06 |
| Compound (10 μM) b | Inhibition (%) |
|---|---|
| 3 | 88.36 ± 10.93 |
| 6 | 82.11 ± 9.31 |
| 7 | 88.36 ± 10.93 |
| 8 | 77.78 ± 4.24 |
| 9 | 77.73 ± 4.85 |
| 11 | 92.42 ± 9.50 |
| Control | 100.00 ± 9.90 |
| Untreated Control | 41.91 ± 0.04 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.Y.; Lee, S.-H.; Tai, B.H.; Jang, H.-D.; Kim, Y.H. Antioxidant and Anti-Osteoporosis Activities of Chemical Constituents of the Stems of Zanthoxylum piperitum . Molecules 2018, 23, 457. https://doi.org/10.3390/molecules23020457
Yang SY, Lee S-H, Tai BH, Jang H-D, Kim YH. Antioxidant and Anti-Osteoporosis Activities of Chemical Constituents of the Stems of Zanthoxylum piperitum . Molecules. 2018; 23(2):457. https://doi.org/10.3390/molecules23020457
Chicago/Turabian StyleYang, Seo Young, Sang-Hyun Lee, Bui Huu Tai, Hae-Dong Jang, and Young Ho Kim. 2018. "Antioxidant and Anti-Osteoporosis Activities of Chemical Constituents of the Stems of Zanthoxylum piperitum " Molecules 23, no. 2: 457. https://doi.org/10.3390/molecules23020457