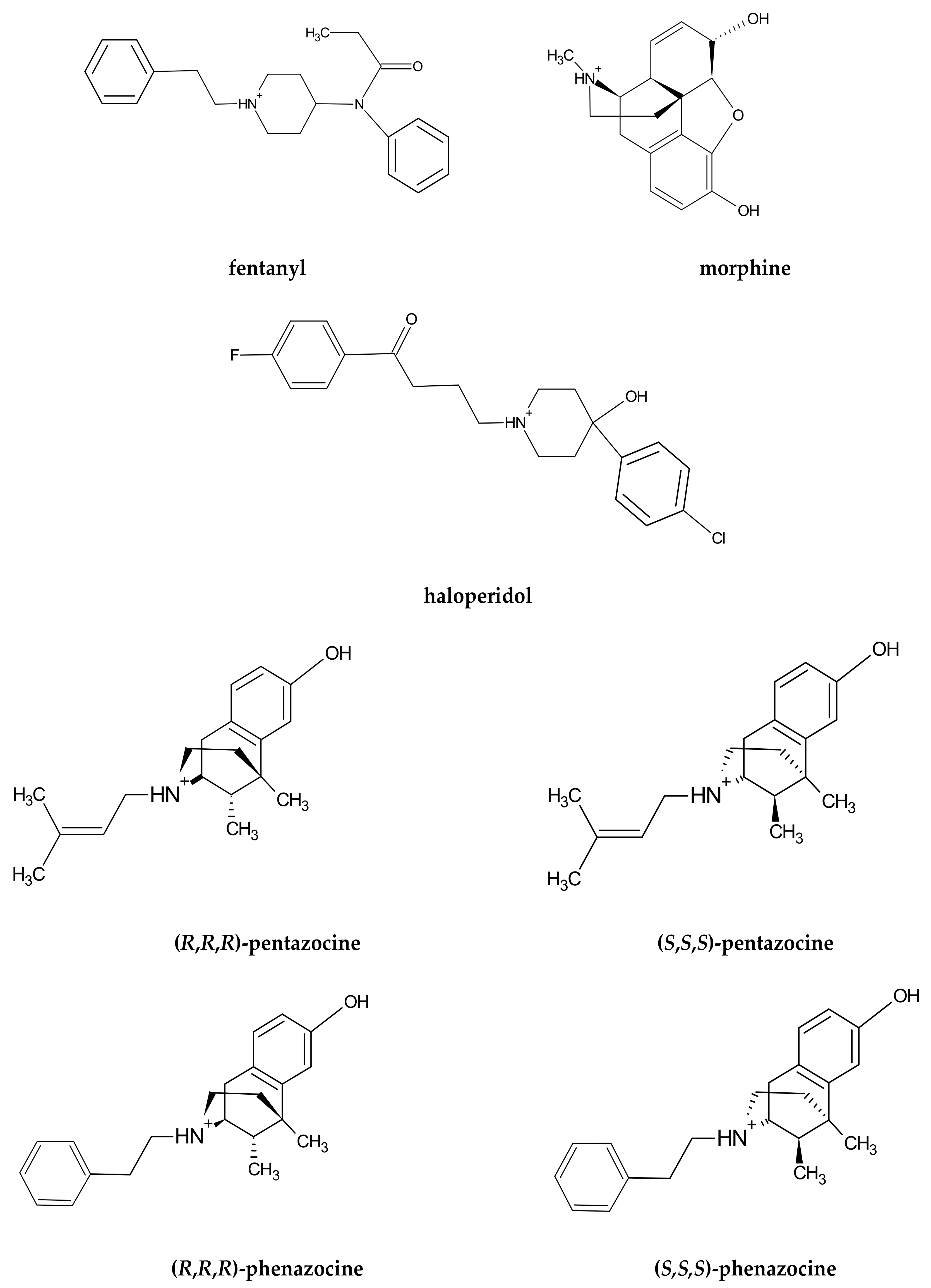
Structural Insights into σ1 Receptor Interactions with Opioid Ligands by Molecular Dynamics Simulations



Abstract
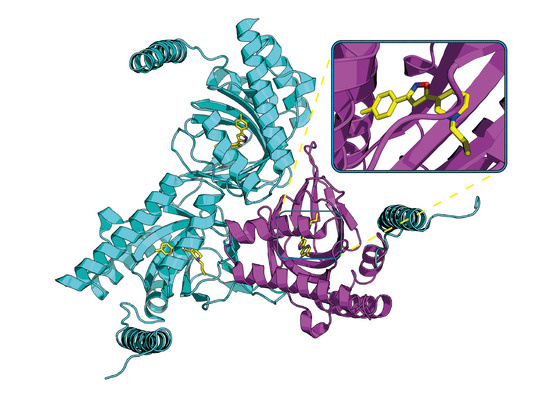
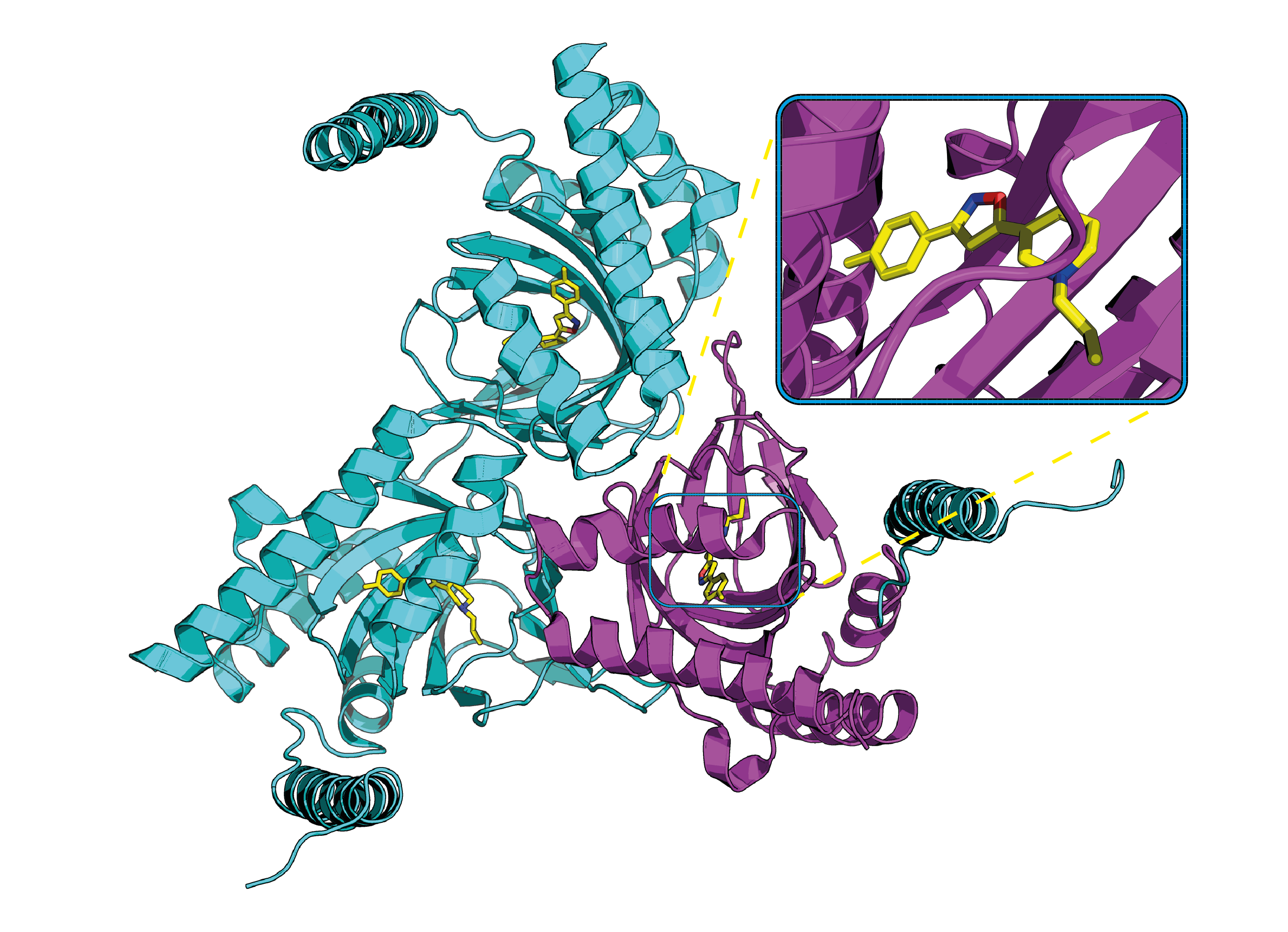
:
1. Introduction
- (i)
- Carrying out independent MD simulations for the top-ranked poses of each ligand;
- (ii)
- Examining the frequency of contacts and patterns between different ligand moieties and the receptor; ligand and different receptor residues; maps of different ligand moieties vs. different receptor residues;
- (ii)
- Determining the binding energies (ΔGbind) and decomposing them according to the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) approach to identify the most important contributions from polar and nonpolar contacts. Decomposition of ΔGbind refers to terms such as ionic bond (or salt bridge) and hydrogen bond where the latter contains also attractive van der Waals and hydrophobic interactions essential for stabilizing the structure;
- (iv)
- Searching for a relationship/trend between ΔGbind and hydrophobicity/hydrophilicity of the residues through the linear regressions based on the studied ligands being treated as a training set.
2. Method
2.1. Ligand Preparation
2.2. Receptor Preparation
2.3. Molecular Docking of Ligands to the σ1 Receptor
2.4. Molecular Dynamics Simulation of the Receptor–Ligand Complex
2.5. The Formal Analysis of the MD Simulations and Presentation
2.6. MM/PBSA Calculations for Binding Free Energy
2.7. Simulation of the PD144418 Ligand
3. Results and Discussions
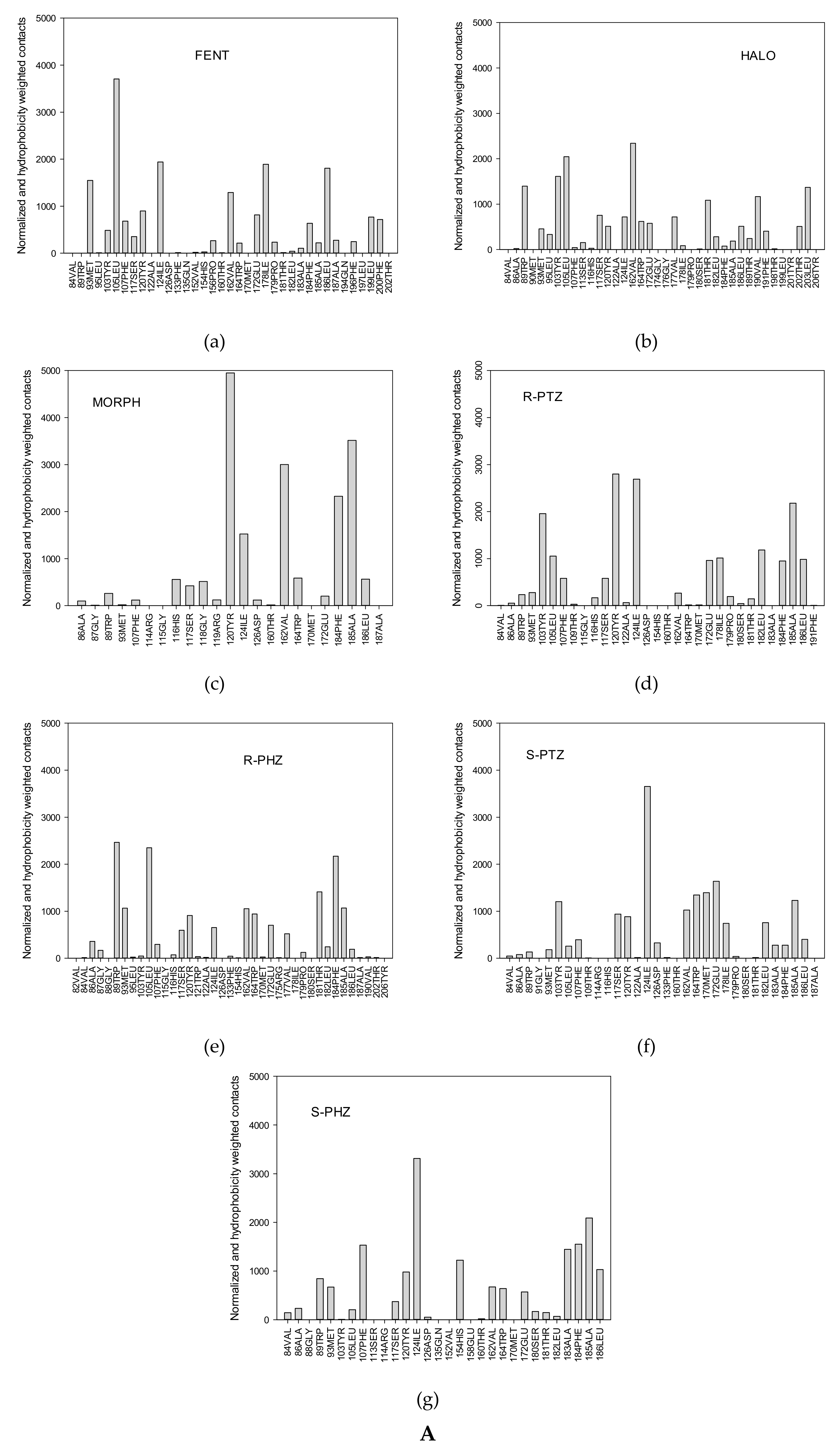
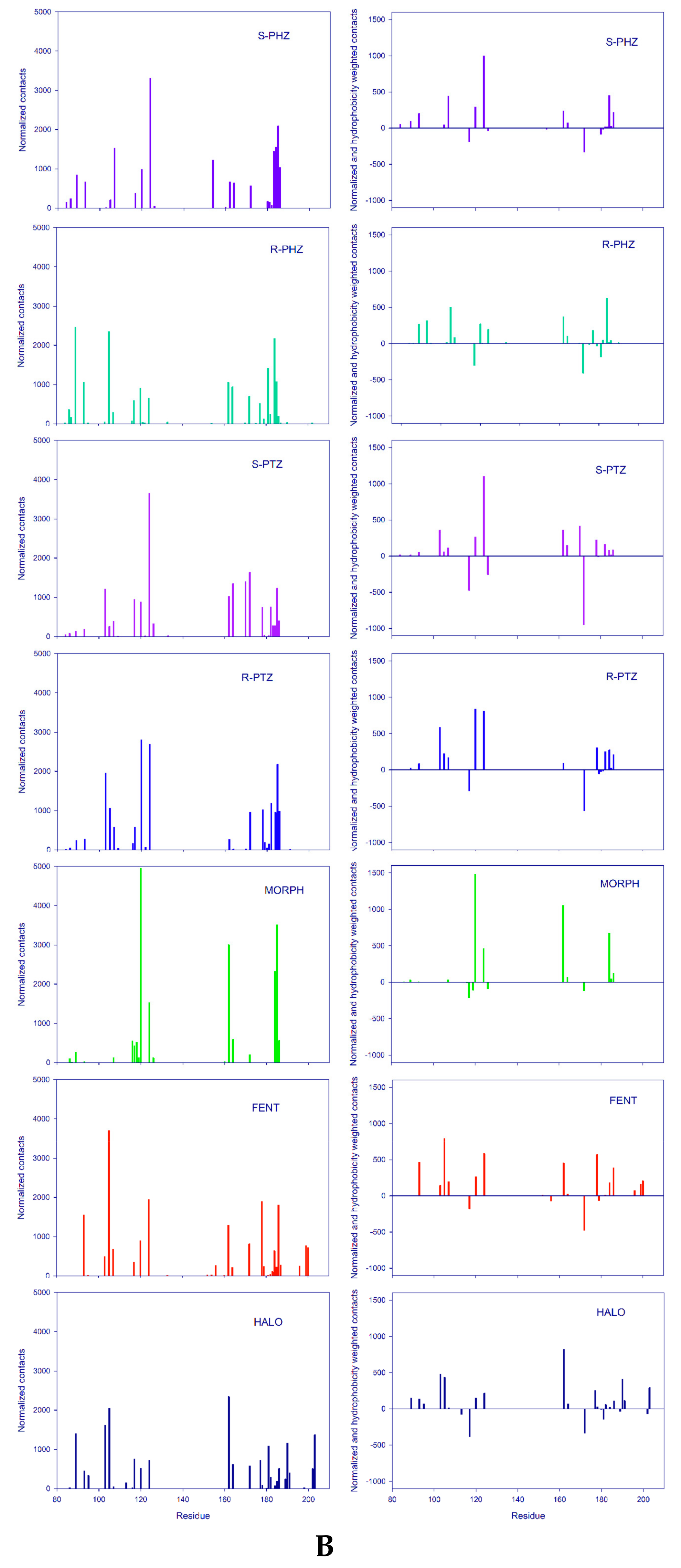
3.1. Dynamical Properties of the σ1 Receptor–Ligand Interactions
- (i)
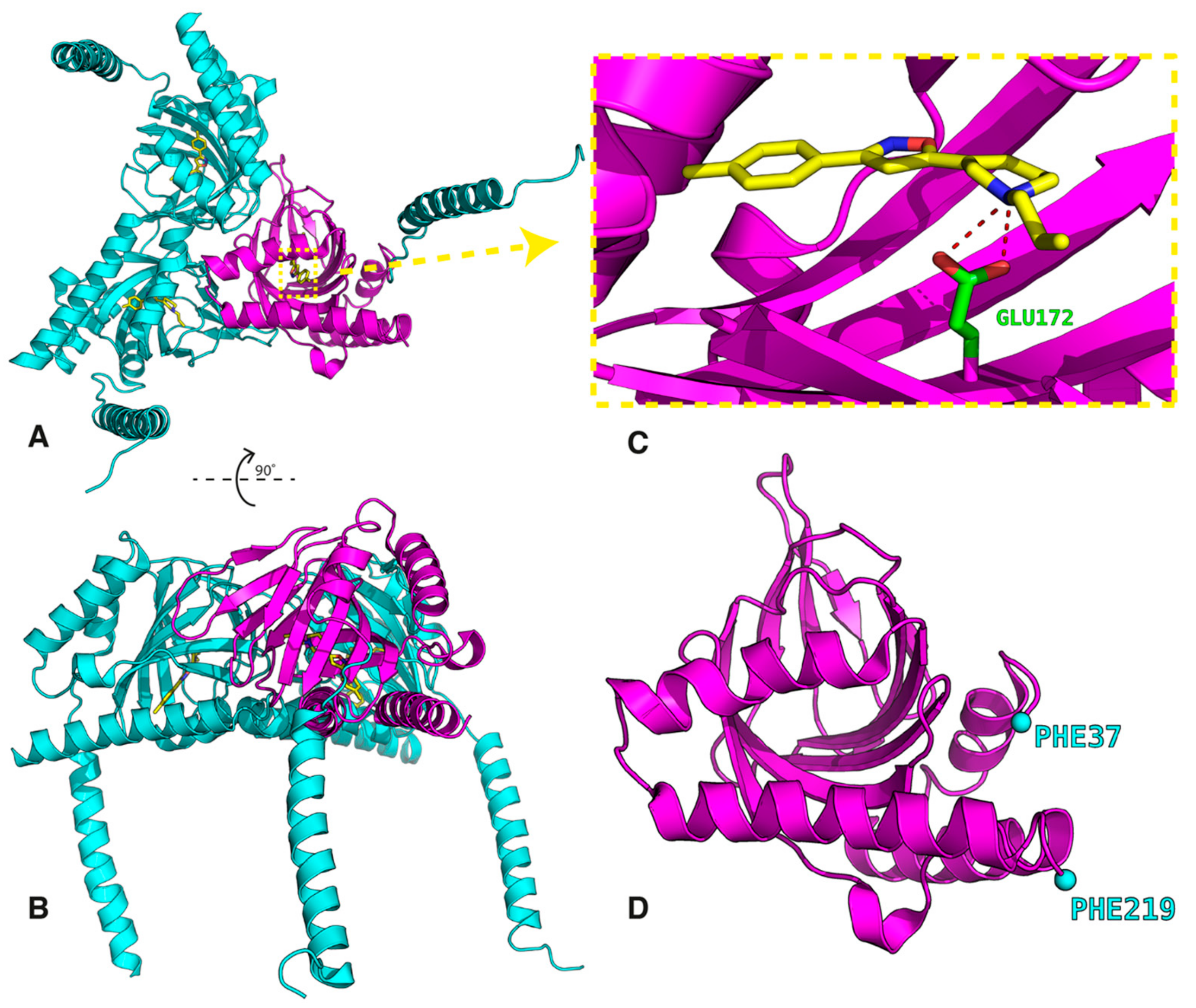
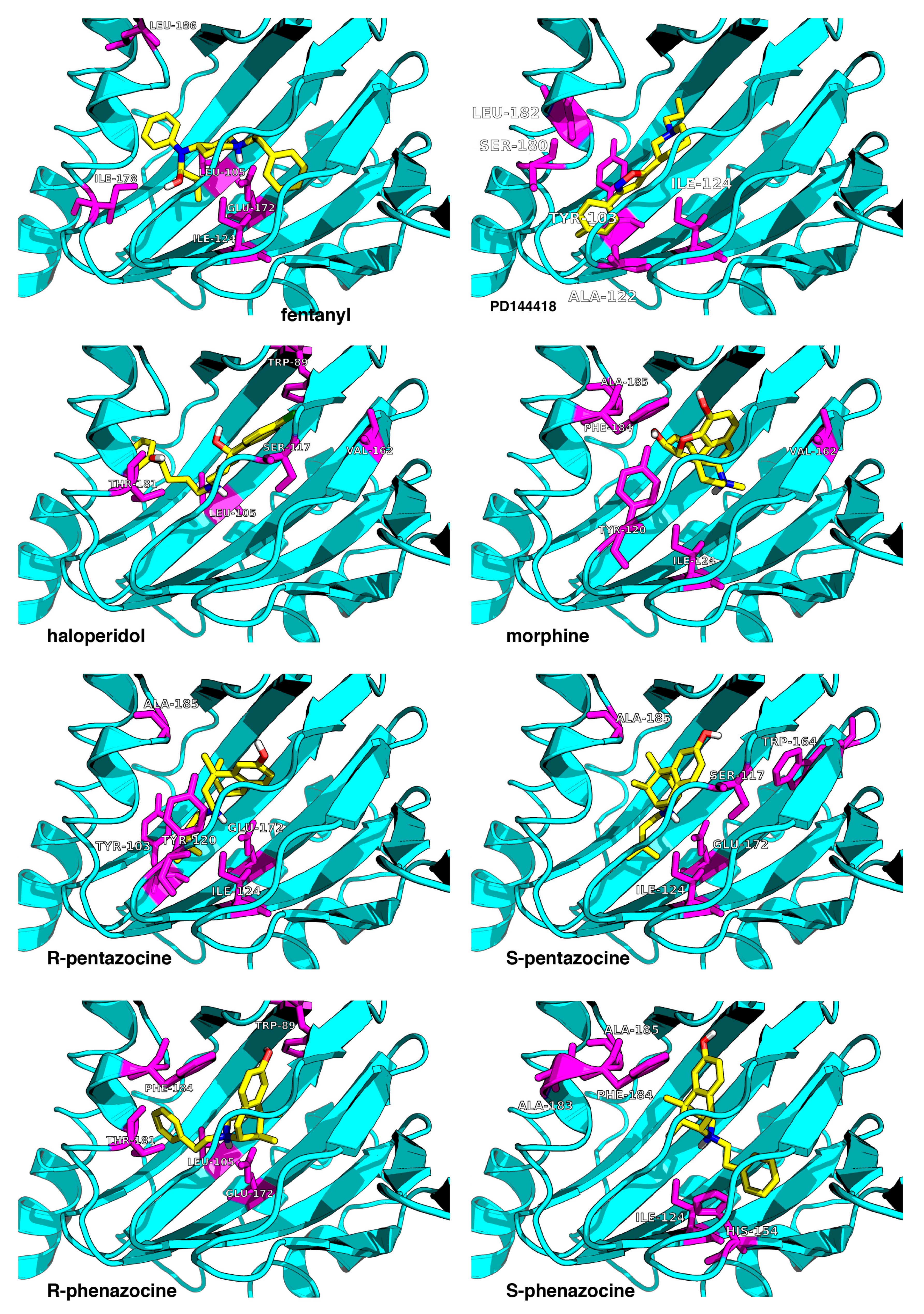
- The first group is localized around Asp126 and forms the intermolecular salt bridge between the protonated nitrogen atom (N-H+) in the ligands and the carboxyl (COO−) group of the receptor Glu172 residue. We include also the stable hydrogen bonds between the carboxyl C=O group of Thr181 with the OH groups of the ligands in this region.
- (ii)
- The second region is placed around the Tyr103 residue and plays a determining role in the π-π stacking interactions between Tyr103 and the aromatic/heteroaromatic rings of FENT, HALO, MORPH, PTZ, and PHZ.
- (iii)
- It is noticeable that the residues (Phe107, Trp164, Ile178, Val162, Leu105, Leu182, and Ala185) from the third group belong to the hydrophobic pocket. They yield the required van der Waals and hydrophobic interactions, arranging appropriately the hydrophobic moieties of the ligands.
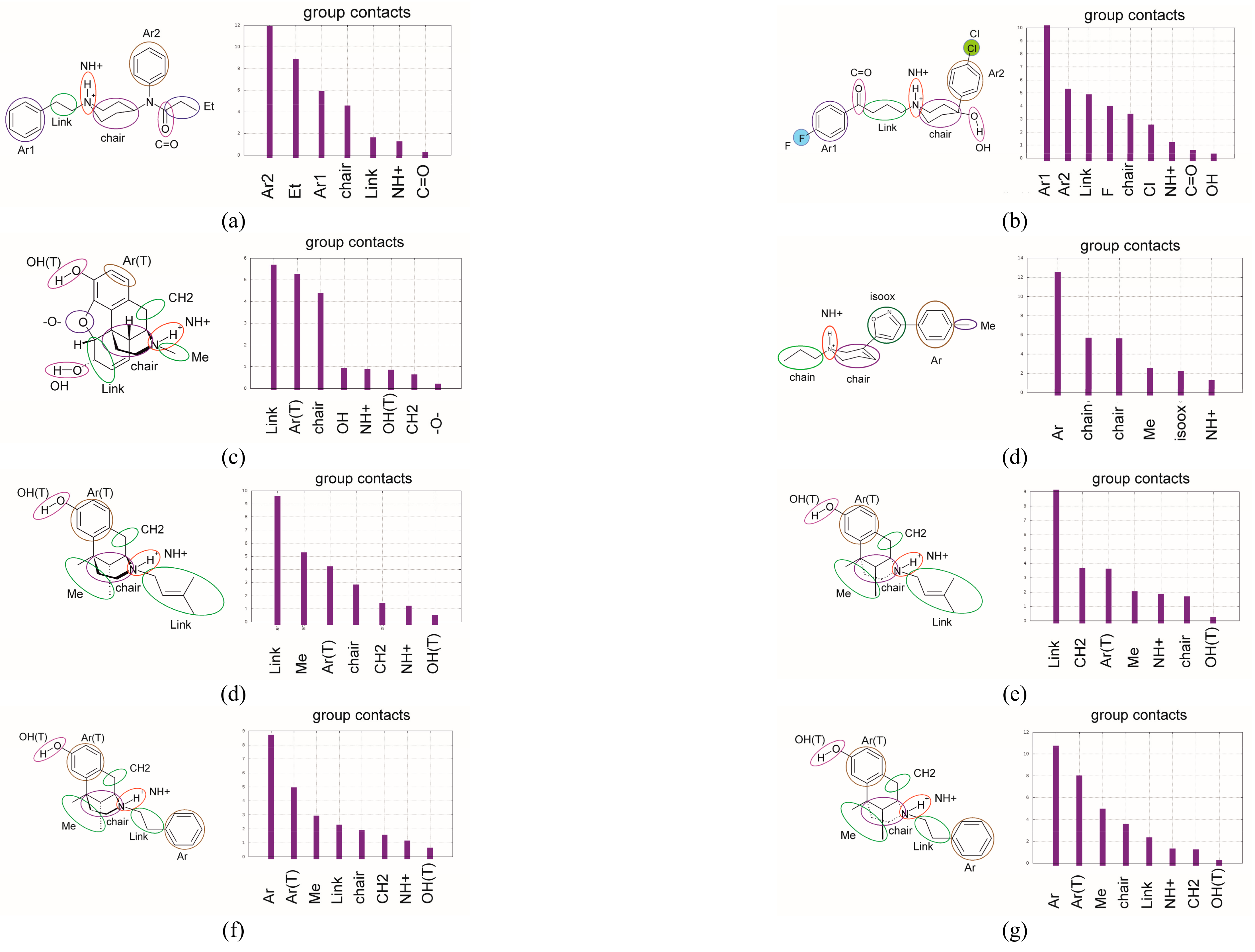
3.2. Ligand–Receptor Binding in Terms of the Frequency of Ligand–Receptor Contacts
- (i)
- the ligand moieties with the receptor;
- (ii)
- the receptor residues with the whole ligand;
- (iii)
- the ligand–residue contact maps averaged over the MD trajectory.
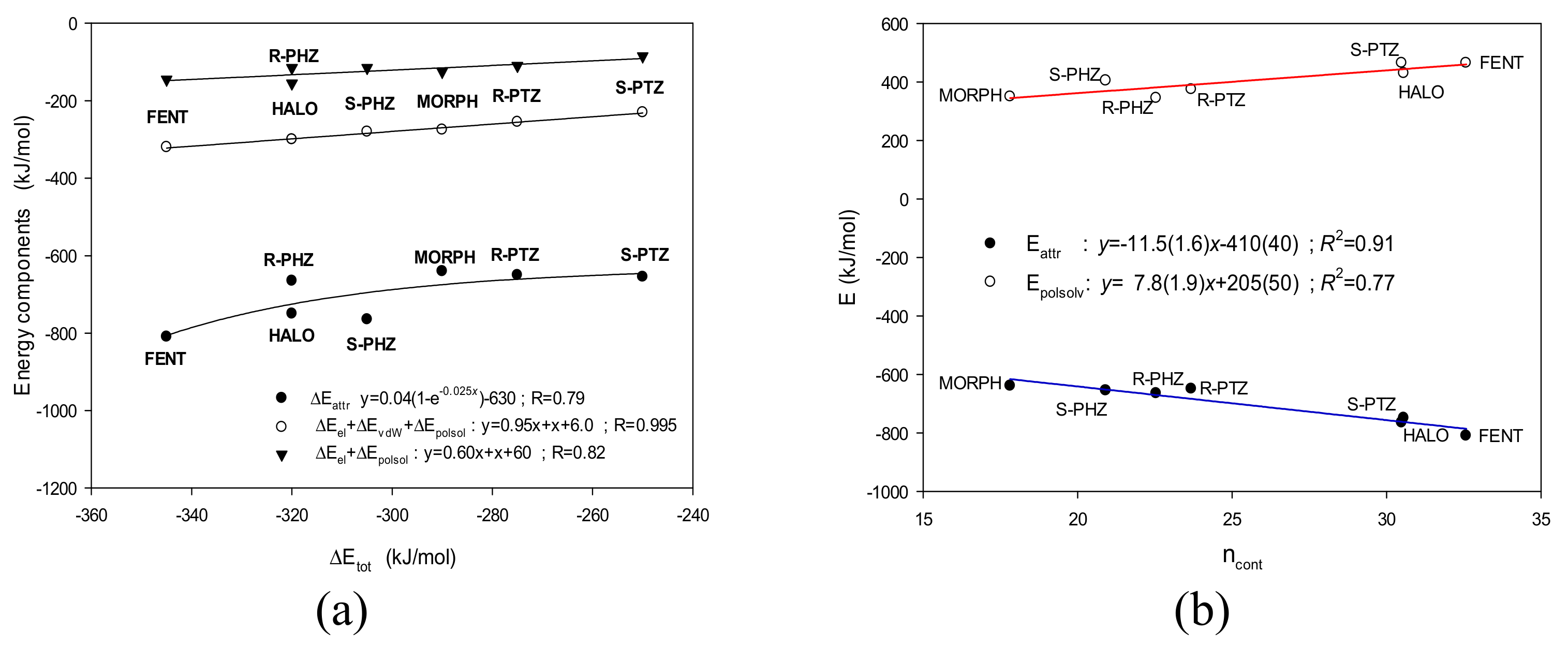
3.3. The Binding Energy Analysis
- (A)
- Hence, the highest total binding energy is found for FENT, while the smallest is for S-PTZ. Moreover, even if the errors are taken into account and one considers only the low limits, the above conclusions remain true.
- (B)
- On the other hand, the values of total interaction energies of the most strongly interacting ligands—FENT, HALO, R-PHZ, and S-PHZ—are within the error bars of each other. The same holds true for the most weakly interacting ligands—S-PTZ, R-PTZ, and MORPH—for which the total interaction energies are also within the error bars of each other.
- (C)
- The high result for FENT is supported by both the highest electrostatic term and one of the highest van der Waals. FENT exhibits the largest attractive contribution and despite the fact that the repulsive term for it is also one of the two largest, the difference is still the biggest.
- (i)
- (ii)
- (iii)
- On the other hand, the HALO ligand, which is the strongest σ1 receptor antagonist considered here (Ki = 0.65 nM [13]) exhibits the second highest number of contacts and the attractive energy term.
- (iv)
- Unfortunately, the computational predictions of the FENT binding energy were not successful. For fentanyl, the experimental Ki was estimated to be greater than 1000 nM [48,49], whereas in our calculations both the number of contacts and energy terms indicate FENT to interact very strongly (Figure 5b).
- (v)
- The other studied ligands, R-PHZ, R-PTZ and S-PTZ, are located somewhere between the extreme interactions. However, they fit the predicted tendencies (Figure 5b).
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schmidt, H.R.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Brune, S.; Pricl, S.; Wünsch, B. Miniperspective. Structure of σ1 receptor and its ligand binding site. J. Med. Chem. 2013, 56, 9809–9819. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, C.G.; Greene, S.F. Sigma receptors: Biology in normal and diseased states. J. Recept. Signal. Transduct. Res. 2015, 36, 327–388. [Google Scholar] [CrossRef] [PubMed]
- Brune, S.; Schepmann, D.; Klempnauer, K-H.; Marson, D.; Dal Col, V.; Laurini, E. ; Fermeglia, M.; Wünsch, B.; Pricl, S. The sigma enigma: In vitro/in silico site-directed mutagenesis studies unveil σ1 receptor ligand binding. Biochemistry 2014, 53, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C. Structural insights into sigma1 function. In Handbook of Experimental Pharmacology; Springer International Publishing AG: Basel, Switzerland, 2016. [Google Scholar] [CrossRef]
- Van Waarde, A.; Rybczyńska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R.A. Potential application for sigma receptor ligands in cancer diagnosis and therapy. Biochem. Biophys. Acta 2015, 1848, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Su, T.-P. Sigma receptors: Their role in disease and as therapeutic targets. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2017. [Google Scholar]
- Guitart, X.; Codony, X.; Monray, X. Sigma Receptors: Biology and therapeutic potential. Psychopharmacology 2004, 174, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Hashimoto, K. The Role of Sigma-1 Receptor in the Pathophysiology of Neuropsychiatric Diseases. J. Recept. Ligand Channel Res. 2010, 3, 25–36. [Google Scholar]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chapetones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Cobos, E.J.; Entrena, J.M.; Nieto, F.R.; Cendan, C.M.; Del Pozo, E. Pharmacology and therapeutic potential of sigma-1 receptor ligands. Curr. Neuropharm. 2008, 6, 344–366. [Google Scholar] [CrossRef] [PubMed]
- Banister, S.D.; Kassiou, M. The therapeutic potential of sigma receptor for the treatment of central nervous system diseases: evaluation of the evidence. Curr. Pharm. Des. 2012, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Bhat, R.; Mesangegau, C.; Poupaert, J.H.; McCurdy, C.R. Early development of sigma-receptor ligands. Future Med. Chem. 2011, 3, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Almansa, C.; Vela, J.M. Selective sigma-1 receptor antagonists for the treatment of pain. Future Med. Chem. 2014, 6, 1179–1199. [Google Scholar] [CrossRef] [PubMed]
- Megalizzi, V.; Le Mercier, M. Sigma receptors and their ligands in cancer biology: Overview and new perspectives for cancer therapy. Med. Res. Rev. 2012, 32, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptor. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Chu, U.B.; Ruoho, A.E. Sigma receptor binding assays. Curr. Protoc. Element. Pharmacol. 2015, 71, 1.34.1–1.34.21. [Google Scholar]
- Kourrich, S.; Su, T.P.; Fujimoto, M.; Bonci, A. The sigma-1 receptor: Roles in neuronal plasticity and disease. Trends Neurosci. 2012, 35, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Lucke-Wold, B.; Mookerjee, S.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of sigma-1 receptors in neurodegenerative disease. J. Pharmacol. Sci. 2015, 127, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor ligands: Potential in the treatment of neuropsychiatric disorders. CNS Drugs 2004, 18, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Wünsch, B. Pharmacophore models and development of spirocyclic ligands for sigma-1 receptor. Curr. Pharm. Des. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Fontanilla, D.; Johannessen, M.; Hajipour, A.R.; Cozii, N.V.; Jackson, M.B.; Ruoho, A.E. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 2009, 323, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Arena, E.; Sanchez-Fernandez, C.; Turnaturi, R.; Parenti, C.; Marrazzo, A.; Catalano, R.; Amata, E.; Pasquinucci, L.; Cobos, E.J. (+)-and (–)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/sigma1 antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 2017, 125, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Wünsch, B. The σ1 Receptor Antagonist S1RA Is a Promising Candidate for the Treatment of Neurogenic Pain. J. Med. Chem. 2012, 55, 8209–8210. [Google Scholar] [CrossRef] [PubMed]
- Laurini, E.; Dal Col, V.; Mamolo, M.G.; Zampieri, D.; Posocco, P.; Fermeglia, M.; Vio, L.; Pricl, S. Homology model and docking-based virtual screening for ligands of the σ1 receptor. ACS Med. Chem. Lett. 2011, 22, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Laurini, E.; Marson, D.; Dal Col, V.; Fermeglia, M.; Mamolo, M.G.; Zampieri, D.; Vio, L.; Pricl, S. Another brick in the wall. Validation of the σ1 receptor 3D model by computer-assisted design, synthesis, and activity of new σ1 ligands. Mol. Pharm. 2012, 9, 3107–3126. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Brune, S.; Orgel, F.B.; Korpis, K.; Lange, C.; Bednarski, P.; Laurini, E.; Fermegia, M.; Prici, S.; Schepmann, D.; et al. Rigidity versus flexibility is this an issue in sigma-1 receptor ligand affinity and activity. J. Med. Chem. 2016, 259, 5505–5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Schepman, D.; Yanagisawa, S.; Yamaguchi, J.; Dal Col, V.; Laurini, E.; Itami, K.; Pricl, S.; Wünsch, B. Pd-catalyzed direct C-H bond functionalization of spirocyclic σ1 ligands: Generation of a pharmacophore model and analysis of the reverse binding mode by docking into a 3D homology model of the σ1 receptor. J. Med. Chem. 2012, 55, 8047–8065. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Pedrali, A.; Gaggeri, R.; Marra, A.; Pignataro, L.; Laurini, E.; Dal Col, V.; Fermeglia, M.; Pricl, S.; Schepmann, D.; et al. Chemical, Pharmacological and in vivo metabolic stability studies on enantiomerically pure RC-33 compounds: promising neuroprotective agents acting as σ1 receptor agonists. ChemMedChem 2013, 8, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Marra, A.; Picconi, P.; Serra, M.; Catenacci, L.; Sorrenti, M.; Laurini, E.; Fermeglia, M.; Pricl, S.; Brambilla, S.; et al. Identification of RC-33 as a potent and selective σ1 receptor agonist potentiating NGF-induced neurite outgrowth in PC12 cells. Part 2: g-scale synthesis, physicochemical characterization and in vitro metabolic stability. Bioorg. Med. Chem. 2013, 21, 2577–2586. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Marra, A.; Rui, M.; Laurini, E.; Fermeglia, M.; Pricl, S.; Schepmann, D.; Wünsch, B.; Peviani, M.; Curti, D.; et al. A step forward in the sigma enigma: A role for chirality in the sigma receptors-ligand interaction. Med. Chem. Commun. 2015, 6, 138–146. [Google Scholar] [CrossRef]
- Zampieri, D.; Laurini, E.; Vio, L.; Fermeglia, M.; Pricl, S.; Wünsch, B.; Schepmann, D.; Mamolo, M.G. Improving selectivity preserving affinity: New piperidine-4-carboxamide derivatives as effective sigma-1-ligands. Eur. J. Med. Chem. 2015, 90, 797–808. [Google Scholar]
- Franchini, S.; Battisti, U.M.; Prandi, A.; Tait, A.; Borsari, C.; Cichero, E.; Fossa, P.; Cilia, A.; Prezzavento, O.; Ronsisvalle, S.; et al. Scounting new sigma receptor ligands: Synthesis, pharmacological evaluation and molecular modeling of 1,3-dioxolane-based structures and derivatives. Eur. J. Med. Chem. 2016, 112, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.A.; Wünsch, B. Novel spiropiperidines as highly potent and subtype selective sigma receptor ligands. Part I. J. Med. Chem. 2002, 45, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and repository: version 1.0. J. Chem. Theory Comp. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comp. Physics Comm. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2014, 47, 5.6.1–5.6.32. [Google Scholar]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for Ligand-Receptor Docking. Curr. Protoc. Bioinform. 2008, 8, 8.14.1–8.14.40. [Google Scholar]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopuo, A.D. Molecular Docking and Structure Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.; Ducrot, P.; Raimbaud, E.; Rognan, D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model 2013, 53, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R. Open Source Drug Discovery Consortium, and Lynn A.G_mmpbsa-a GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J. Dispersive interactions in solution complexes. Acc. Chem. Res. 2015, 248, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Schauperl, M.; Podewitz, M.; Waldner, B.J.; Liedl, K.R. Enthalpic and entropic contributions to hydrophobicity. J. Comp. Theory Chem. 2012, 12, 4600–4610. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the MM/PBSA method. J. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Moret, M.A.; Zebende, G.F. Amino acid hydrophobicity and accessible surface area. Phys. Rev. E 2007, 75, 011920. [Google Scholar] [CrossRef] [PubMed]
- Sguazzini, E.; Schidtm, H.R.; Iyer, K.A.; Kruse, A.C.; Dukat, M. Reevaluation of fenpropimorph as a receptor sigma ligand: structure-affinity relationship studies as human σ1 receptor. Bio. Med. Chem. Lett. 2017, 27, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Largent, B.L.; Wikstrom, H.; Gundlach, A.L.; Snyder, S.H. Structural determination of sigma receptor affinity. Mol. Pharm. 1987, 32, 772–784. [Google Scholar]
- Chen, J.C.; Smith, E.R.; Cahen, M.; Fishman, J.B. The opioid receptor binding of dezocine, morphine, fentanyl, butorphanol, nalbuphine. Life Sci. 1992, 52, 389–397. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C. Structural basis for G protein-coupled receptor activation. Biochemistry 2017, 56, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand ∆E | FENT | HALO | MORPH | R-PTZ | S-PTZ | R-PHZ | S-PHZ |
|---|---|---|---|---|---|---|---|
| ΔEtot | −345 (35) | −320 (20) | −290 (20) | −275 (20) | −250 (25) | −320 (25) | −305 (25) |
| ΔEvdW | −175 (20) | −185 (15) | −150 (15) | −145 (15) | −145 (15) | −145 (15) | −165 (15) |
| ΔEelect | −610 (40) | −545 (20) | −475 (20) | −485 (25) | −490 (25) | −500 (50) | −580 (30) |
| ΔEnonpolarsol | −22 (1) | −22 (1) | −16 (1) | −18 (1) | −18 (1) | −18 (1) | −19 (1) |
| ΔEpolarsol | 465 (35) | 430 (15) | 350 (20) | 375 (25) | 405 (40) | 345 (60) | 465 (25) |
| ΔEattr | −810 | −750 | −640 | −650 | −655 | −665 | −765 |
| ΔEvdW/ΔEtot(%) | 51 | 58 | 52 | 53 | 58 | 45 | 54 |
| ΔEelect/ΔEtot (%) | 177 | 170 | 164 | 176 | 196 | 156 | 190 |
| −ΔEpolarsol/ΔEtot (%) | 135 | 134 | 121 | 136 | 162 | 108 | 152 |
| ΔEvdW/ΔEattr (%) | 22 | 24 | 23 | 22 | 22 | 22 | 22 |
| ΔEelect/ΔEattr (%) | 76 | 73 | 74 | 75 | 75 | 75 | 76 |
| −ΔEpolarsol/ΔEattr (%) | 58 | 57 | 55 | 58 | 62 | 52 | 60 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurciński, M.; Jarończyk, M.; Lipiński, P.F.J.; Dobrowolski, J.C.; Sadlej, J. Structural Insights into σ1 Receptor Interactions with Opioid Ligands by Molecular Dynamics Simulations. Molecules 2018, 23, 456. https://doi.org/10.3390/molecules23020456
Kurciński M, Jarończyk M, Lipiński PFJ, Dobrowolski JC, Sadlej J. Structural Insights into σ1 Receptor Interactions with Opioid Ligands by Molecular Dynamics Simulations. Molecules. 2018; 23(2):456. https://doi.org/10.3390/molecules23020456
Chicago/Turabian StyleKurciński, Mateusz, Małgorzata Jarończyk, Piotr F. J. Lipiński, Jan Cz. Dobrowolski, and Joanna Sadlej. 2018. "Structural Insights into σ1 Receptor Interactions with Opioid Ligands by Molecular Dynamics Simulations" Molecules 23, no. 2: 456. https://doi.org/10.3390/molecules23020456