Synthesis, Characterization, and Computational Modeling of N-(1-Ethoxyvinyl)pyridinium Triflates, an Unusual Class of Pyridinium Salts

 , , , and
, , , and
Abstract
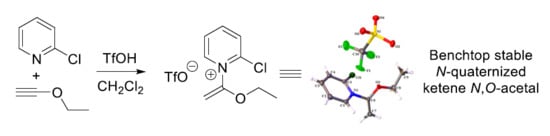
:
1. Introduction
Biological and Synthetic Significance of N-Substituted Pyridinium Salts
2. Results and Discussion
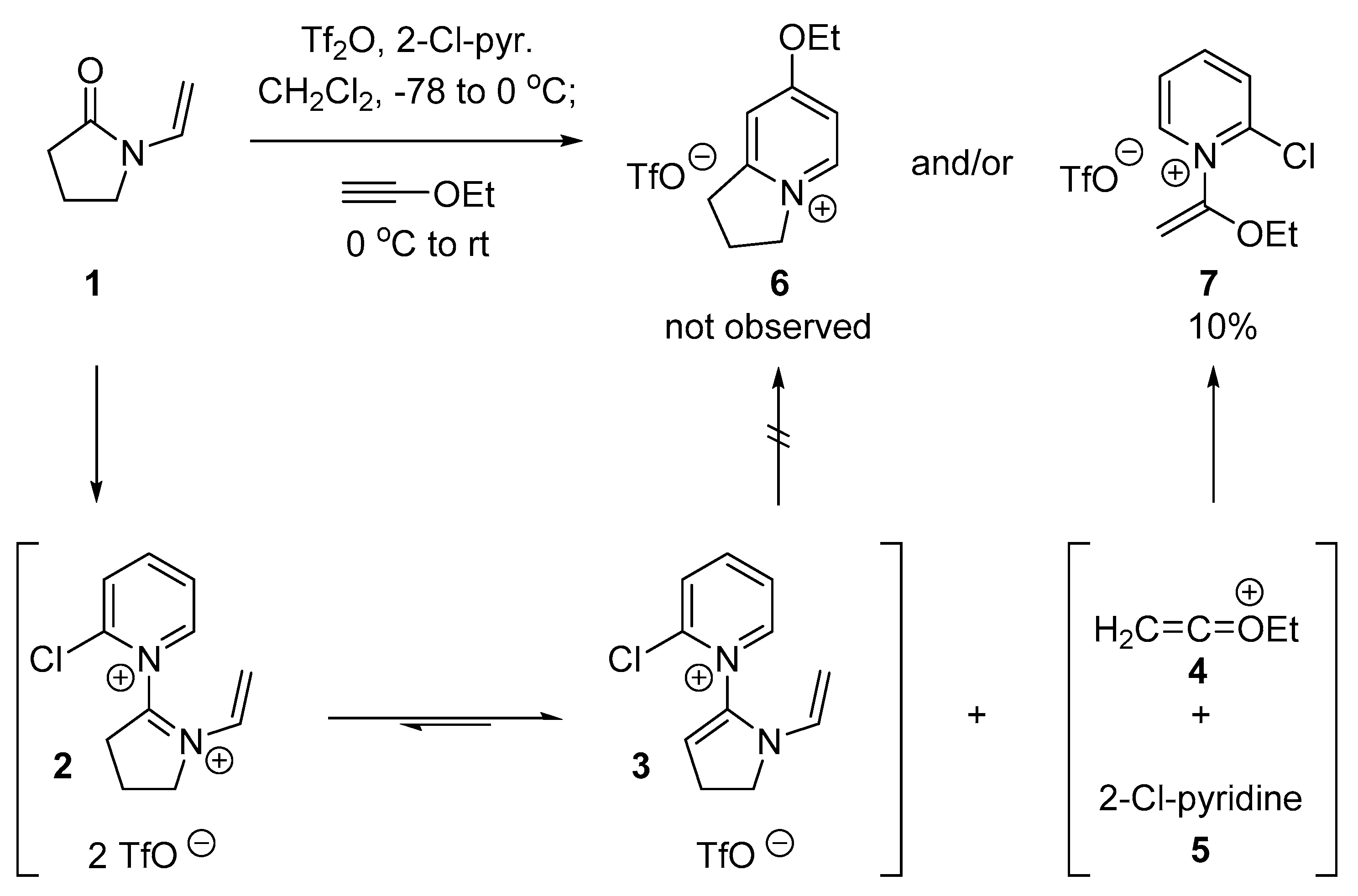
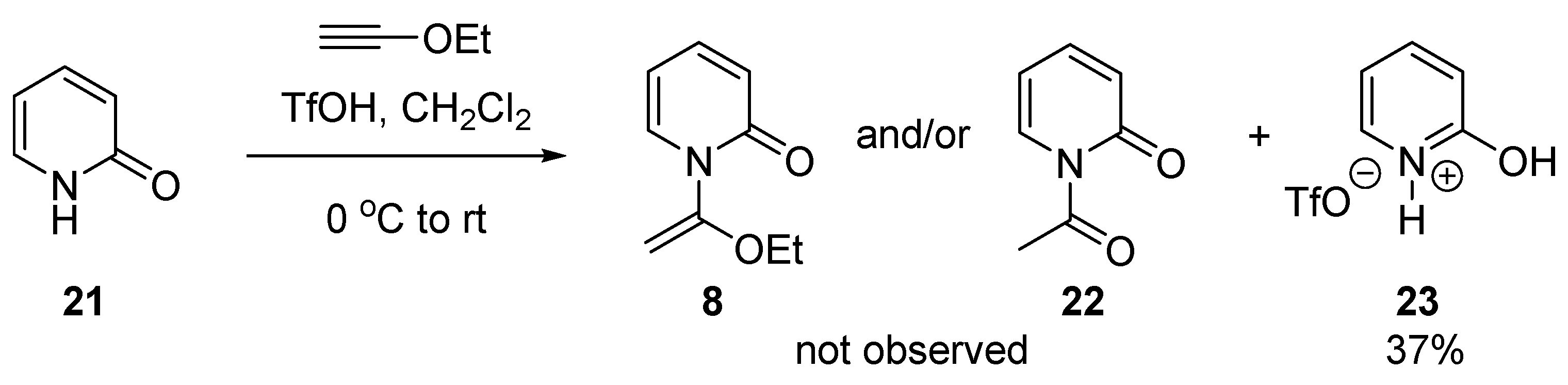
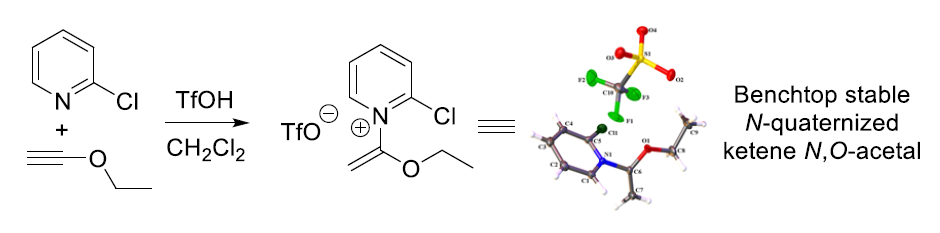
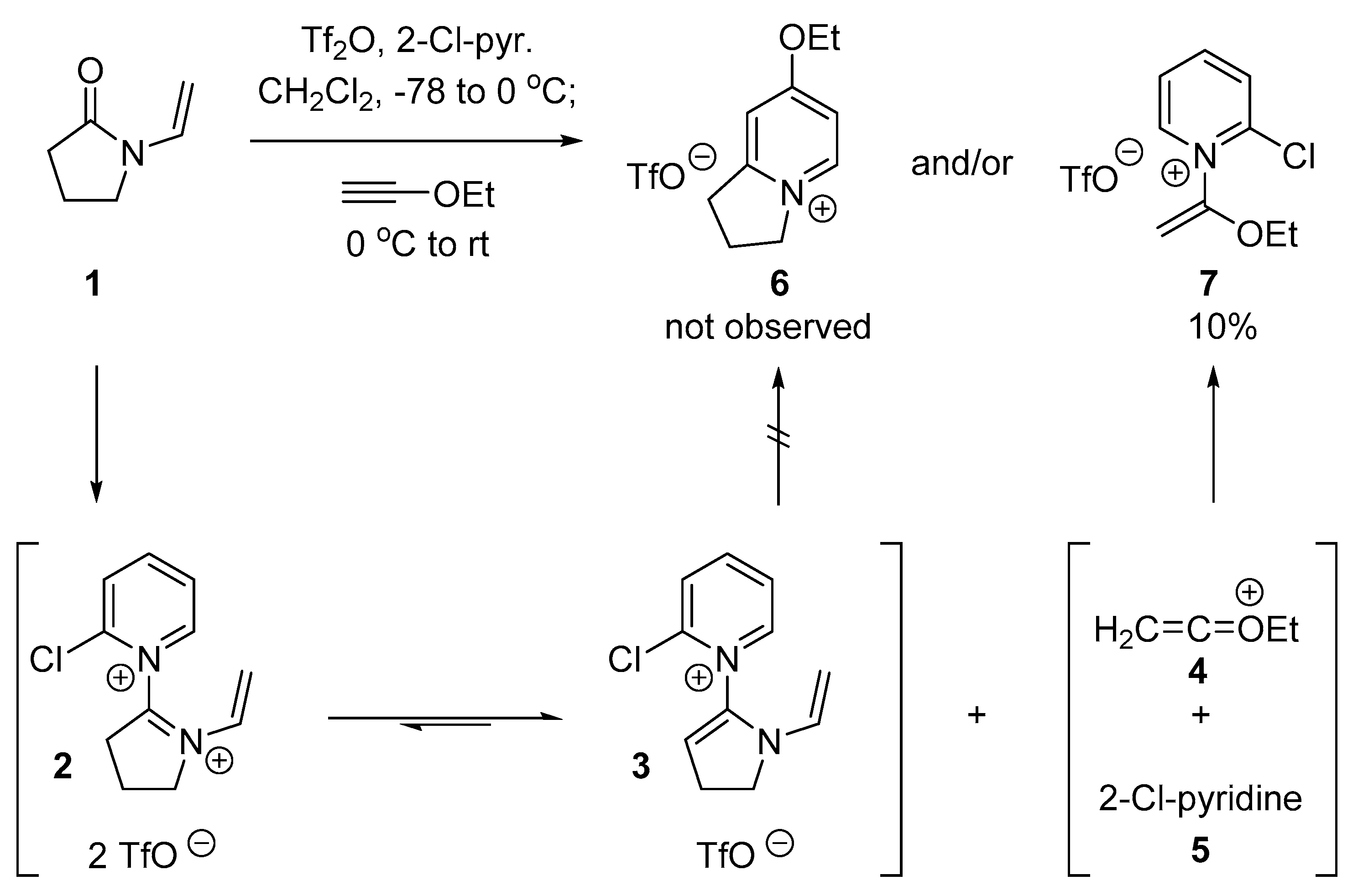
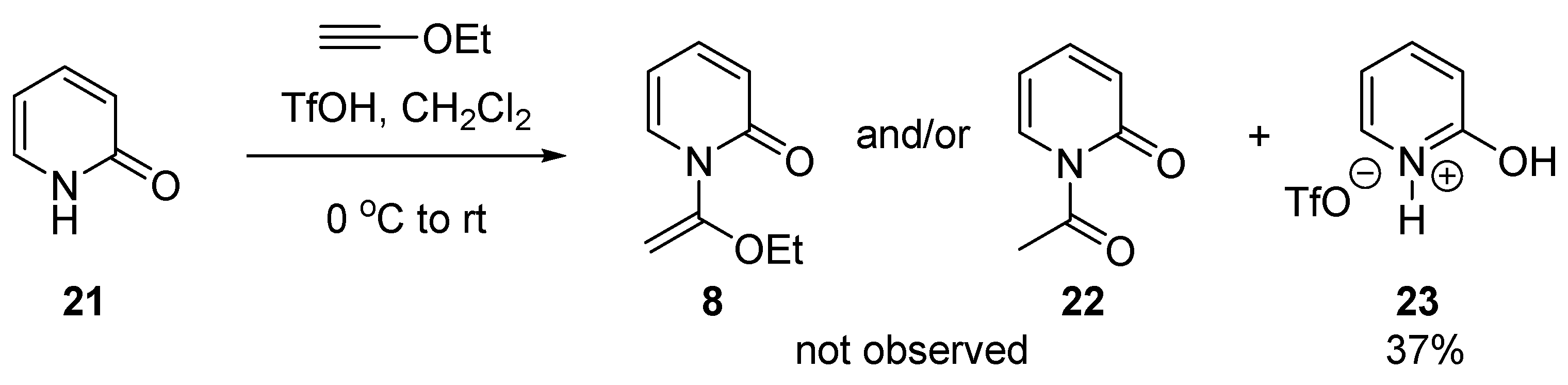
2.1. Discovery of 2-Chloro-N-(1-ethoxyvinyl)pyridine-1-ium Triflate
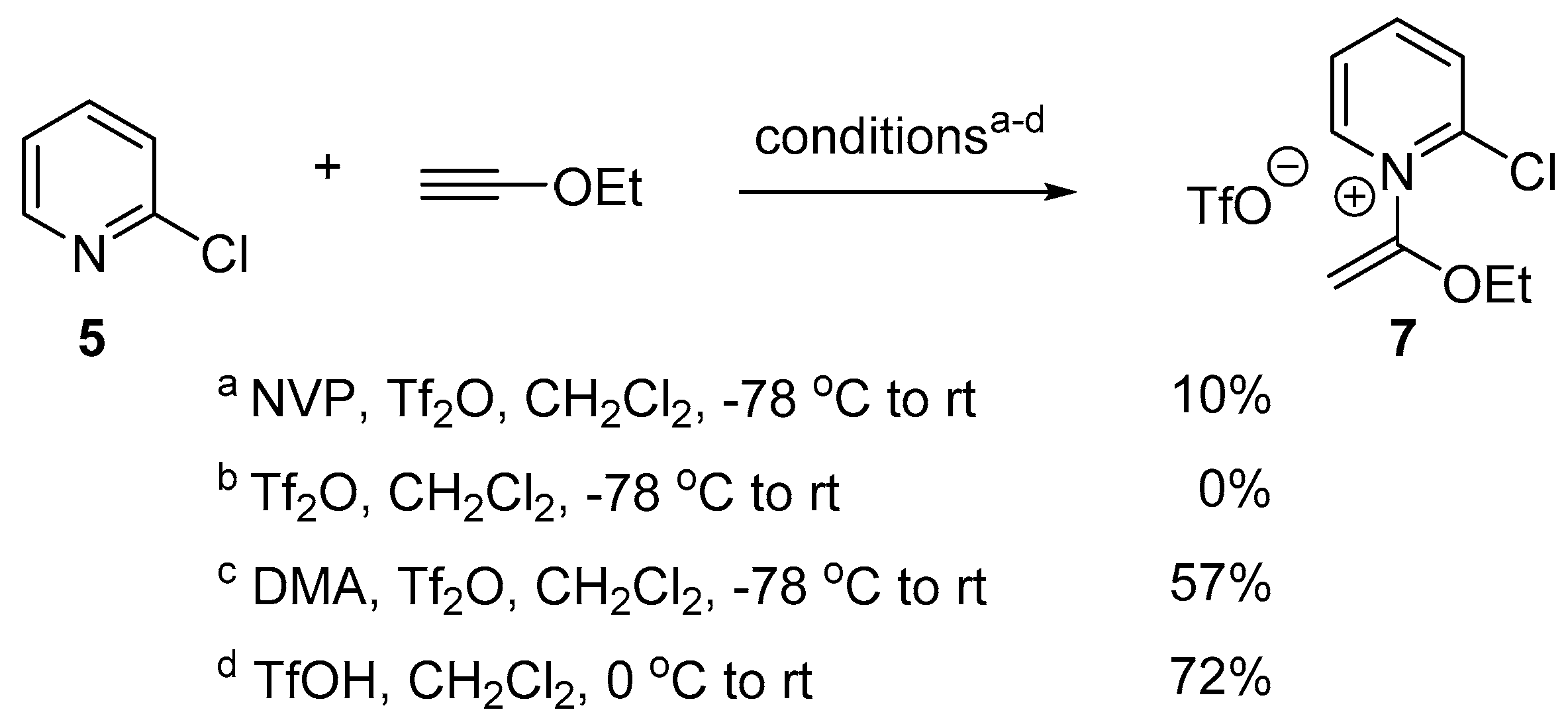
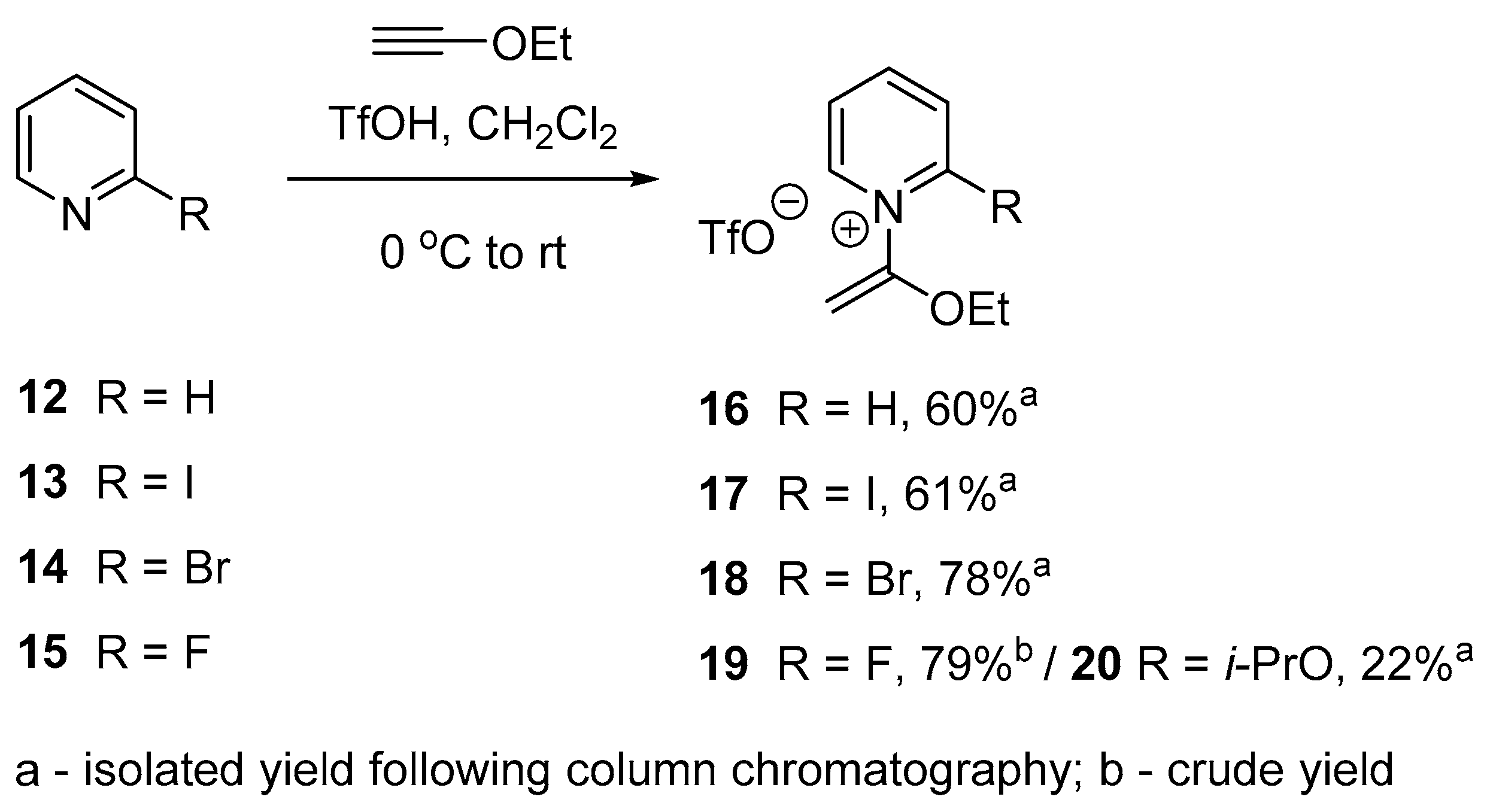
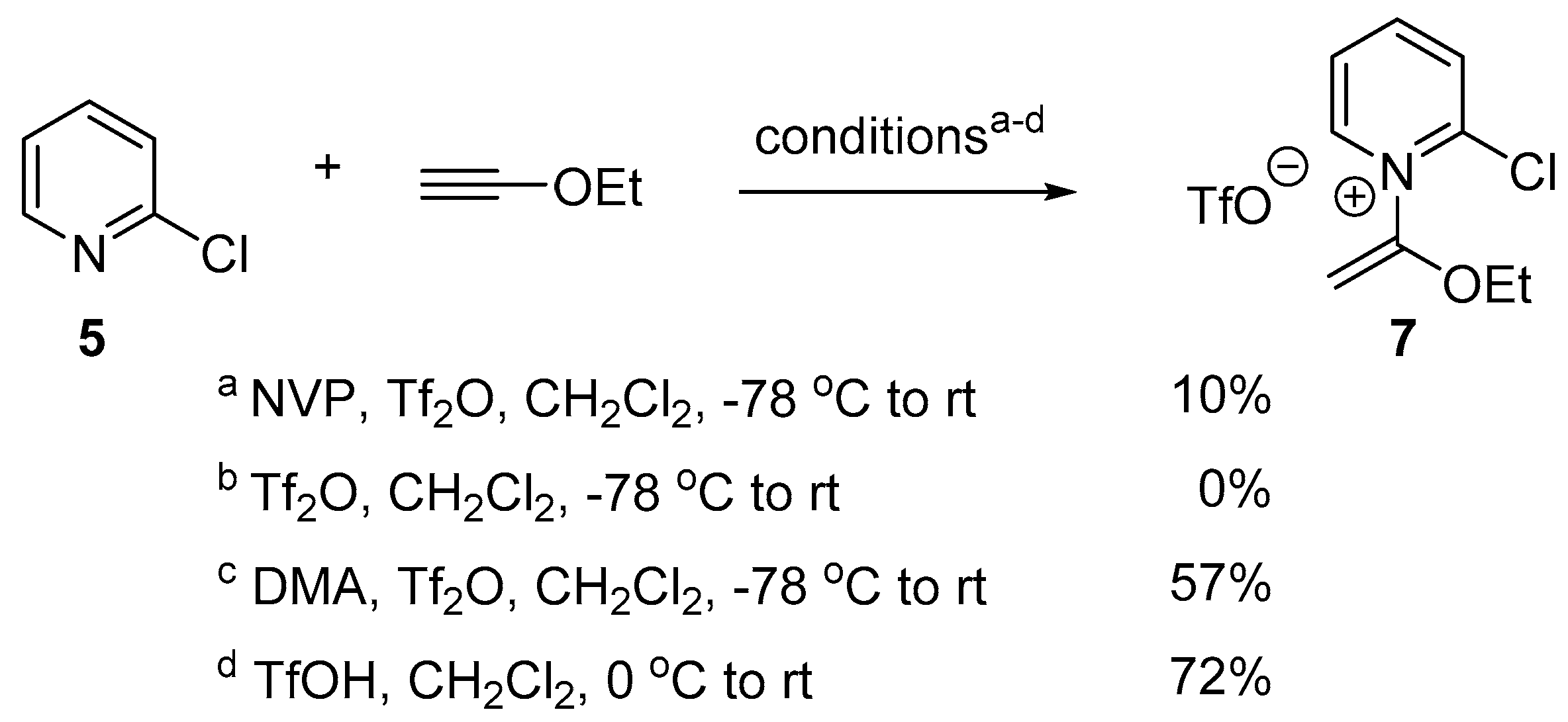
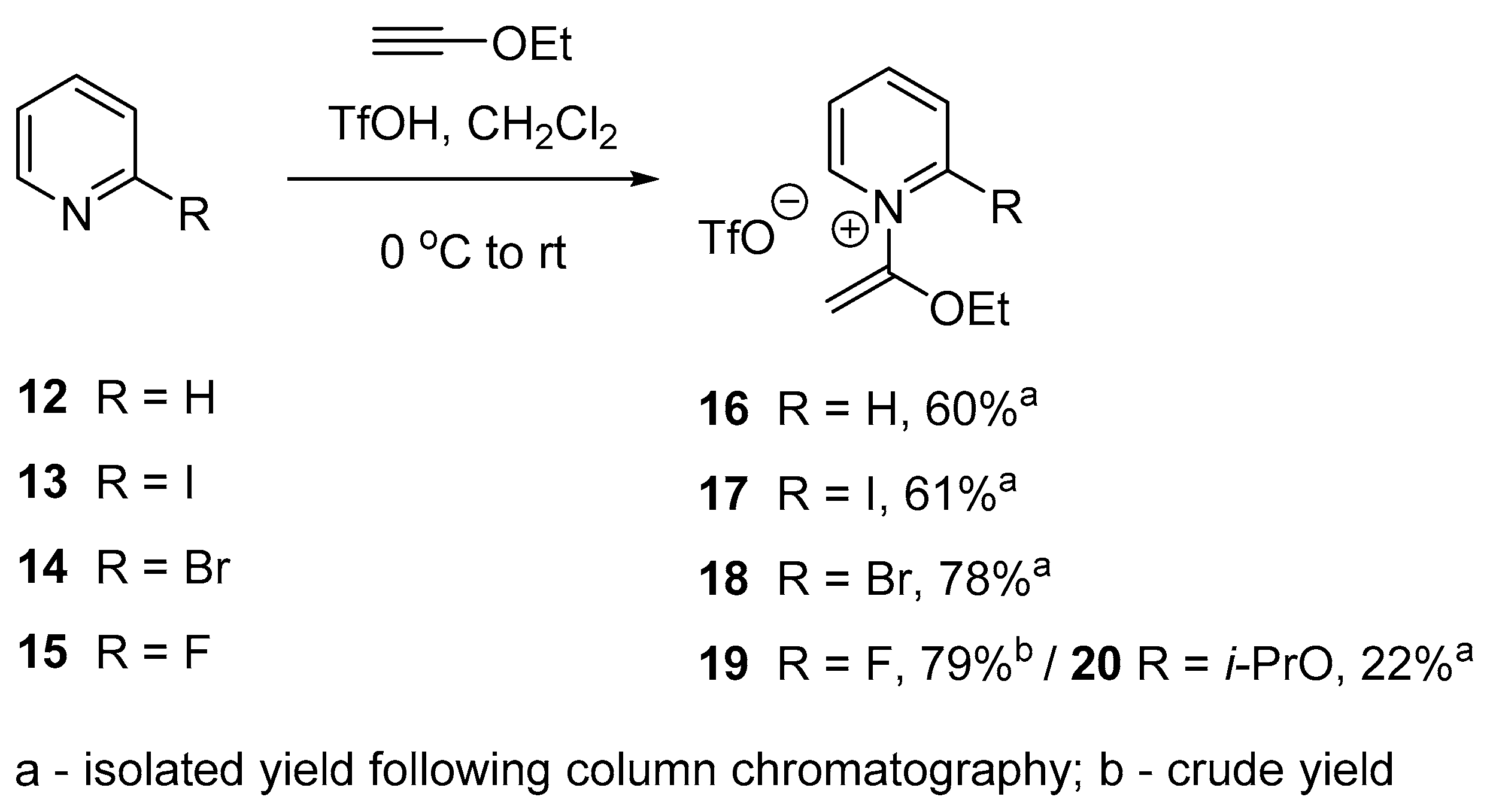
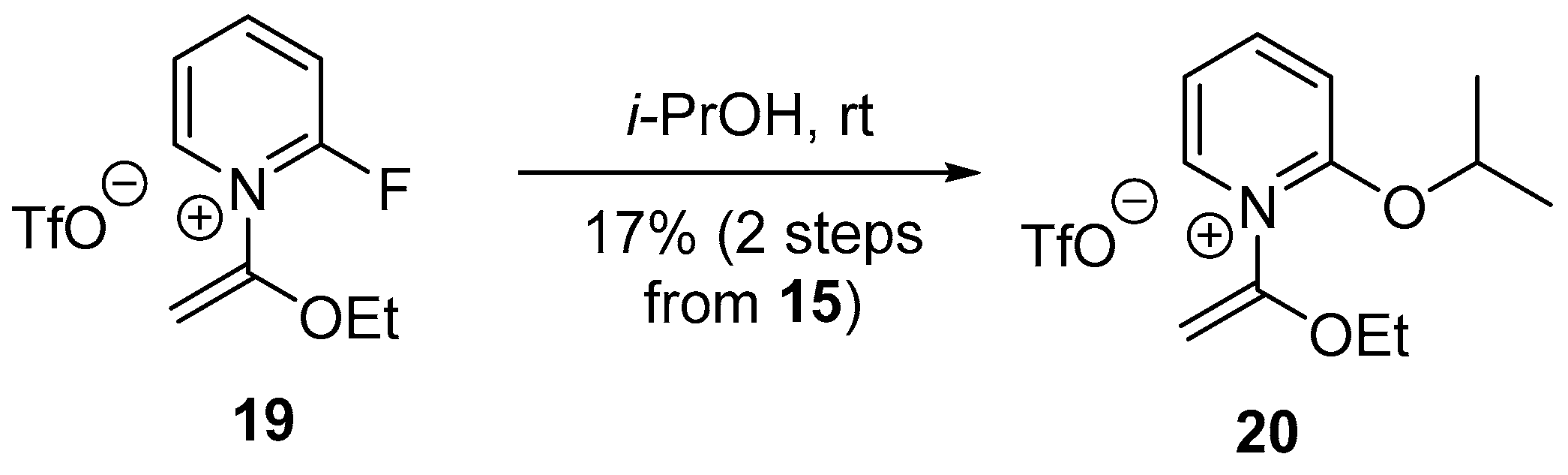
2.2. Optimized Synthesis of N-(1-Ethoxyvinyl)pyridinium Triflates
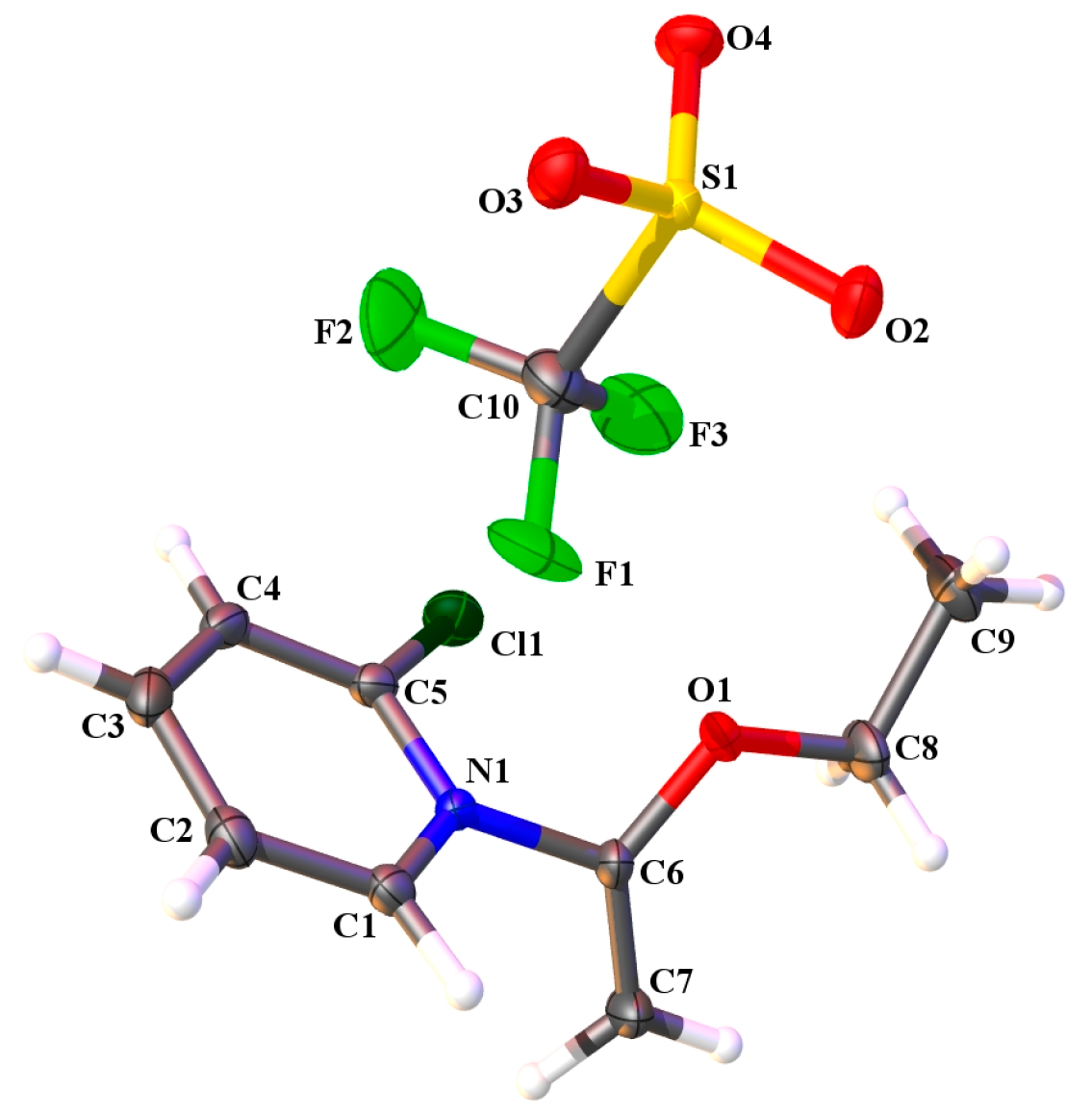
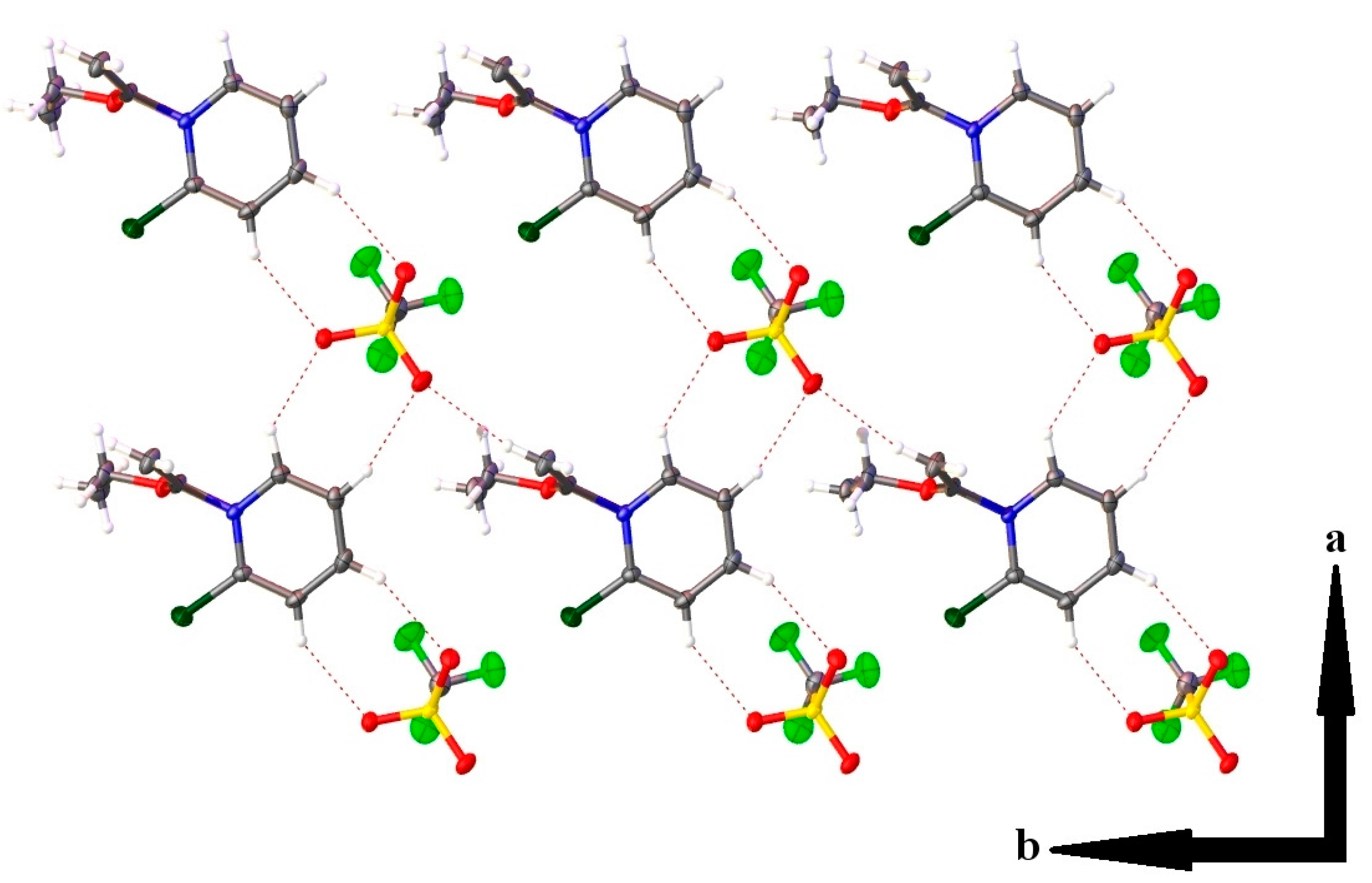
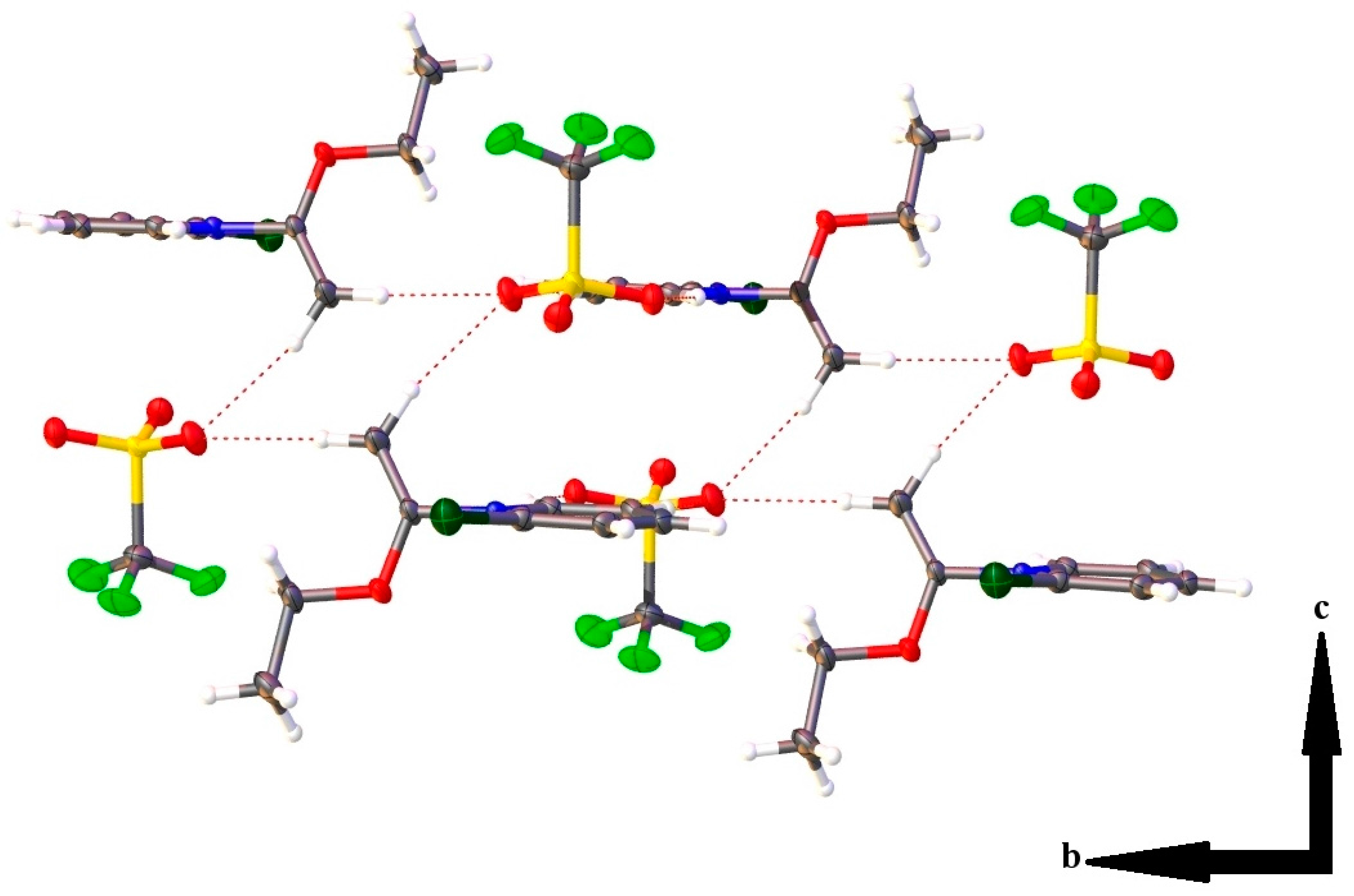
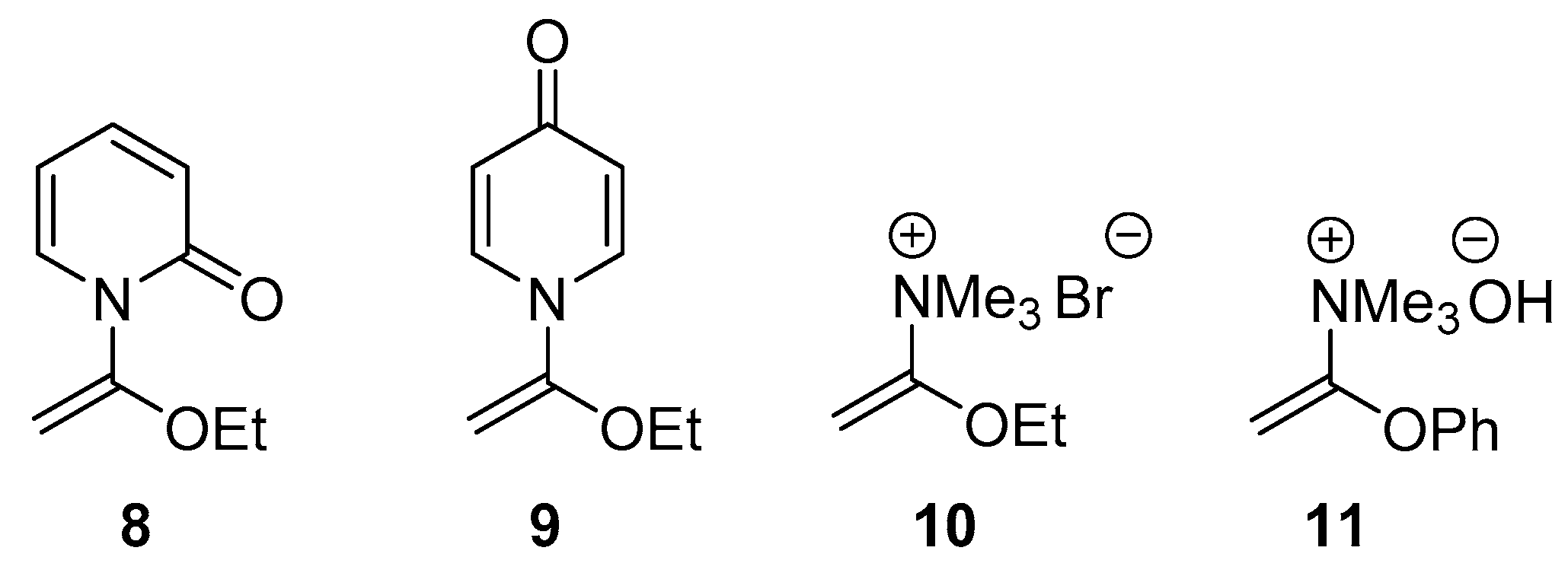
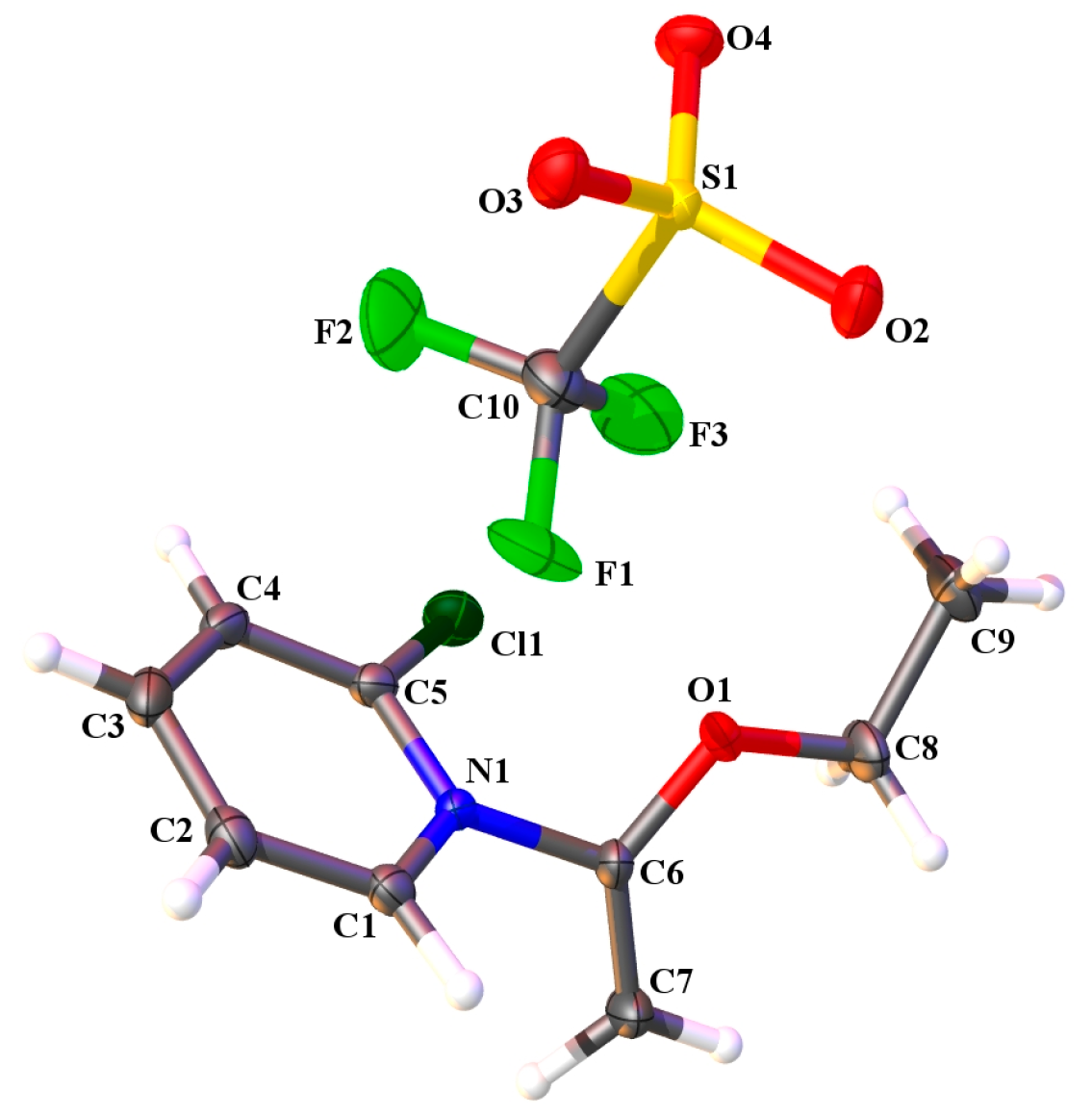
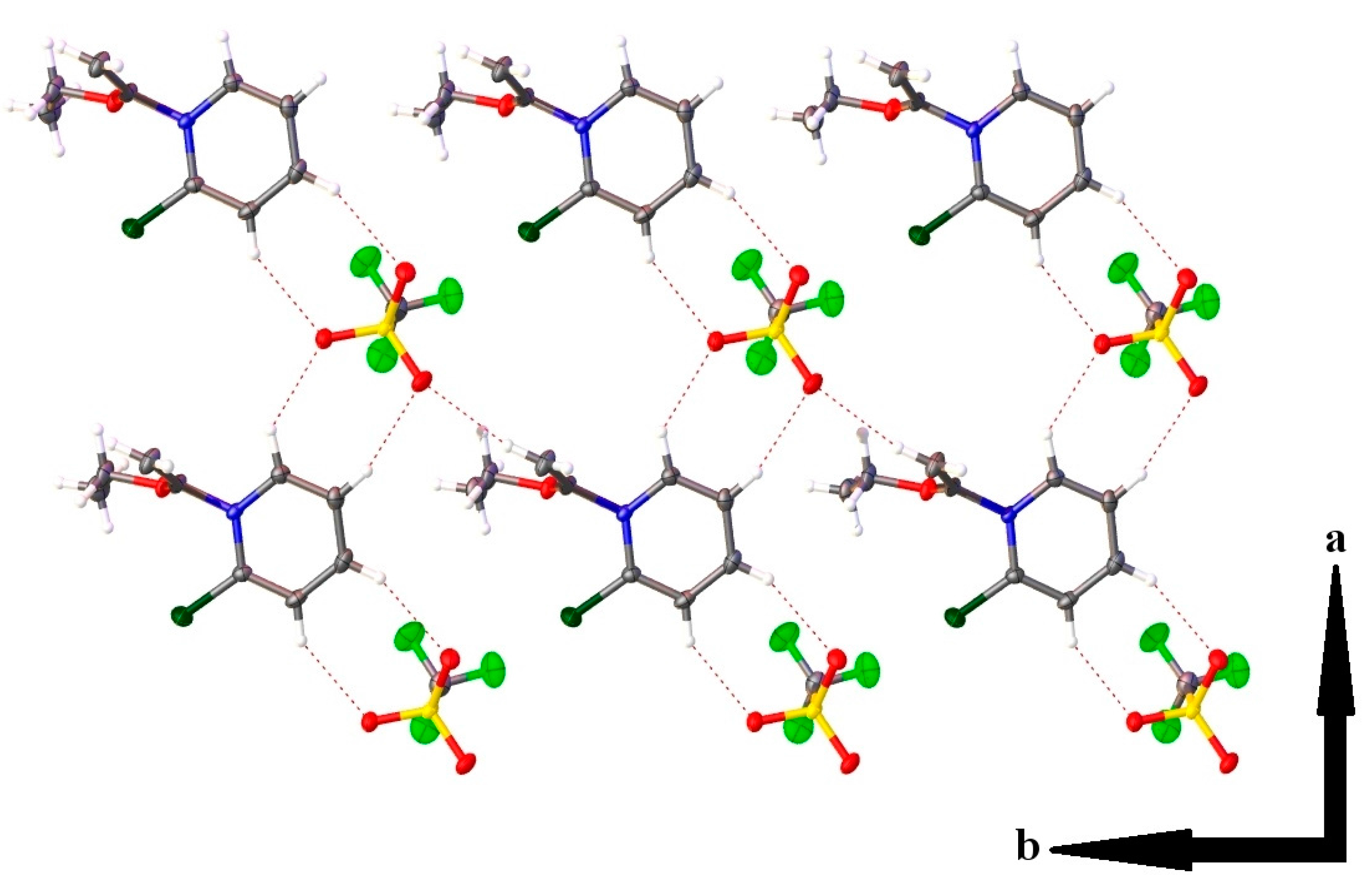
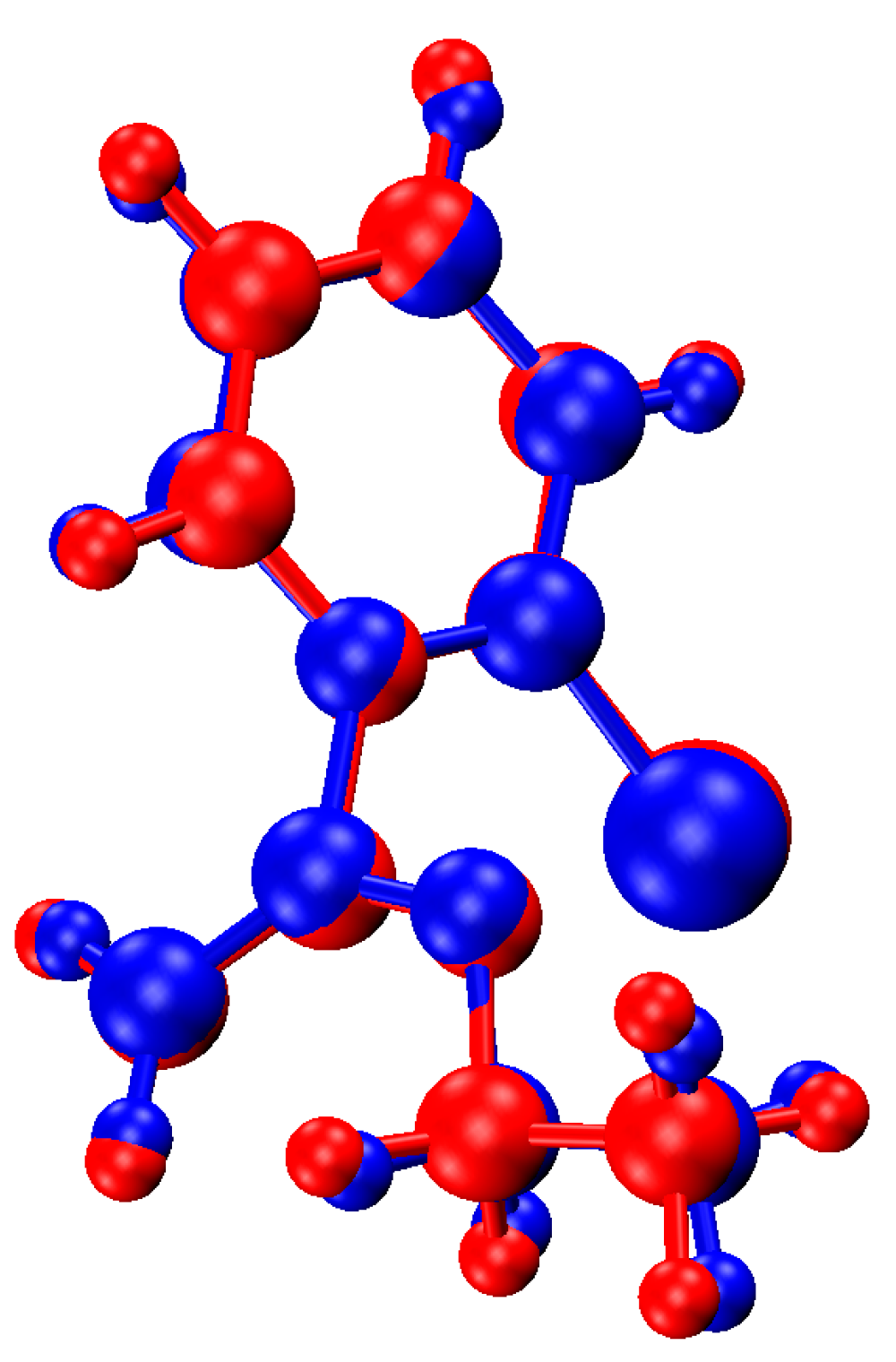
2.3. Single Crystal X-ray Crystallography
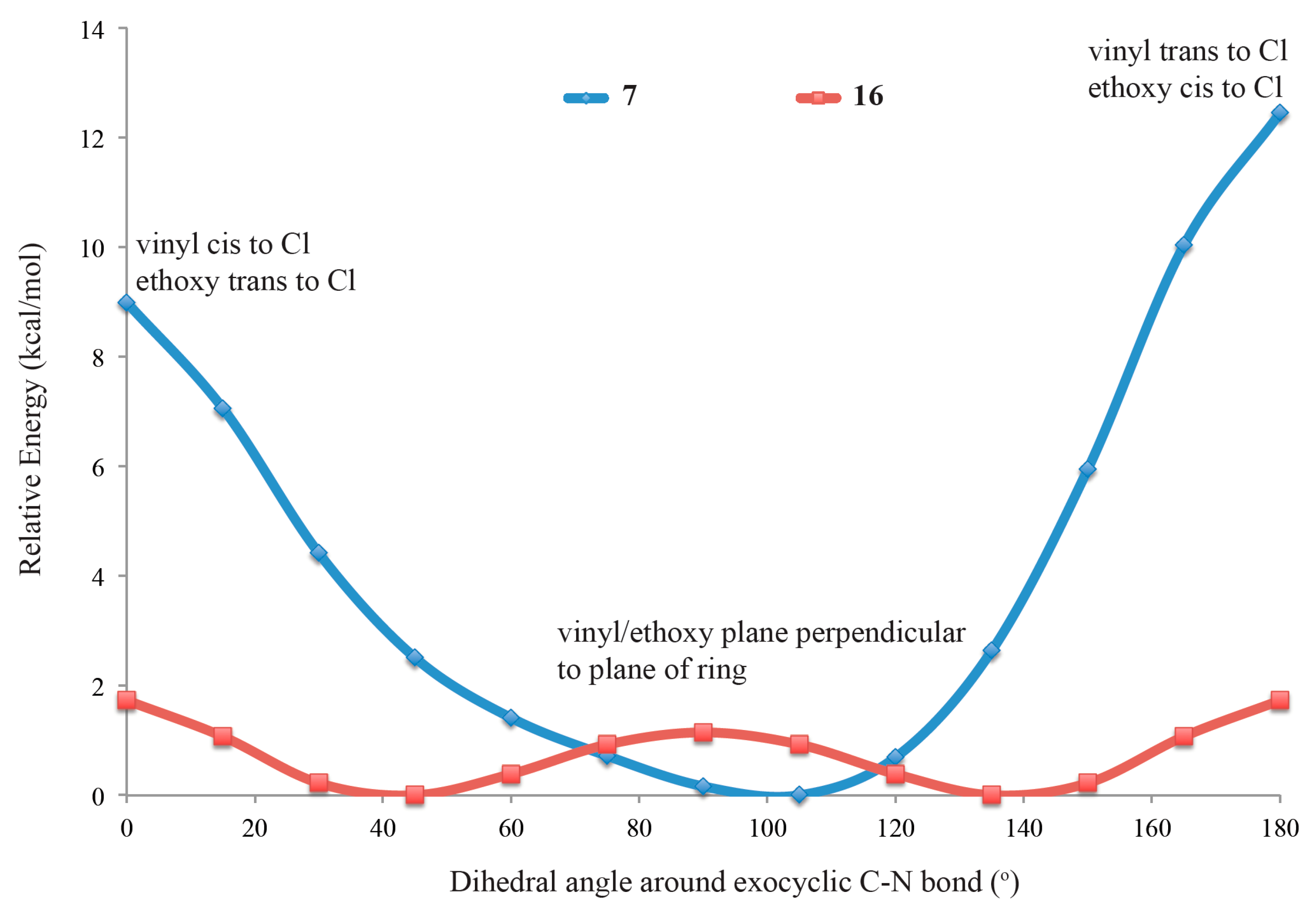
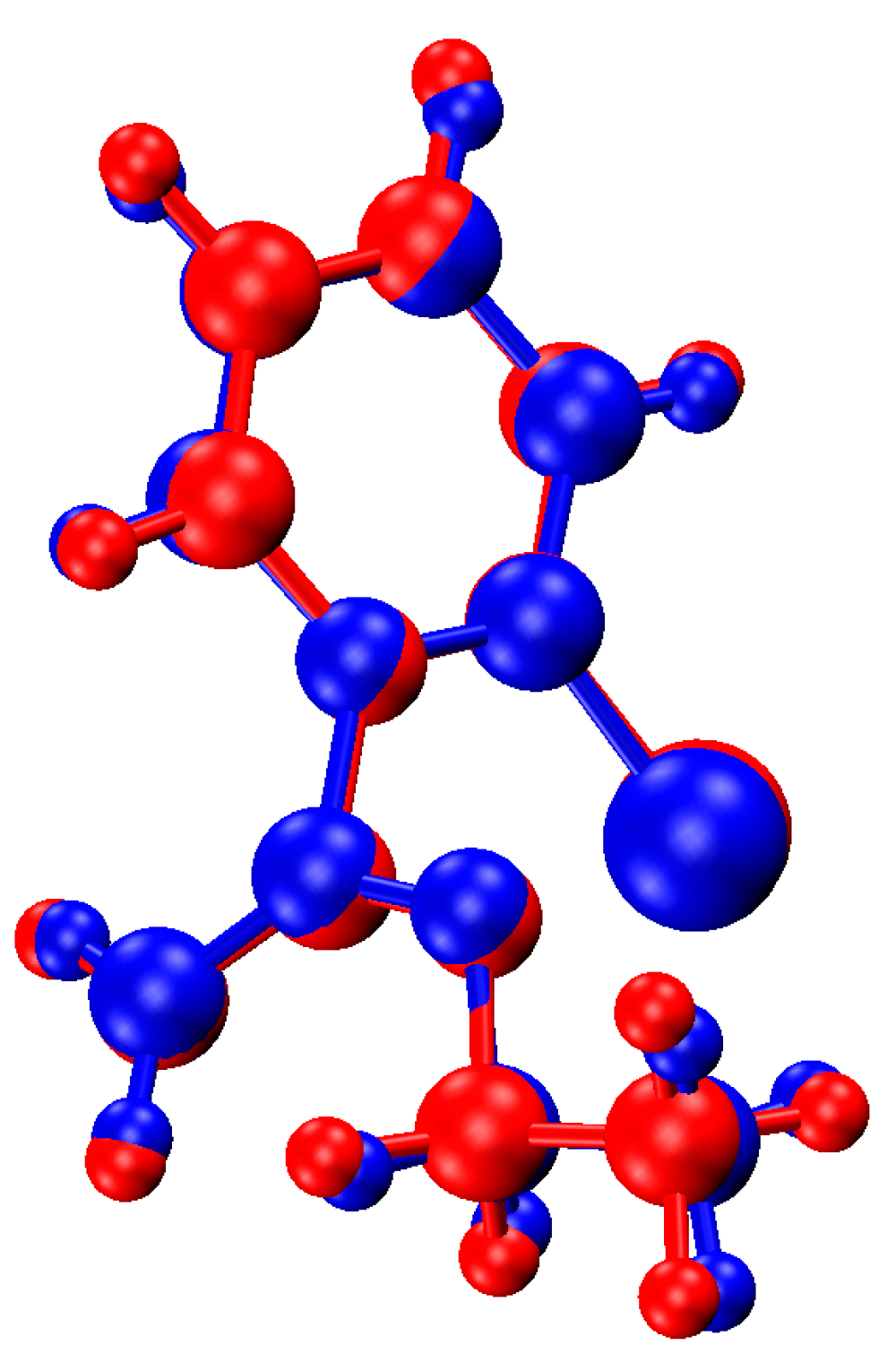
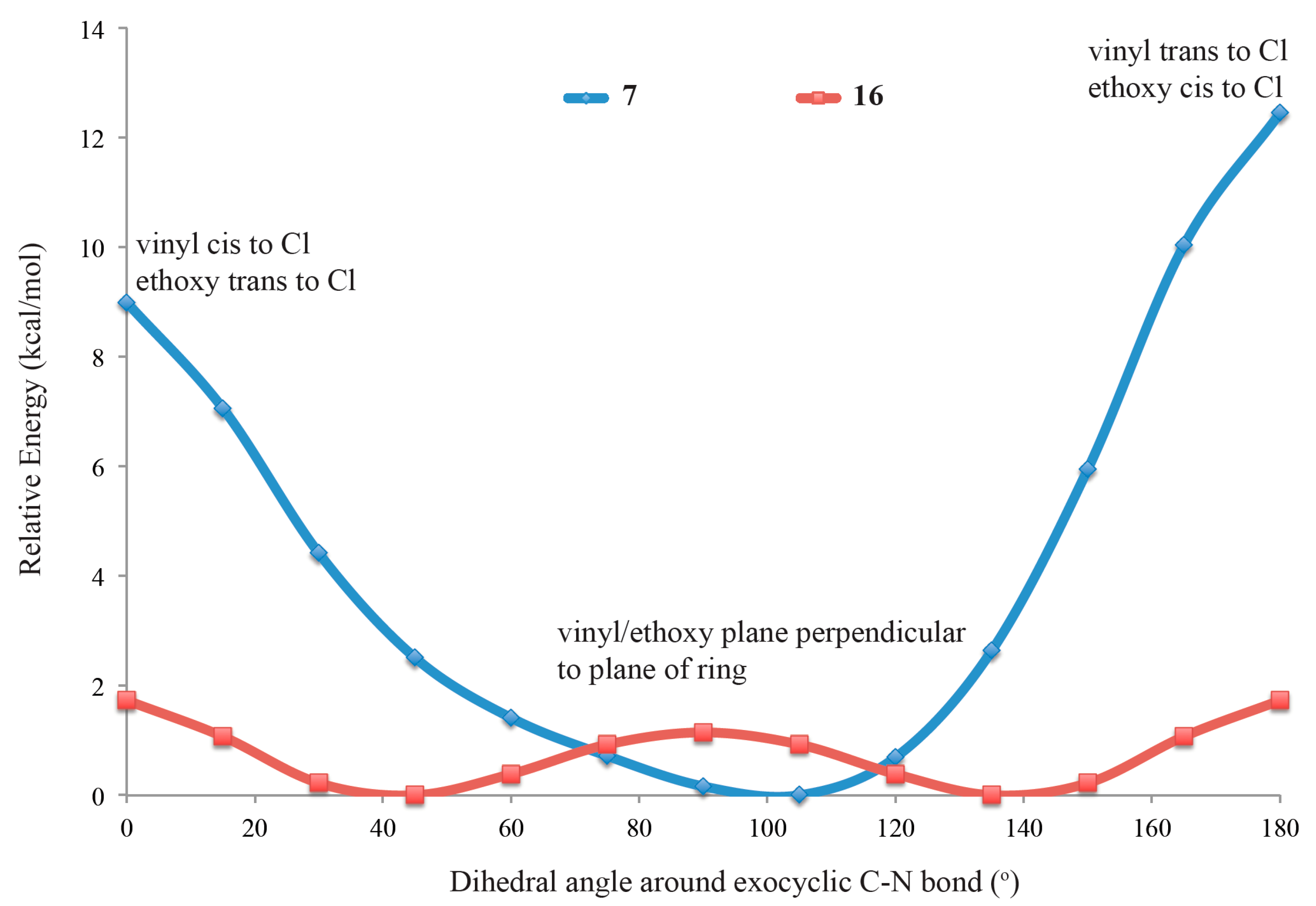
2.4. Computational Modeling
3. Materials and Methods
3.1. General Experimental Methods
3.2. Synthesis of N-(1-Ethoxyvinyl)pyridinium Triflates
3.2.1. Synthesis of 2-Chloro-1-(1-ethoxyvinyl)pyridine-1-ium Trifluoromethanesulfonate (7)
3.2.2. Synthesis of 1-(1-Ethoxyvinyl)pyridine-1-ium Trifluoromethanesulfonate (16)
3.2.3. Synthesis of 2-Iodo-1-(1-ethoxyvinyl)pyridine-1-ium Trifluoromethanesulfonate (17)
3.2.4. Synthesis of 2-Bromo-1-(1-ethoxyvinyl)pyridine-1-ium Trifluoromethanesulfonate (18)
3.2.5. Synthesis of 2-Fluoro-1-(1-ethoxyvinyl)pyridine-1-ium Trifluoromethanesulfonate (19)
3.2.6. Synthesis of 1-(1-Ethoxyvinyl)-2-isopropoxypyridin-1-ium Trifluoromethanesulfonate (20)
3.2.7. Synthesis of 2-Hydroxypyridin-1-ium Trifluoromethanesulfonate (23)
3.3. X-ray Diffraction Methodology
3.4. Computational Methodology
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Jennings, M.C.; Minbiole, K.P.C.; Wuest, W.M. Quaternary ammonium compounds: An antibacterial mainstay and platform for innovation to address bacterial resistance. ACS Infect. Dis. 2015, 1, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.C.; Buttaro, B.A.; Minbiole, K.P.C.; Wuest, W.M. Bioorganic investigation of multicationic antimicrobials to combat QAC-resistant Staphylococcus aureus. ACS Infect. Dis. 2015, 1, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Krátky, M.; Vinŝová, J. Antimycobacterial activity of quaternary pyridinium salts and pyridinium N-oxides—Review. Curr. Pharm. Des. 2013, 19, 1343–1355. [Google Scholar] [PubMed]
- Sowmiah, S.; Esperança, J.M.S.S.; Rebelo, L.P.N.; Afonso, C.A.M. Pyridinium salts: From synthesis to reactivity and applications. Org. Chem. Front. 2018, 5, 453–493. [Google Scholar] [CrossRef]
- Madaan, P.; Tyagi, V.K. Quaternary pyridinium salts: A review. J. Oleo Sci. 2008, 58, 197–215. [Google Scholar] [CrossRef]
- Śliwa, W. Quaternary salts of pyridines and related compounds. Curr. Org. Chem. 2003, 7, 995–1048. [Google Scholar] [CrossRef]
- Pendelton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed. Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Śliwa, W. N-Substituted pyridinium salts. Heterocycles 1989, 29, 557–595. [Google Scholar] [CrossRef]
- Śliwa, W. The reactivity of N-substituted pyridinium salts. Heterocycles 1986, 24, 181–219. [Google Scholar] [CrossRef]
- Katritzky, A.R. Summary of Katritzky research group scientific results (1954–1993). Heterocycles 1994, 37, 3–60. [Google Scholar] [CrossRef]
- Mukaiyama, T. New synthetic reactions based on the onium salts of aza-arenes. Angew. Chem. Int. Ed. Engl. 1979, 18, 707–721. [Google Scholar] [CrossRef]
- Armstrong, A.; Wang, Y.; Wang, P. 2-Chloro-1-methylpyridinium iodide. e-EROS 2008. [Google Scholar] [CrossRef]
- Novosjolova, I. The Mukaiyama reagent: An efficient condensation agent. Synlett 2013, 24, 135–136. [Google Scholar] [CrossRef]
- Sutherland, J.K.; Widdowson, D.A. Use of 2-iodo-1-methylpyridinium iodide in amide synthesis. J. Chem. Soc. 1964, 4650–4651. [Google Scholar]
- Bald, E.; Saigo, K.; Mukaiyama, T. A facile synthesis of carboxamides by using 1-methyl-2-halopyridinium iodides as coupling reagents. Chem. Lett. 1975, 4, 1163–1166. [Google Scholar] [CrossRef]
- Saigo, K.; Usui, M.; Kikushi, K.; Shimada, E.; Mukaiyama, T. New method for the preparation of carboxylic esters. Bull. Chem. Soc. Jpn. 1977, 50, 1863–1866. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falgue, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Mukaiyama, T.; Masahiro, U.; Kazuhiko, S. The facile synthesis of lactones. Chem. Lett. 1976, 5, 49–50. [Google Scholar] [CrossRef]
- Narasaka, K.; Maruyama, K.; Mukaiyama, T. A useful method for the synthesis of macrocyclic lactone. Chem. Lett. 1978, 7, 885–888. [Google Scholar] [CrossRef]
- Parenty, A.; Moreau, X.; Campagne, J.-M. Macrolactonizations in the total synthesis of natural products. Chem. Rev. 2006, 106, 911–939. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.K.; Mills, S.G.; Reamer, R.A.; Askin, D.; Desmond, R.; Volante, R.P.; Shinkai, I. Total synthesis of immunosuppressant (−)-FK-506. J. Am. Chem. Soc. 1989, 111, 1157–1159. [Google Scholar] [CrossRef]
- Nakatsuka, M.; Ragan, J.A.; Smmakia, T.; Smith, D.B.; Uehling, D.E.; Schreiber, S.L. Total synthesis of FK506 and an FKBP probe reagent, [C8,C9-13C2]-FK506. J. Am. Chem. Soc. 1990, 112, 5583–5601. [Google Scholar] [CrossRef]
- Roush, W.R.; Coffey, D.S.; Madar, D.J. Total synthesis of (+)-damavaricin D. J. Am. Chem. Soc. 1997, 119, 11331–11332. [Google Scholar] [CrossRef]
- Smith, A.B., III; Wan, Z. Total synthesis of the ansamycin antibiotic (+)-thiazinotrienomycin E. J. Org. Chem. 2000, 65, 3738–3753. [Google Scholar] [CrossRef] [PubMed]
- Vandromme, L.; Teulade-Fichou, M.-P. Beneficial effect of Mukaiyama reagent on macrobislactamization reactions. Synlett 2006, 3423–3426. [Google Scholar] [CrossRef]
- Romo, D.; Abbasov, M.E. The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis. Nat. Prod. Rep. 2014, 31, 1318–1327. [Google Scholar]
- Huang, H.; Iwasawa, N.; Mukaiyama, T. A convenient method for construction of β-lactam compounds from β-amino acids using 2-chloro-1-methylpyridinium iodide as condensing reagent. Chem. Lett. 1984, 13, 1465–1466. [Google Scholar] [CrossRef]
- Pitts, C.R.; Lectka, T. Chemical synthesis of β-lactams: Asymmetric catalysis and other recent advances. Chem. Rev. 2014, 114, 7930–7953. [Google Scholar] [CrossRef] [PubMed]
- Baraznenok, I.L.; Nenajdenko, V.G.; Balenkova, E.S. Chemical transformations induced by triflic anhydride. Tetrahedron 2000, 56, 3077–3119. [Google Scholar] [CrossRef]
- Madelaine, C.; Valerio, V.; Maulide, N. Revisiting keteniminium salts: More than the nitrogen analogs of ketenes. Chem. Asian J. 2011, 6, 2224–2239. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Maulide, N. Making the least reactive electrophile the first in class: Domino electrophilic activation of amides. J. Org. Chem. 2016, 81, 4421–4428. [Google Scholar] [CrossRef] [PubMed]
- Movassaghi, M.; Hill, M.D.; Ahmad, O.K. Direct synthesis of pyridine derivatives. J. Am. Chem. Soc. 2007, 129, 10096–10097. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, O.K.; Medly, J.N.; Coste, A.; Movassaghi, M. Direct synthesis of azaheterocycles from N-aryl/vinyl amides. Synthesis of 4-(methylthio)-2-phenylquinazoline and 4-(4-methoxyphenyl)-2-phenylquinoline. Org. Synth. 2012, 89, 549–561. [Google Scholar] [PubMed]
- Charette, A.B.; Grenon, M. Spectroscopic studies of the electrophilic activation of amides with triflic anhydride and pyridine. Can. J. Chem. 2001, 79, 1694–1703. [Google Scholar] [CrossRef]
- Weinstein, B.; Brattesani, D.N. Heterocyclic compounds. VIII. Reaction of ethoxyacetylene with 2- and 4-pyridone. J. Org. Chem. 1967, 32, 4107–4108. [Google Scholar] [CrossRef]
- Kantlehner, N. Product Subclass 9: 1-Nitrogen-functionalized 1-(organooxy)alk-1-enes (ketene O,N-acetals). In Science of Synthesis; de Meijere, A., Ed.; Thieme: Stuttgart, Germany, 2005; Volume 24, pp. 337–440 and references therein. [Google Scholar]
- Barnes, H.M.; Kundiger, D.; McElvain, S.M. Ketene acetals. V. The reaction of ketene diethylacetal with various compounds containing an active hydrogen. J. Am. Chem. Soc. 1940, 62, 1281–1287. [Google Scholar] [CrossRef]
- Klages, F.; Drerup, E. Über 1-amino-vinyläther. Liebigs Ann. 1941, 547, 65–72. [Google Scholar] [CrossRef]
- Arens, J.F.; Bouman, J.G.; Koerts, D.H. The chemistry of acetylenic ethers XV. The reaction of ethoxyacetylene with aqueous solutions of tertiary aliphatic amines. Recl. Trav. Chim. Pays-Bas 1955, 74, 1040–1044. [Google Scholar] [CrossRef]
- Organon, N.V. Unsaturated Quaternary Ammonium Compounds. U.S. Patent NL 79,439, 15 October 1955. [Google Scholar]
- Lehn, J.M.; Seher, R. Nuclear spin-spin interactions. 14N-1H spin-spin coupling in quaternary enammonium salts. Chem. Commun. 1966, 847–849. [Google Scholar] [CrossRef]
- Otsuru, M.; Tori, K.; Lehn, J.-M.; Seher, R. 14N,1H and 1H,1H spin couplings in quaternary enammonium salts. J. Am. Chem. Soc. 1969, 91, 1187–1194. [Google Scholar] [CrossRef]
- Herkes, F.E.; Simmons, H.E. Mono- and disubstituted vinyltrialkylammonium compounds. Synthesis and stereochemistry. J. Org. Chem. 1973, 38, 2845–2851. [Google Scholar] [CrossRef]
- Filippova, A.K.; Kashik, T.V.; Lyashenko, G.S.; Ponomareva, S.M.; Kalikhman, I.D.; Vyazankin, N.S. Quaternary ammonium bases and salts from aromatic acetylene ethers. Zh. Obshch. Khim. 1983, 53, 1141–1145. [Google Scholar]
- Filippova, A.K.; Kashik, T.V.; Lyashenko, G.S.; Voronkov, M.G. Vinyl quaternary ammonium bases and salts based on aromatic acetylene ethers and sulfides. In Proceedings of the 5th Tezisy Dokl.—Vses. Konf. Khim. Atsetilena, Tiflis, USSR; 1975; p. 362. [Google Scholar]
- In particular, compounds 7, 17, 18 and 20 were all found to be benchtop stable when stored as a solid for >3 months under ambient temperature and atmosphere. However, for best results we suggest storing these compounds under a dry, inert atmosphere due to slight hygroscopicity.
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999; ISBN 9780198509707. [Google Scholar]
- Rigaku Oxford Diffraction. CrysAlisPro, Version 171.39.33b; Rigaku Oxford Diffraction: Oxford, UK, 2017. [Google Scholar]
- Rigaku Oxford Diffraction. SCALE3 ABSPACK—A Rigaku Oxford Diffraction Program for Absorption Corrections; Rigaku Oxford Diffraction: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
Sample Availability: Samples of N-(1-ethoxyvinyl)pyridinium triflates 7, 16–18, and 20 are available from the corresponding author upon request. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 7 |
|---|---|
| CCDC Code | 1815230 |
| Formula | C10H11ClF3NO4S |
| Formula weight | 333.71 |
| Temp. | 100(2) |
| Space group | P-1 |
| a, Å | 8.5347(2) |
| b, Å | 9.0911(2) |
| c, Å | 9.2950(2) |
| α, deg | 85.871(2) |
| β, deg | 80.449(2) |
| γ, deg | 82.656(2) |
| Volume, Å3 | 704.41(3) |
| Z | 2 |
| Density (calculated), mg/m3 | 1.573 |
| μ, mm−1 | 4.259 |
| Scan | ω scan |
| θ range for data collection, deg | 6.582–67.716 |
| Reflections measured | 5893 |
| Independent observed reflns. | 2522 |
| Independent reflns. [I > 2σ] | 2467 |
| Data/restraints/parameters | 2522/0/217 |
| Rint | 0.0194 |
| Final R Indices [I > 2σ] | R1 = 0.0322, |
| wR2 = 0.0858 | |
| R Indices (all data) | R1 = 0.0329, |
| wR2 = 0.0864 | |
| Goodness-of-fit on F2 | 1.085 |
| Hydrogen Bonding Interaction | H•••O/C•••O Bond Distances (Å) | C–H•••O Bond Angle (°) | Symmetry Operation |
|---|---|---|---|
| C(1)–H(1)•••O(2) | 2.37(2)/3.139(2) | 134.0(16) | -x, 2-y, 1-z |
| C(2)–H(2)•••O(4) | 2.35(2)/3.268(2) | 163.5(18) | -x, 2-y, 1-z |
| C(3)–H(3)•••O(3) | 2.41(2)/3.276(2) | 156.8(18) | 1-x, 2-y, 1-z |
| C(4)–H(4)•••O(2) | 2.46(2)/3.259(2) | 144.5(17) | 1-x, 2-y, 1-z |
| C(7)–H(7A)•••O(4) | 2.46(2)/3.390(2) | 156.6(17) | x, y, z-1 |
| C(7)–H(7B)•••O(4) | 2.42(3)/3.350(2) | 168.1(19) | -x, 1-y, 1-z |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapiro, J.D.; Sonberg, J.C.; Schafer, B.C.; Williams, C.C.; Ferris, H.R.; Reinheimer, E.W.; Van Wynsberghe, A.W.; Kriley, C.E.; Majireck, M.M. Synthesis, Characterization, and Computational Modeling of N-(1-Ethoxyvinyl)pyridinium Triflates, an Unusual Class of Pyridinium Salts. Molecules 2018, 23, 413. https://doi.org/10.3390/molecules23020413
Shapiro JD, Sonberg JC, Schafer BC, Williams CC, Ferris HR, Reinheimer EW, Van Wynsberghe AW, Kriley CE, Majireck MM. Synthesis, Characterization, and Computational Modeling of N-(1-Ethoxyvinyl)pyridinium Triflates, an Unusual Class of Pyridinium Salts. Molecules. 2018; 23(2):413. https://doi.org/10.3390/molecules23020413
Chicago/Turabian StyleShapiro, Jonathan D., Justin C. Sonberg, Benjamin C. Schafer, Christopher C. Williams, Hannah R. Ferris, Eric W. Reinheimer, Adam W. Van Wynsberghe, Charles E. Kriley, and Max M. Majireck. 2018. "Synthesis, Characterization, and Computational Modeling of N-(1-Ethoxyvinyl)pyridinium Triflates, an Unusual Class of Pyridinium Salts" Molecules 23, no. 2: 413. https://doi.org/10.3390/molecules23020413
APA StyleShapiro, J. D., Sonberg, J. C., Schafer, B. C., Williams, C. C., Ferris, H. R., Reinheimer, E. W., Van Wynsberghe, A. W., Kriley, C. E., & Majireck, M. M. (2018). Synthesis, Characterization, and Computational Modeling of N-(1-Ethoxyvinyl)pyridinium Triflates, an Unusual Class of Pyridinium Salts. Molecules, 23(2), 413. https://doi.org/10.3390/molecules23020413