Aethiopinolones A–E, New Pregnenolone Type Steroids from the East African Basidiomycete Fomitiporia aethiopica



Abstract

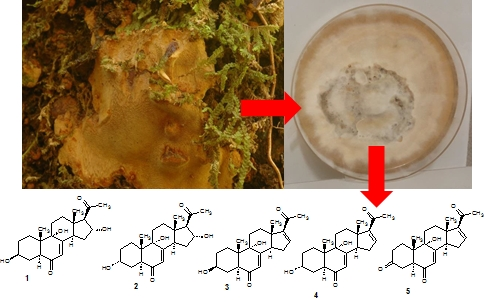
1. Introduction
2. Results and Discussion
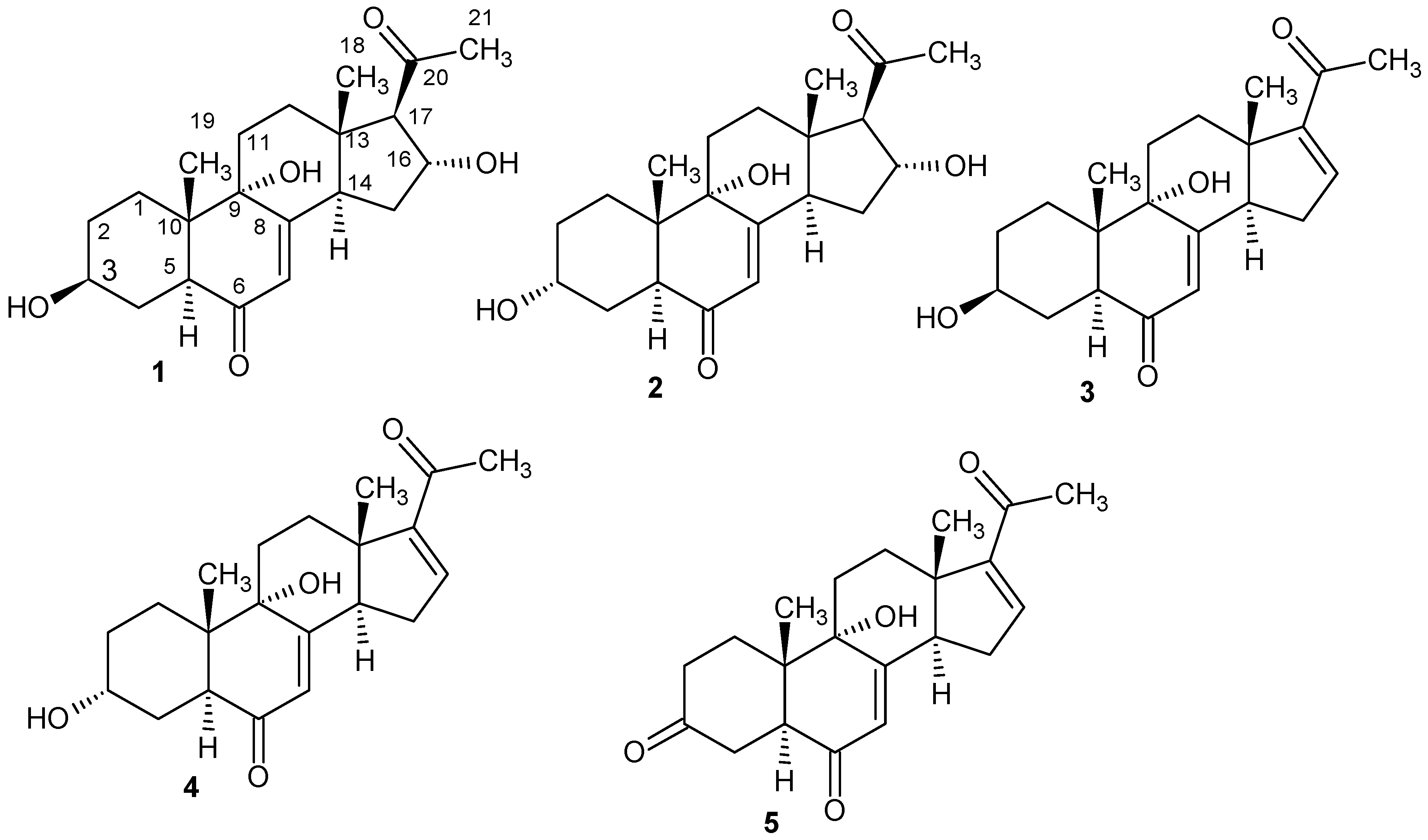
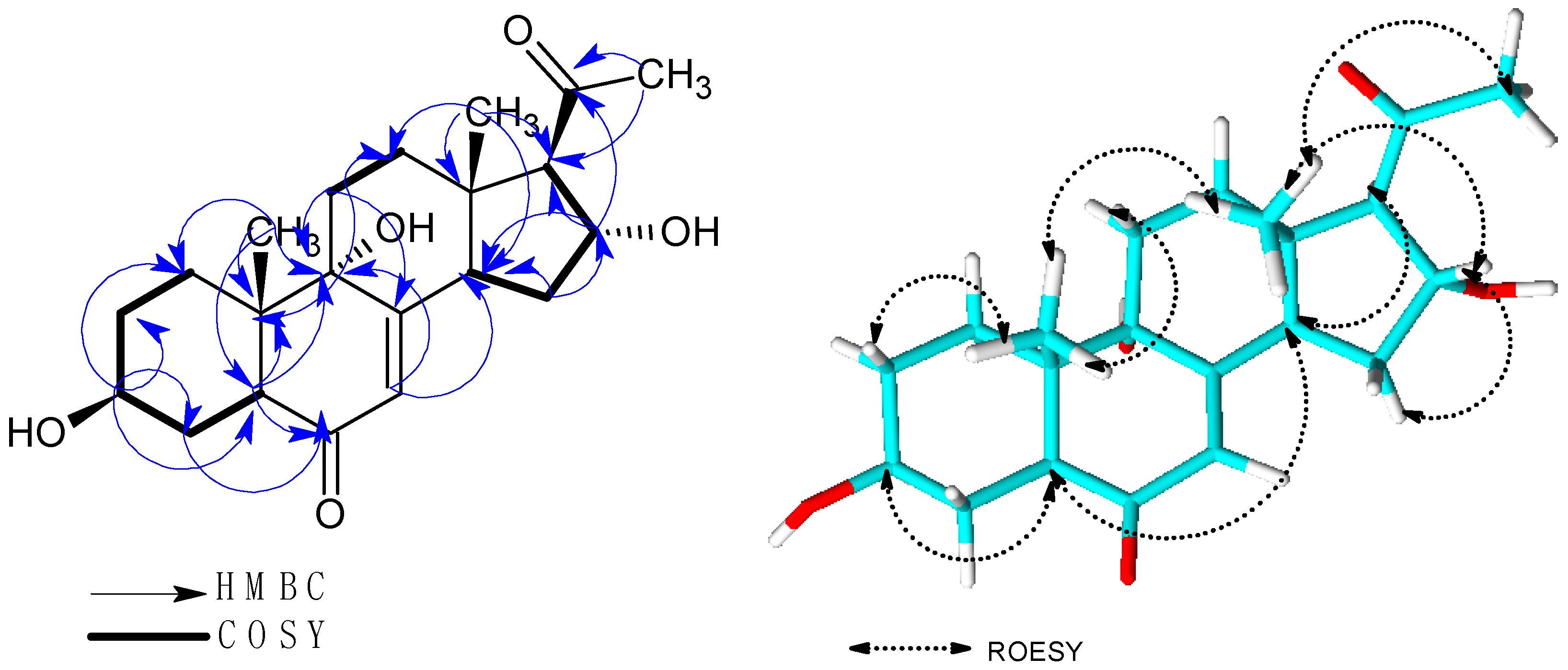
2.1. Structure Elucidation
2.2. Biological Activities
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation
3.4. Extraction
3.5. Isolation and Physico-Chemical Characteristics of Compounds 1–5
3.6. Preparation of the (R)- and (S)-MTPA Ester Derivatives
3.7. Antimicrobial Assay
3.8. Cytotoxicity Assay
3.9. Nematicidal Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karwehl, S.; Stadler, M. Exploitation of fungal biodiversity for discovery of novel antibiotics. Curr. Top. Microbiol. Immunol. 2016, 398, 303–338. [Google Scholar] [CrossRef] [PubMed]
- De Silva, D.D.; Rapior, S.; Sudarman, E.; Stadler, M.; Xu, J.; Aisyah, S.A.; Hyde, K.D. Bioactive metabolites from macrofungi: Ethnopharmacology, biological activities and chemistry. Fungal Divers 2013, 62, 1–40. [Google Scholar] [CrossRef]
- Chepkirui, C.; Matasyoh, J.C.; Decock, C.; Stadler, M. Two cytotoxic triterpenes from cultures of a Kenyan Laetiporus sp. (BasiDOImycota). Phytochem. Lett. 2017, 20, 106–110. [Google Scholar] [CrossRef]
- Chepkirui, C.; Richter, C.; Matasyoh, J.C.; Stadler, M. Monochlorinated calocerins A–D and 9-oxostrobilurin derivatives from the basiDOImycete Favolaschia calocera. Phytochemistry 2016, 132, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Mudalungu, C.M.; Richter, C.; Wittstein, K.; Abdalla, A.M.; Matasyoh, J.C.; Stadler, M.; Süssmuth, R.D. Laxitextines A and B, Cyathane xylosides from the tropical fungus Laxitextum incrustatum. J. Nat. Prod. 2015, 79, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Decock, C.; Bitew, A.; Castillo, G. Fomitiporia tenuis and Fomitiporia aethiopica (BasiDOImycetes, Hymenochaetales), two undescribed species from the Ethiopian highlands: Taxonomy and phylogeny. Mycologia 2005, 97, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Amalfi, M.; Yombiyeni, P.; Decock, C. Fomitiporia in sub-Saharan Africa: Morphology and multigene phylogenetic analysis support three new species from the Guineo-Congolian rainforest. Mycologia 2010, 102, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Cloete, M.; Fischer, M.; Mostert, L.; Halleen, F. A novel Fomitiporia species associated with esca on grapevine in South Africa. Mycol. Prog. 2014, 13, 303–311. [Google Scholar] [CrossRef]
- Buckingham, J. Dictionary of Natural Products on DVD; Chapman & Hall, Chemical Database, CRC: Boca Raton, FL, USA, 2017. [Google Scholar]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Bao, H.Y.; Cui, B.K. Chemical constituents of Fomitiporia ellipsoidea fruiting bodies. Mycosystema 2011, 30, 459–463. [Google Scholar]
- Cui, B.-K.; Decock, C. Phellinus castanopsidis sp. nov. (Hymenochaetaceae) from southern China, with preliminary phylogeny based on rDNA sequences. Mycol. Prog. 2013, 12, 341–351. [Google Scholar] [CrossRef]
- Fiasson, J.; Niemelä, T. The Hymenochaetales: A revision of the European poroid taxa. Karstenia 1984, 24, 14–28. [Google Scholar] [CrossRef]
- Fiasson, J. Distribution of styrylpyrones in the basiDOIcarps of various Hymenochaetaceae. Biochem. Syst. Ecol. 1982, 10, 289–296. [Google Scholar] [CrossRef]
- Hilaire, V.K.W.; Hartl, A.; Trinh, T.K.; Hertweck, C. Inotilone and related phenylpropanoid polyketides from Inonotus sp. and their identification as potent COX and XO inhibitors. Org. Biomol. Chem. 2006, 4, 2545–2548. [Google Scholar] [CrossRef]
- Ye, S.D.; Ying, S.H.; Chen, C.; Feng, M.G. New solid-state fermentation chamber for bulk production of aerial conidia of fungal biocontrol agents on rice. Biotechnol. Lett. 2006, 28, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Kuephadungphan, W.; Helaly, S.E.; Daengrot, C.; Phongpaichit, S.; Luangsa-ard, J.J.; Rukachaisirikul, V.; Stadler, M. Akanthopyrones A–D, α-pyrones bearing a 4-O-Methyl-β-D-glucopyranose moiety from the spider-associated ascomycete Akanthomyces novoguineensis. Molecules 2017, 22, 1202. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, S.; Helaly, S.E.; Schroers, H.J.; Stadler, M.; Richert-Poeggeler, K.R.; Dababat, A.A.; Maier, W. Ijuhya vitellina sp. nov., a novel source for chaetoglobosin A, is a destructive parasite of the cereal cyst nematode Heterodera filipjevi. PLoS ONE 2017, 12, e0180032. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Zhao, Z.; Ji, X.; Dong, Z.; Li, Z.; Feng, T.; Liu, J. Steroids and sesquiterpenes from cultures of the fungus Phellinus igniarius. Nat. Prod. Bioprospect. 2015, 5, 17–22. [Google Scholar] [CrossRef]
- Mahmoud, F.E.; Kehraus, S.; König, G.M. Caught between triterpene- and steroid-metabolism: 4a-Carboxylic pregnane-derivative from the marine alga-derived fungus Phaeosphaeria spartinae. Steroids 2013, 78, 880–883. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–5 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| Position | ∂C, Type | ∂H (J in Hz) | ∂C, Type | ∂H (J in Hz) |
| 1 | 30.5, CH2 | β:1.31, m b; α:1.48, m b | 24.2, CH2 | β:1.01, m b; α:2.27, m b |
| 2 | 31.6, CH2 | β:1.32, m b; α:1.77, m b | 27.4, CH2 | α:1.52, m b, β:1.40, m b |
| 3 | 70.3, CH | 3.50, tt, (4.5, 11.3) | 63.1, CH | 3.91, m |
| 4 | 31.5, CH2 | β:2.05, m b; α:2.12 m b | 27.9, CH2 | β:1.35, m b; α:1.80, m b |
| 5 | 47.2, CH | 2.90, dd, (12.2, 3.8) | 41.4, CH | 3.17 dd, (12.05, 3.9) |
| 6 | 199.3, C | 200.7, C | - | |
| 7 | 124.2, CH | 5.48, d, (2.3) | 122.6, CH | 5.39, d(1.9) |
| 8 | 160.8, C | 160.4, C | ||
| 9 | 74.2, C | 72.6, C | ||
| 10 | 42.7, C | 41.9, C | ||
| 11 | 28.5, CH2 | α:1.84, m b; β:2.01, m b | 26.8, CH2 | α:1.69, dd, (13.6, 3.9); β:1.78, dd, (13.6, 4.43) |
| 12 | 35.5, CH2 | β:1.97, m b, α:2.07, m b | 34.0, CH2 | β:1.83, m; α:1.94, m |
| 13 | 46.8, C | 45.6, C | ||
| 14 | 50.1, CH | 3.12, ddd, (12.8, 6.7, 2.3) | 48.8, CH | 3.01, ddd, (12.4, 6.6, 1.9) |
| 15 | 35.1, CH2 | β:1.61, m b; α:2.02, m b | 33.8, CH2 | α:1.47, m b; α:1.88, m b |
| 16 | 71.6, CH | 4.74, bt, (3.0) | 69.9, CH | 4.55, bt, (3.1) |
| 17 | 74.3, CH | 2.71, d, (6.0) | 73.0, CH | 2.63, d, (6.1) |
| 18 | 14.8, CH3 | 0.58, s | 14.2, CH2 | 0.46, s |
| 19 | 17.2, CH3 | 0.93, s | 15.8, CH3 | 0.80, s |
| 20 | 207.6, C | 207.7, C | ||
| 21 | 31.9, CH3 | 2.16, s | 31.6, CH3 | 2.15, s |
| 3 | 4 | 5 | ||||
|---|---|---|---|---|---|---|
| Pos. | ∂C, Type | ∂H (J in Hz) | ∂C, Type | ∂H (J in Hz) | ∂C, Type | ∂H (J in Hz) |
| 1. | 30.4, CH2 | β:1.29, m b; α:1.49, m b | 25.3, CH2 | β:1.16, m b; α:2.47, m b | 32.2, CH2 | β:1.82, m b; α:1.91, m b |
| 2. | 30.4, CH2 | β:1.33 m b; α:1.77, m b | 28.7, CH2 | α:1.60, m b; β:1.65, m b | 37.5, CH2 | β:2.26, m b; α:2.35, m b |
| 3. | 70.3, CH | 3.47, tt, (4.4, 11.3) | 65.0, CH | 4.05, m | 210.1, CH | |
| 4. | 31.5, CH2 | β:2.10, m b; α:2.13 m b | 29.2, CH2 | β:1.54, m b; α:1.97, m b | 37.6, CH2 | α:2.39, m b; β:2.51, m b |
| 5. | 47.3, CH | 2.91, dd, (12.2, 3.8) | 42.7, CH | 3.32, dd, (12.3, 4.1) | 49.0, CH | 3.30, dd, (12.7, 4.9) |
| 6. | 199.4, C | 201.1, C | 198.4, C | |||
| 7. | 123.4, CH | 5.60, d, (2.1) | 123.5, CH | 5.58, d, (2.2) | 123.1, CH | 5.68, d, (2.2) |
| 8. | 160.2, C | 160.0, C | 160.8, C | |||
| 9. | 74.4, C | 74.5, C | 74.6, C | |||
| 10. | 42.8, C | 43.4, C | 43.1, C | |||
| 11. | 31.3, CH2 | α:1.77, m b; β:2.15, m b | 29.4, CH2 | β:1.84, m b; α:2.13, m b | 29.6, CH2 | α:1.92, m b; β:2.24, m b |
| 12. | 32.1, CH2 | β:1.81, m b; α:2.30 m b | 32.1, CH2 | β:1.80, m b; α:2.32, m b | 32.1, CH2 | β:1.82, m b; α:2.35, m b |
| 13. | 46.8, C | 49.0, C | 49.0, C | |||
| 14. | 52.9, CH | 3.11, ddd, (11.6, 6.6, 2.1) | 52.7, CH | 3.13, ddd, (11.6, 6.5, 2.2) | 52.7, CH | 3.14, ddd (11.6, 2.2 Hz, 6.2) |
| 15. | 31.3, CH2 | β:2.39, m; α:2.47, m | 31.3, CH2 | β:2.41, m; α:2.47, m | 31.3, CH2 | β:2.42, m; α:2.51, m |
| 16. | 144.3, CH | 6.91, dd, (1.9, 3.4) | 144.3, CH | 6.91, dd, (1.9, 3.2) | 144.3, CH | 6.92, dd, (3.4, 1.9) |
| 17. | 155.1, C | 155.2, C | 155.1, C | |||
| 18. | 16.4, CH3 | 0.88, s | 16.4, CH3 | 0.88, s | 16.4, CH3 | 0.91, s |
| 19. | 17.2, CH3 | 0.99, s | 16.5, CH3 | 0.99, s | 16.5, CH3 | 1.24, s |
| 196.3, C | 196.3, C | - | 196.3, C | |||
| 27.14, CH3 | 2.25, s | 27.2, CH3 | 2.26, s | 27.1, CH3 | 2.26, s | |
| Cell Lines | Cytotoxicity IC50 (μg/mL) | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | Epothilon B | |
| L929 | 28 | 45 | 40 | 45 | 40 | 0.0014 |
| KB3.1 | 19 | 39 | 35 | 39 | 33 | 0.00022 |
| A431 | 22 | - | 27 | 21 | 14 | 0.0006 |
| A549 | nt | - | 70 | 52 | 43 | 0.005 |
| PC-3 | 8 | - | 45 | 40 | 39 | 0.0002 |
| SKOV-3 | 26 | - | 38 | 36 | 34 | 0.0014 |
| MCF-7 | 20 | - | 18 | 17 | 16 | 0.0004 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chepkirui, C.; Sum, W.C.; Cheng, T.; Matasyoh, J.C.; Decock, C.; Stadler, M. Aethiopinolones A–E, New Pregnenolone Type Steroids from the East African Basidiomycete Fomitiporia aethiopica. Molecules 2018, 23, 369. https://doi.org/10.3390/molecules23020369
Chepkirui C, Sum WC, Cheng T, Matasyoh JC, Decock C, Stadler M. Aethiopinolones A–E, New Pregnenolone Type Steroids from the East African Basidiomycete Fomitiporia aethiopica. Molecules. 2018; 23(2):369. https://doi.org/10.3390/molecules23020369
Chicago/Turabian StyleChepkirui, Clara, Winnie C. Sum, Tian Cheng, Josphat C. Matasyoh, Cony Decock, and Marc Stadler. 2018. "Aethiopinolones A–E, New Pregnenolone Type Steroids from the East African Basidiomycete Fomitiporia aethiopica" Molecules 23, no. 2: 369. https://doi.org/10.3390/molecules23020369
APA StyleChepkirui, C., Sum, W. C., Cheng, T., Matasyoh, J. C., Decock, C., & Stadler, M. (2018). Aethiopinolones A–E, New Pregnenolone Type Steroids from the East African Basidiomycete Fomitiporia aethiopica. Molecules, 23(2), 369. https://doi.org/10.3390/molecules23020369