VSpipe, an Integrated Resource for Virtual Screening and Hit Selection: Applications to Protein Tyrosine Phospahatase Inhibition

Abstract
:
1. Introduction
2. Results
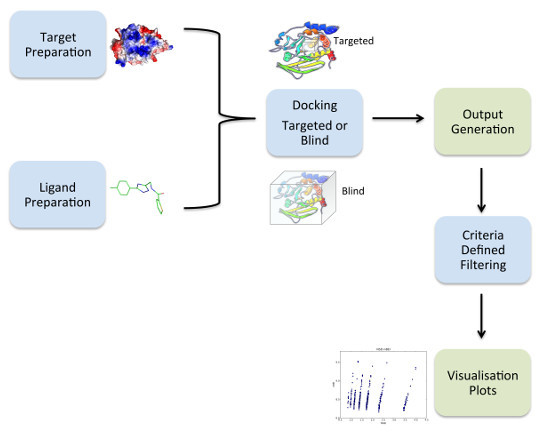
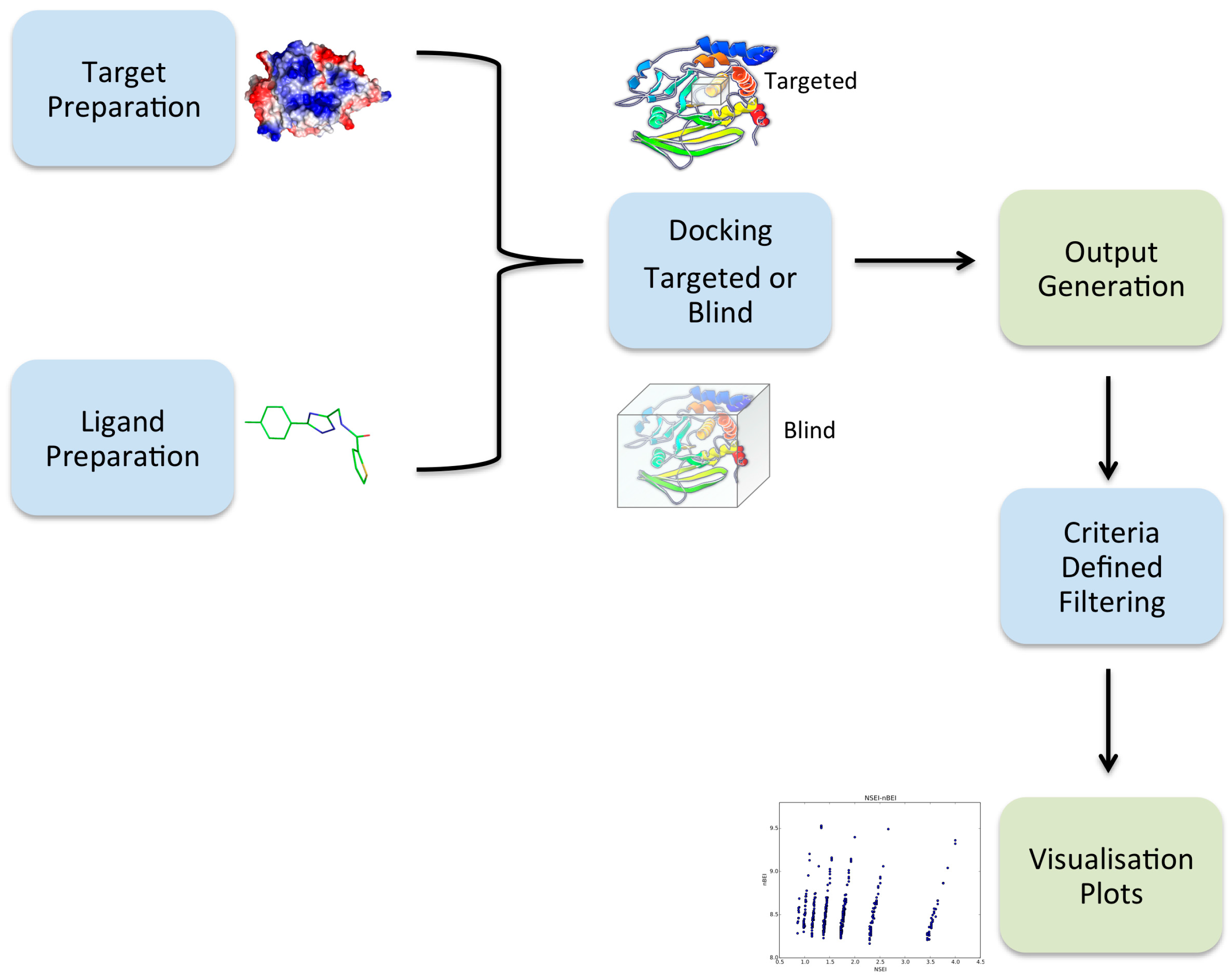
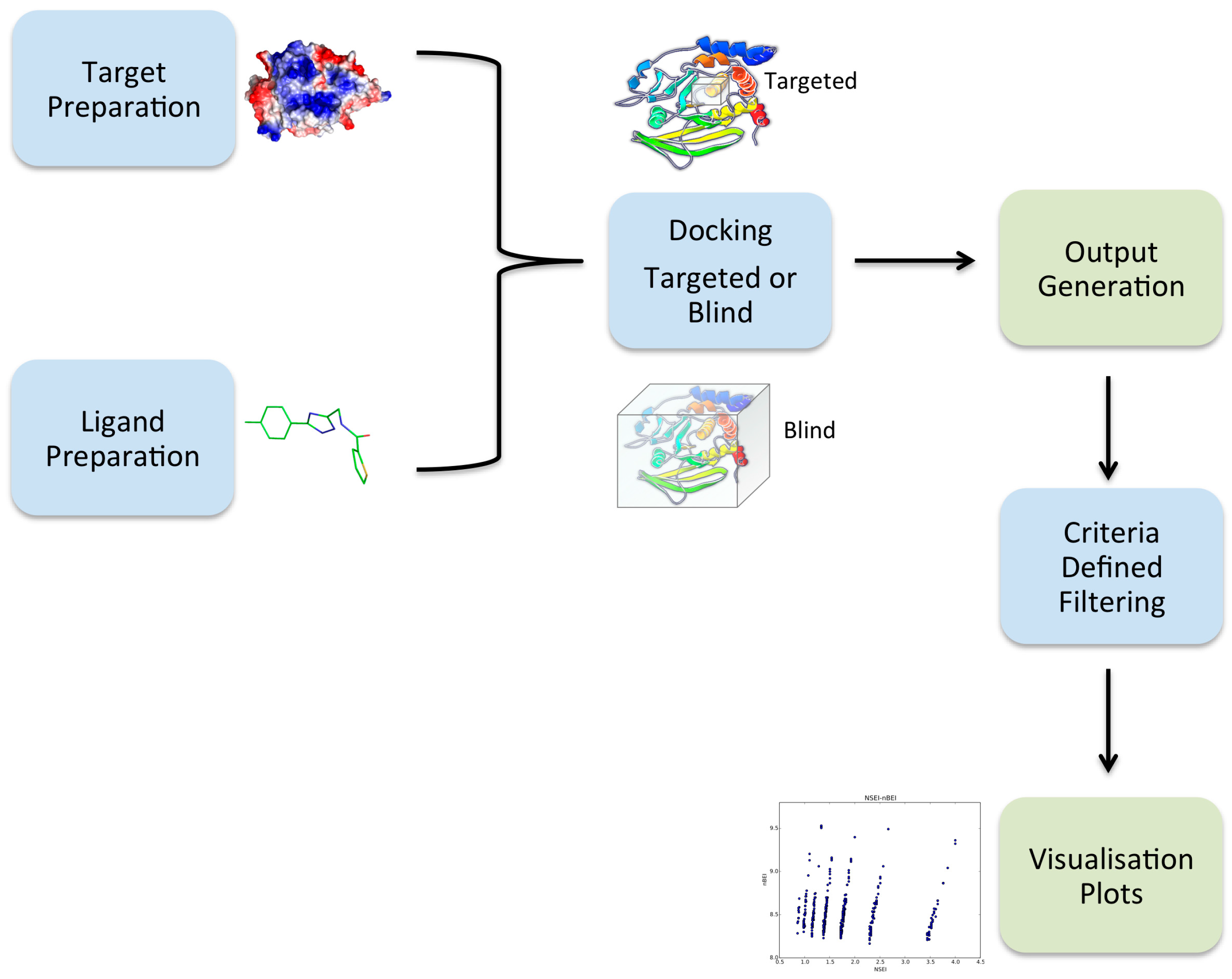
2.1. Overview of VSpipe
2.2. VSpipe-Local Mode
2.3. VSpipe-Cluster Mode
2.4. Docking Module Performance
2.5. Case Study on PTP1B
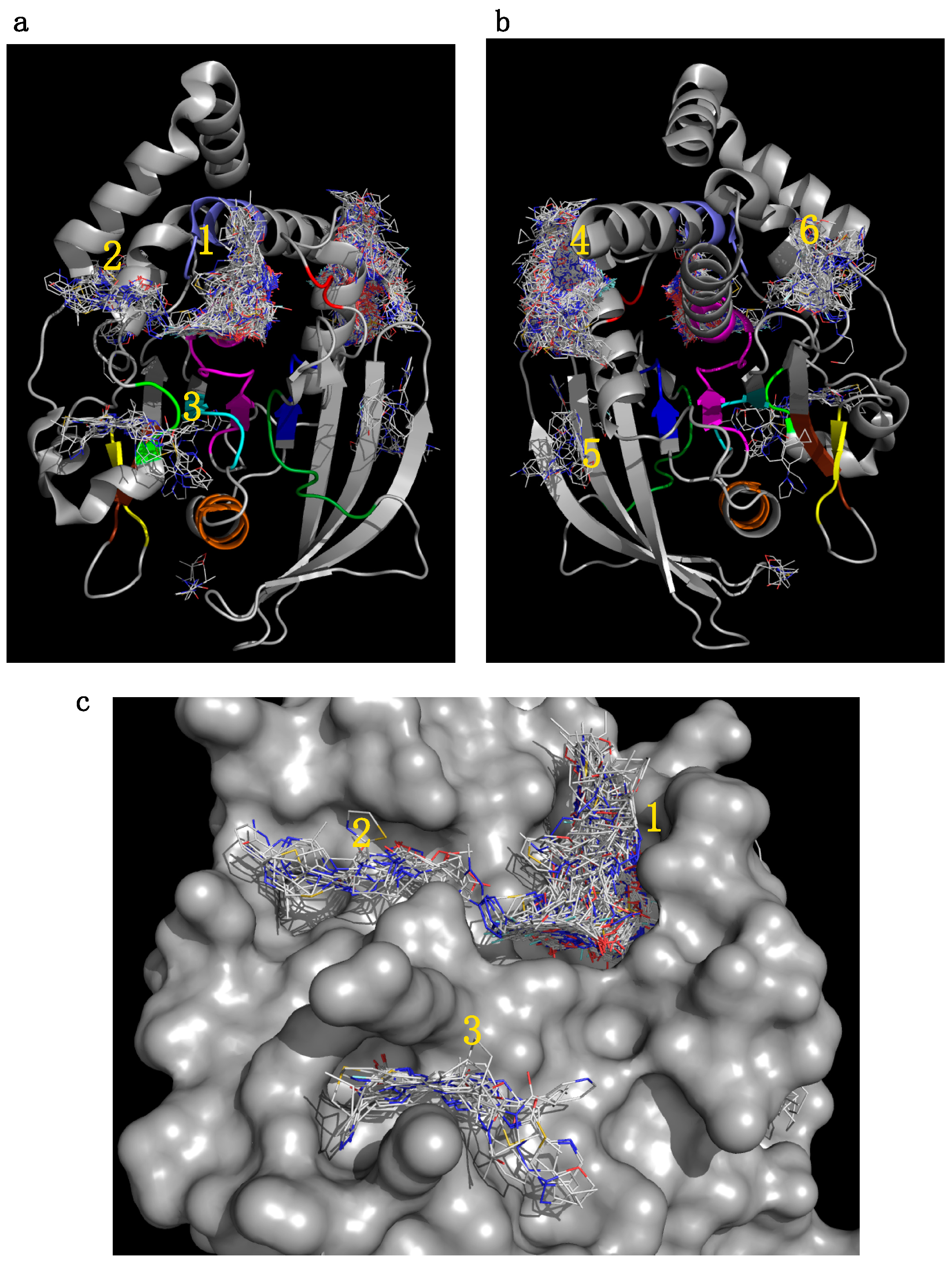
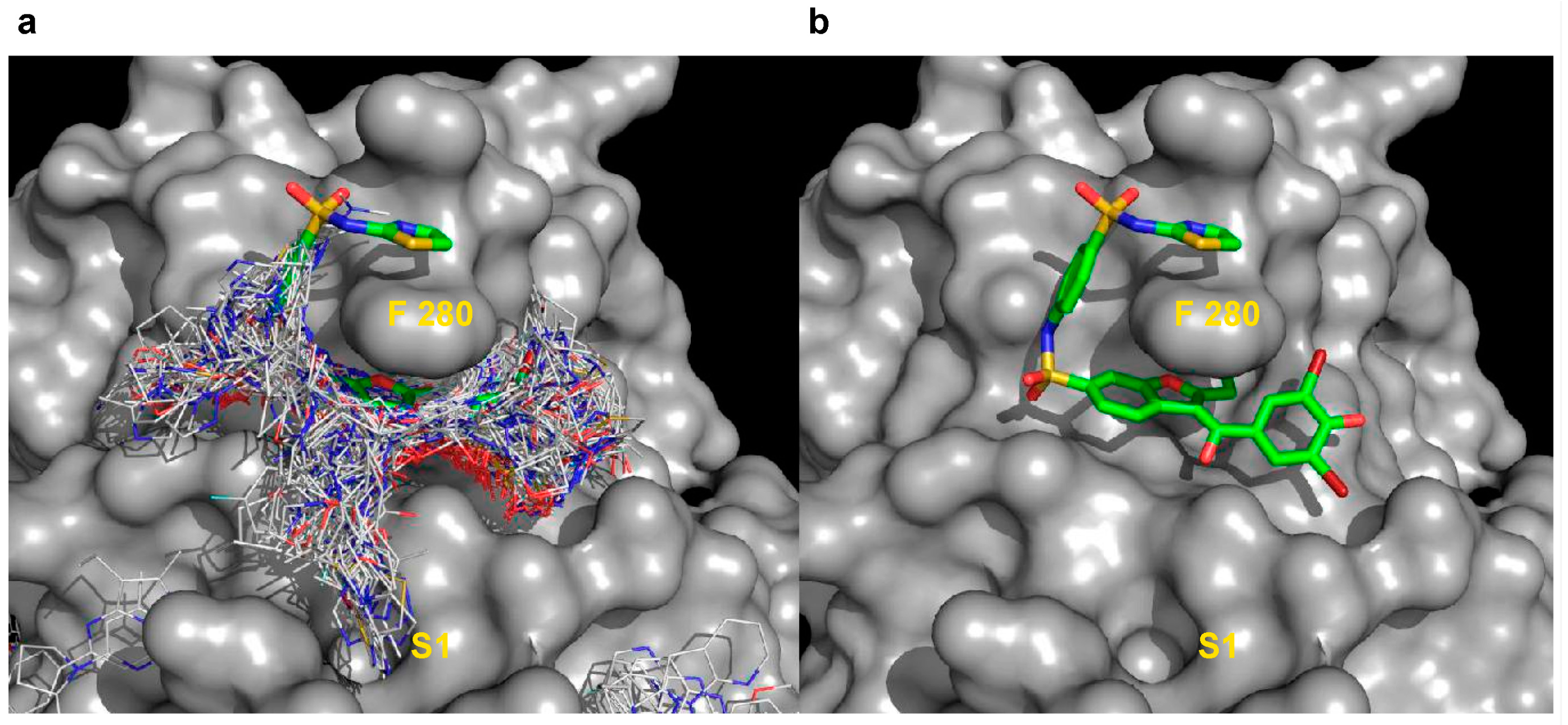
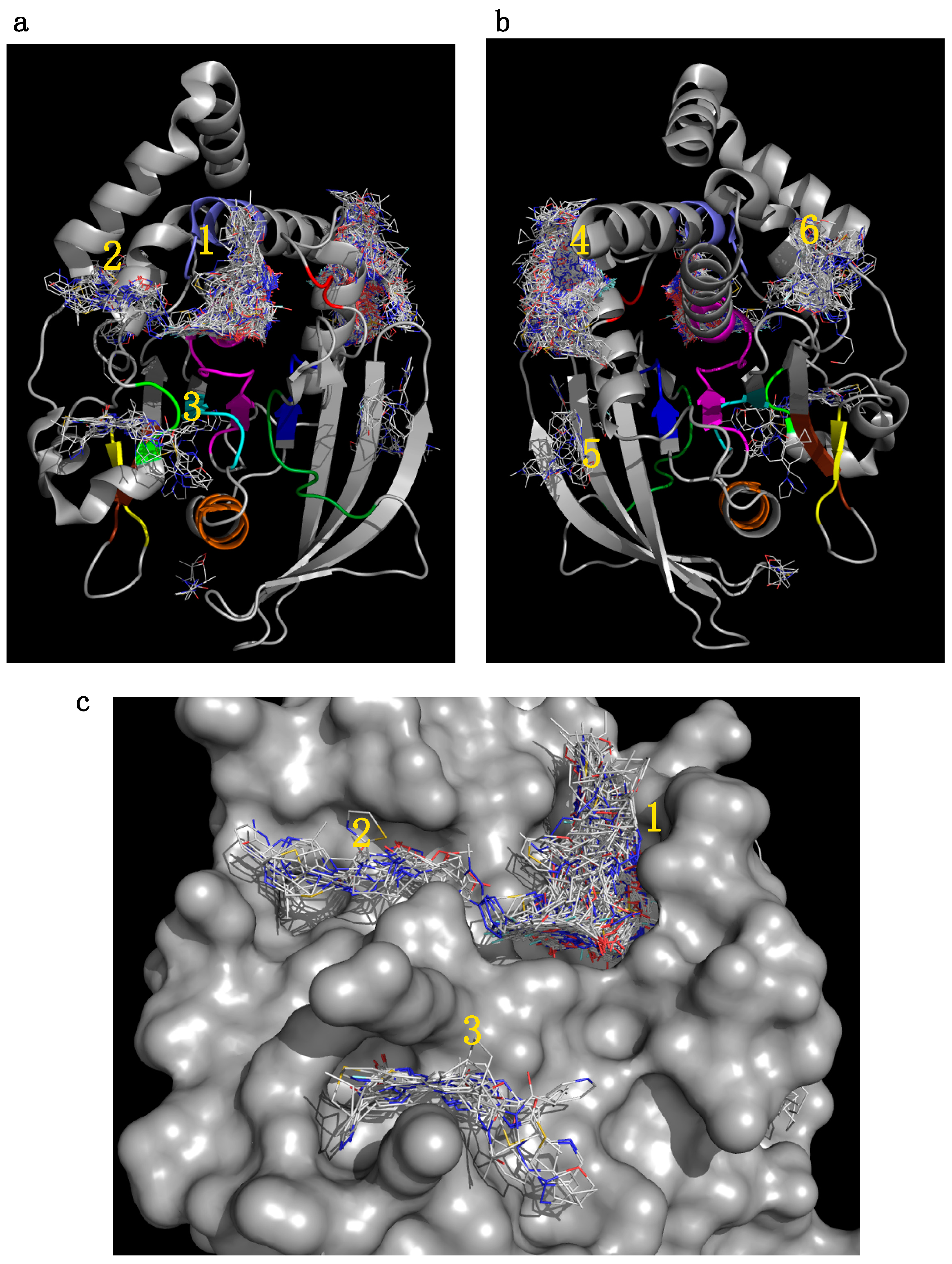
2.5.1. Initial Pocket Identification
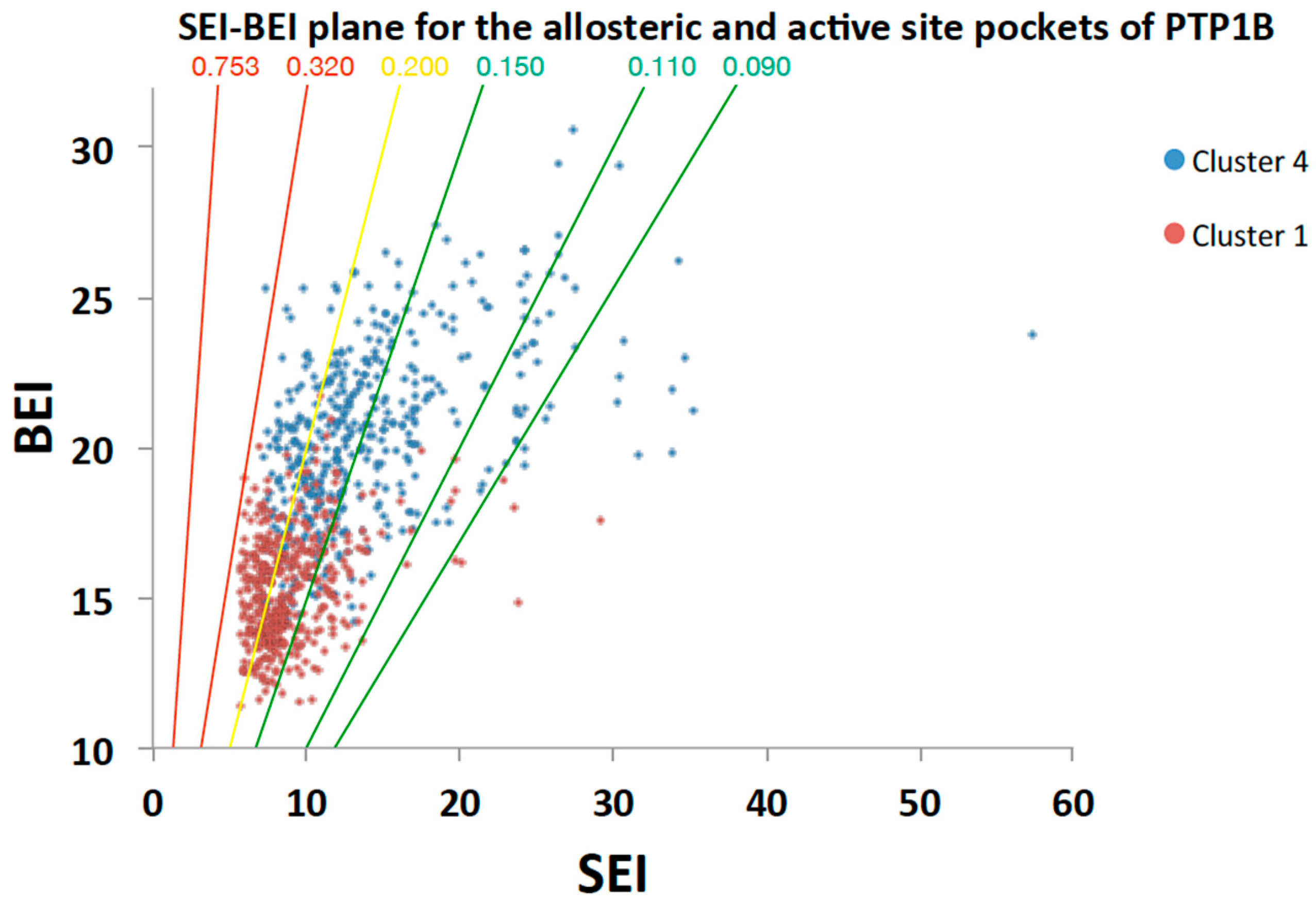
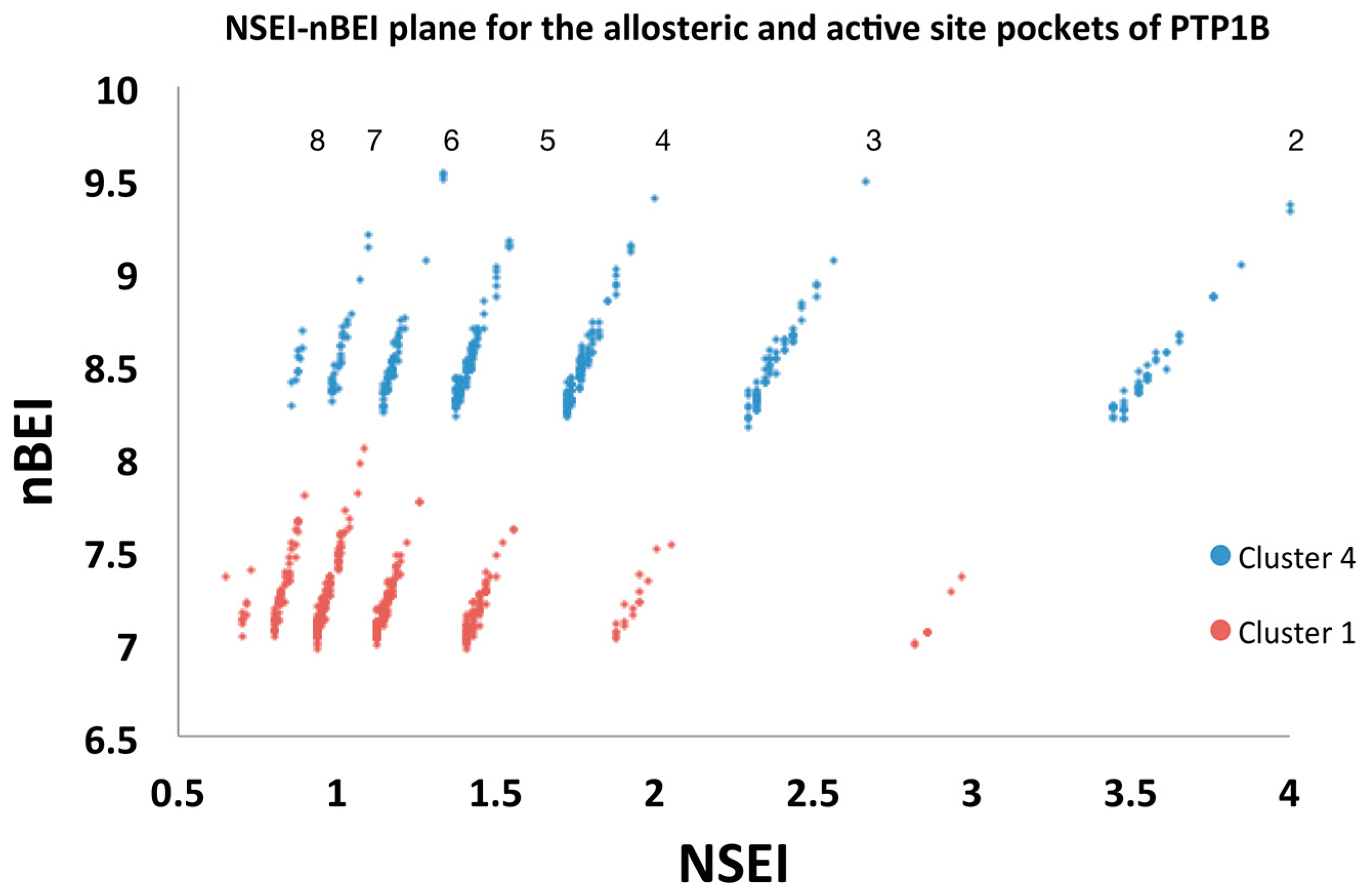
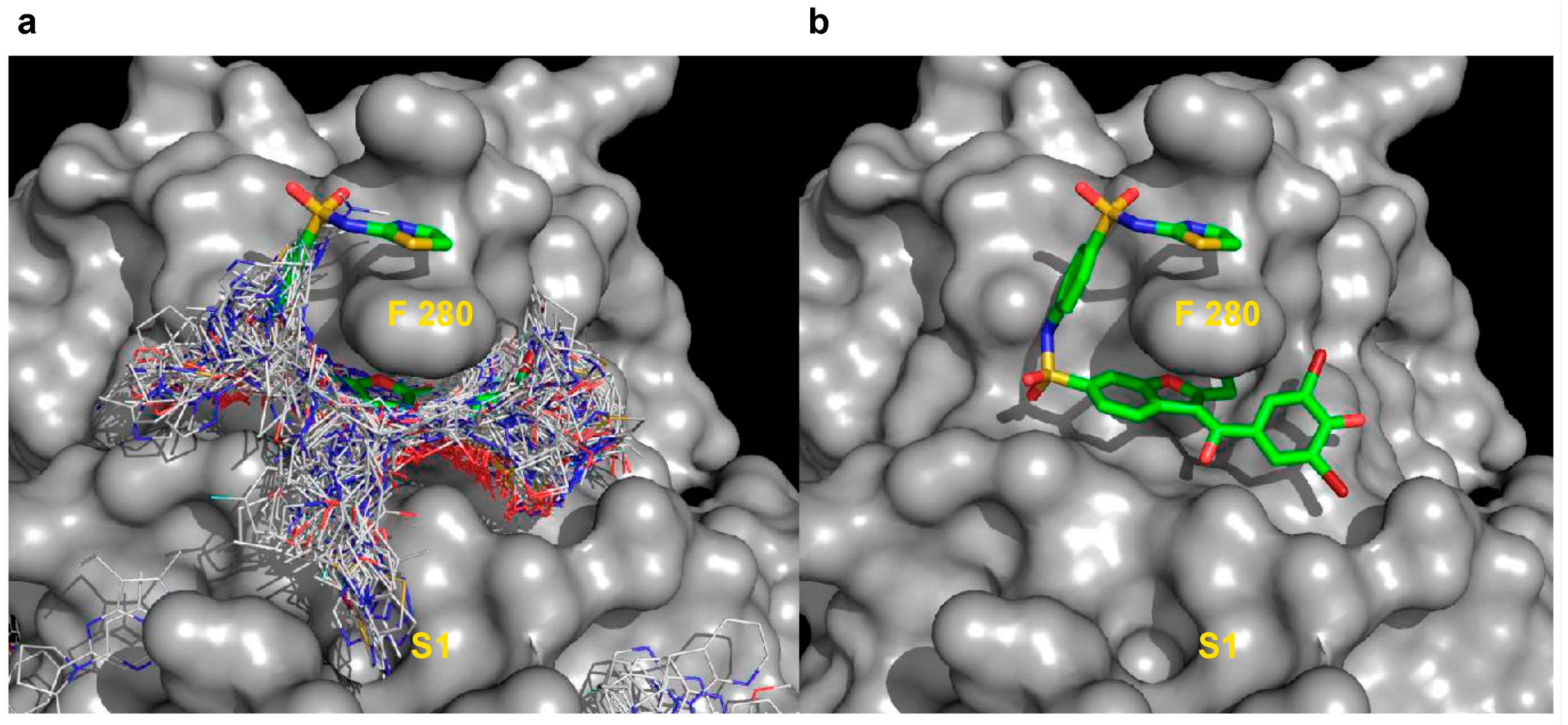
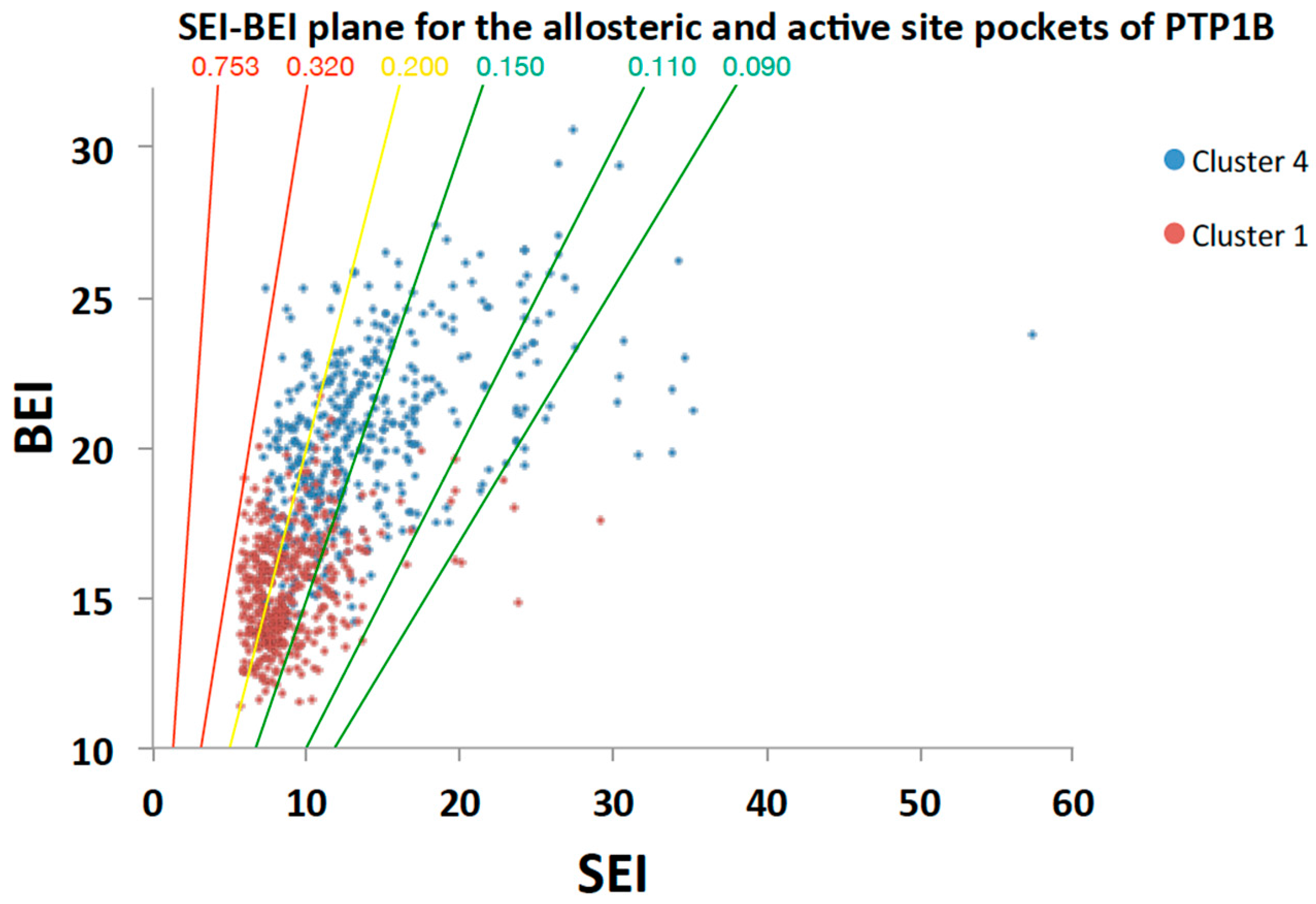
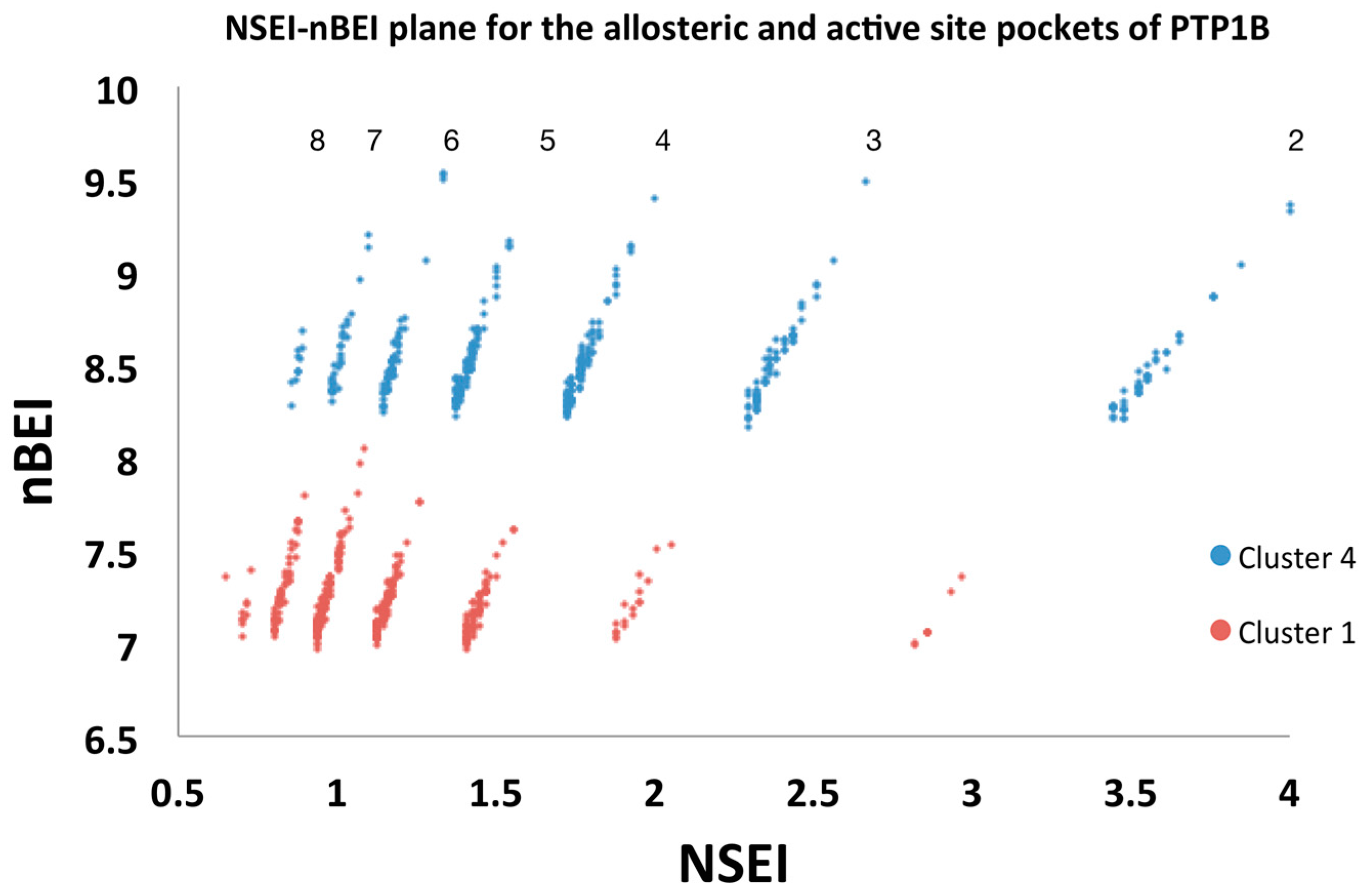
2.5.2. Analysis of Targeted Docking at the Active and Allosteric Sites
3. Discussion
4. Materials and Methods
4.1. Scripting Languages and Working Environment
4.1.1. VSpipe-Local Mode
4.1.2. VSpipe-Cluster Mode
4.2. Testing the Parallelisation of AD4 and Vina
4.3. Target Proteins and Ligands
4.4. Filtering the Results After the VS
4.5. Docking Parameters for PTP1B
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Bialy, L.; Waldmann, H. Inhibitors of protein tyrosine phosphatases: Next-generation drugs? Angew. Chem. Int. Ed. 2005, 44, 3814–3839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Drugging the undruggable: Therapeutic potential of targeting protein tyrosine phosphatases. Acc. Chem. Res. 2017, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Rios, P.; Nunes-Xavier, C.E.; Tabernero, L.; Kohn, M.; Pulido, R. Dual-specificity phosphatases as molecular targets for inhibition in human disease. Antioxid. Redox Signal. 2014, 20, 2251–2273. [Google Scholar] [CrossRef] [PubMed]
- Heneberg, P. Use of protein tyrosine phosphatase inhibitors as promising targeted therapeutic drugs. Curr. Med. Chem. 2009, 16, 706–733. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad-Zapatero, C.; Metz, J.T. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand efficiency indices for effective drug discovery. Expert Opin. Drug Dis. 2007, 2, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J. Local optima avoidance in depot location. J. Oper. Res. Soc. 1981, 32, 815–819. [Google Scholar] [CrossRef]
- Blum, C.; Aguilera, M.J.B.; Roli, A.; Sampels, M. Hybrid Metaheuristics: An Emerging Approach to Optimization Preface; Studies in Computational Intelligence; Springer: Berlin, Germany, 2008; Volume 114, pp. 10–11. [Google Scholar]
- Nocedal, J.; Wright, S.J. Numerical Optimization, 2nd ed.; Springer Series in Operations Research; Springer: Berlin, Germany, 1999; pp. 136–142. [Google Scholar]
- Gilmour, I.J.; Holland, R.; Craig, D.B. Nitrogen analyzer adaptation for use in presence of halogenated anesthetics. Anesthesiology 1975, 43, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1b. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Narang, P.; Xu, Y.; Kauffman, A.L.; Petit, J.; Xu, H.E.; Meurice, N.; MacKeigan, J.P. Identification of small molecule inhibitors of PTPσ through an integrative virtual and biochemical approach. PLoS ONE 2012, 7, e50217. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.T.; Yu, H.B.; Huang, X.F. Selective binding modes and allosteric inhibitory effects of lupane triterpenes on protein tyrosine phosphatase 1b. Sci. Rep. 2016, 6, 20766. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Moller, N.P.H. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal-structure of human protein-tyrosine-phosphatase 1b. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1b: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1b. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of ptp1b with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Nordle, A.K.L.; Rios, P.; Gaulton, A.; Pulido, R.; Attwood, T.K.; Tabernero, L. Functional assignment of MAPK phosphatase domains. Proteins 2007, 69, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.P.; Fedorov, A.A.; Lee, S.Y.; Guo, X.L.; Shen, K.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Crystal structure of ptp1b complexed with a potent and selective bidentate inhibitor. J. Biol. Chem. 2003, 278, 12406–12414. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Wang, L.J.; Shi, D.Y. The design strategy of selective ptp1b inhibitors over TCPTP. Bioorg. Med. Chem. 2016, 24, 3343–3352. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C.; Blasi, D. Ligand efficiency indices (LEIs): More than a simple efficiency yardstick. Mol. Inform. 2011, 30, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Hetenyi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- AD4-Homepage. Available online: http://autodock.scripps.edu (accessed on 4 May 2015).
- Vina-Homepage. Available online: http://vina.scripps.edu (accessed on 4 May 2015).
Sample Availability: The Linux version of VSpipe-local mode and documentation are available at https://github.com/sabifo4/VSpipe. The VSpipe-cluster mode is available upon request. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | VSpipe Version | Docking Time |
|---|---|---|
| (hh:mm) | ||
| AD4 | Local PC | 14:10 |
| AD4 (not parallelised) | Cluster DPSF | 06:37 |
| AD4-OpenMP (8 cores) | Cluster DPSF | 02:04 |
| AD4-OpenMP (16 cores) | Cluster DPSF | 00:45 |
| AD4-MPI (60 cores) | Cluster DPSF | 00:09 |
| Vina | Local PC | 00:48 |
| Vina-OpenMP (8 cores) | Cluster DPSF | 00:21 |
| Vina-OpenMP (16 cores) | Cluster DPSF | 00:13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Carretero, S.; Pavlopoulou, N.; Adams, J.; Gilsenan, J.; Tabernero, L. VSpipe, an Integrated Resource for Virtual Screening and Hit Selection: Applications to Protein Tyrosine Phospahatase Inhibition. Molecules 2018, 23, 353. https://doi.org/10.3390/molecules23020353
Álvarez-Carretero S, Pavlopoulou N, Adams J, Gilsenan J, Tabernero L. VSpipe, an Integrated Resource for Virtual Screening and Hit Selection: Applications to Protein Tyrosine Phospahatase Inhibition. Molecules. 2018; 23(2):353. https://doi.org/10.3390/molecules23020353
Chicago/Turabian StyleÁlvarez-Carretero, Sandra, Niki Pavlopoulou, James Adams, Jane Gilsenan, and Lydia Tabernero. 2018. "VSpipe, an Integrated Resource for Virtual Screening and Hit Selection: Applications to Protein Tyrosine Phospahatase Inhibition" Molecules 23, no. 2: 353. https://doi.org/10.3390/molecules23020353