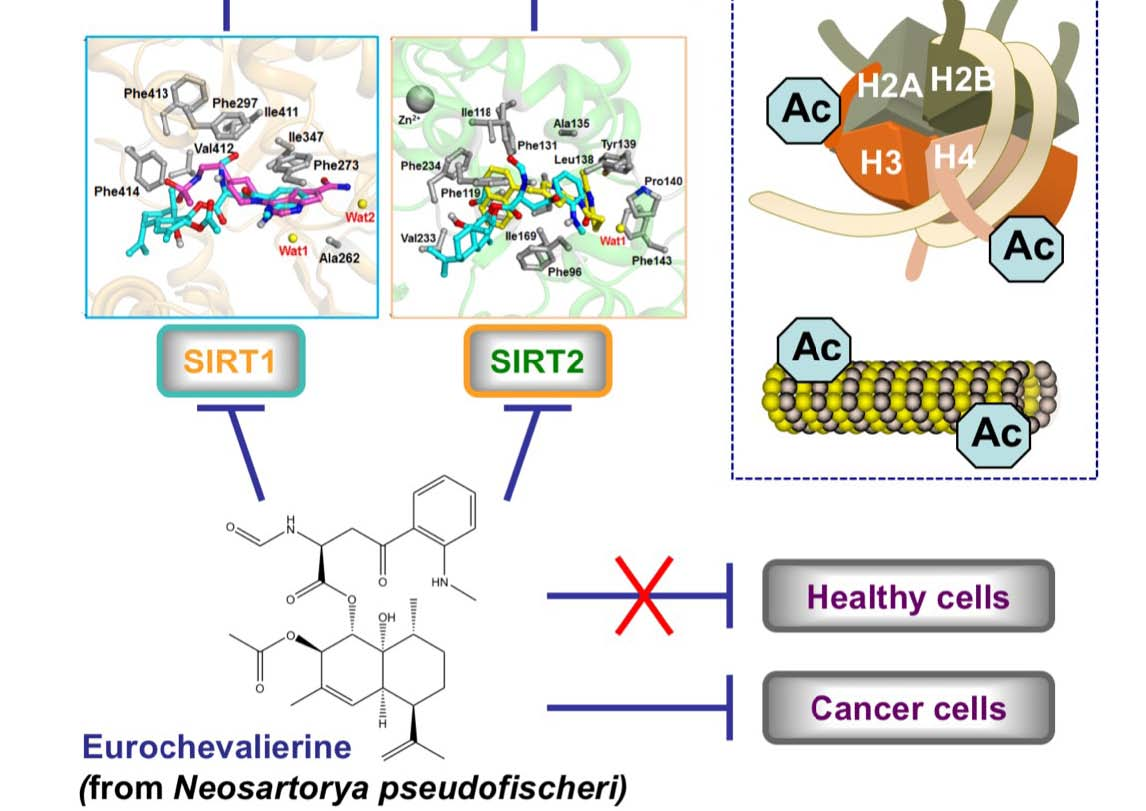
The Fungal Metabolite Eurochevalierine, a Sequiterpene Alkaloid, Displays Anti-Cancer Properties through Selective Sirtuin 1/2 Inhibition

 ,
, 

 ,
,  ,
, 
 and
and 
Abstract
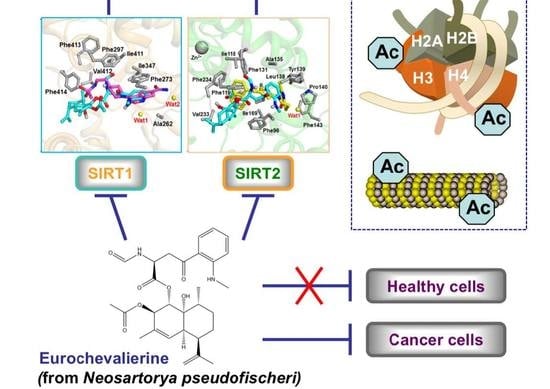
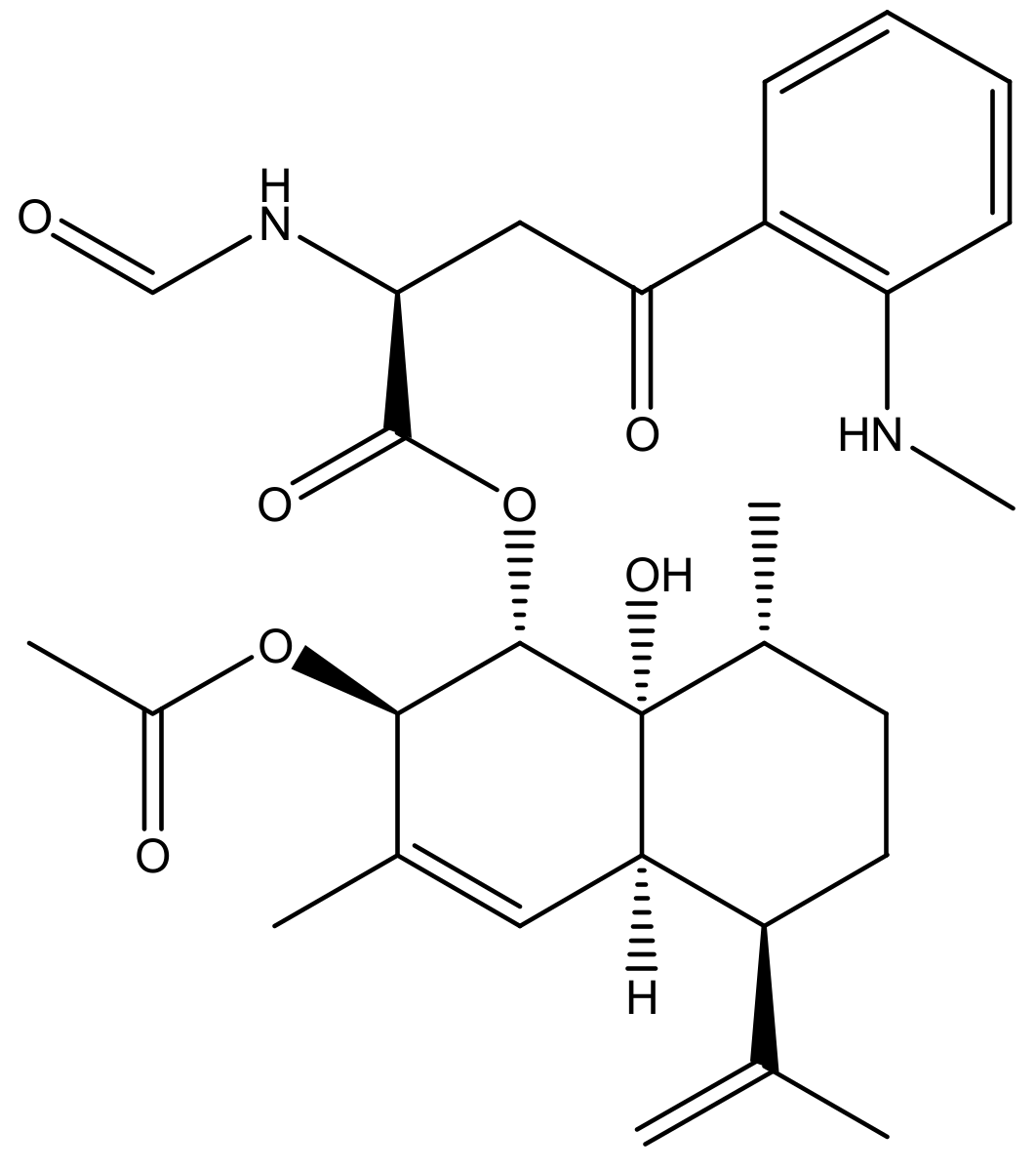
:
1. Introduction
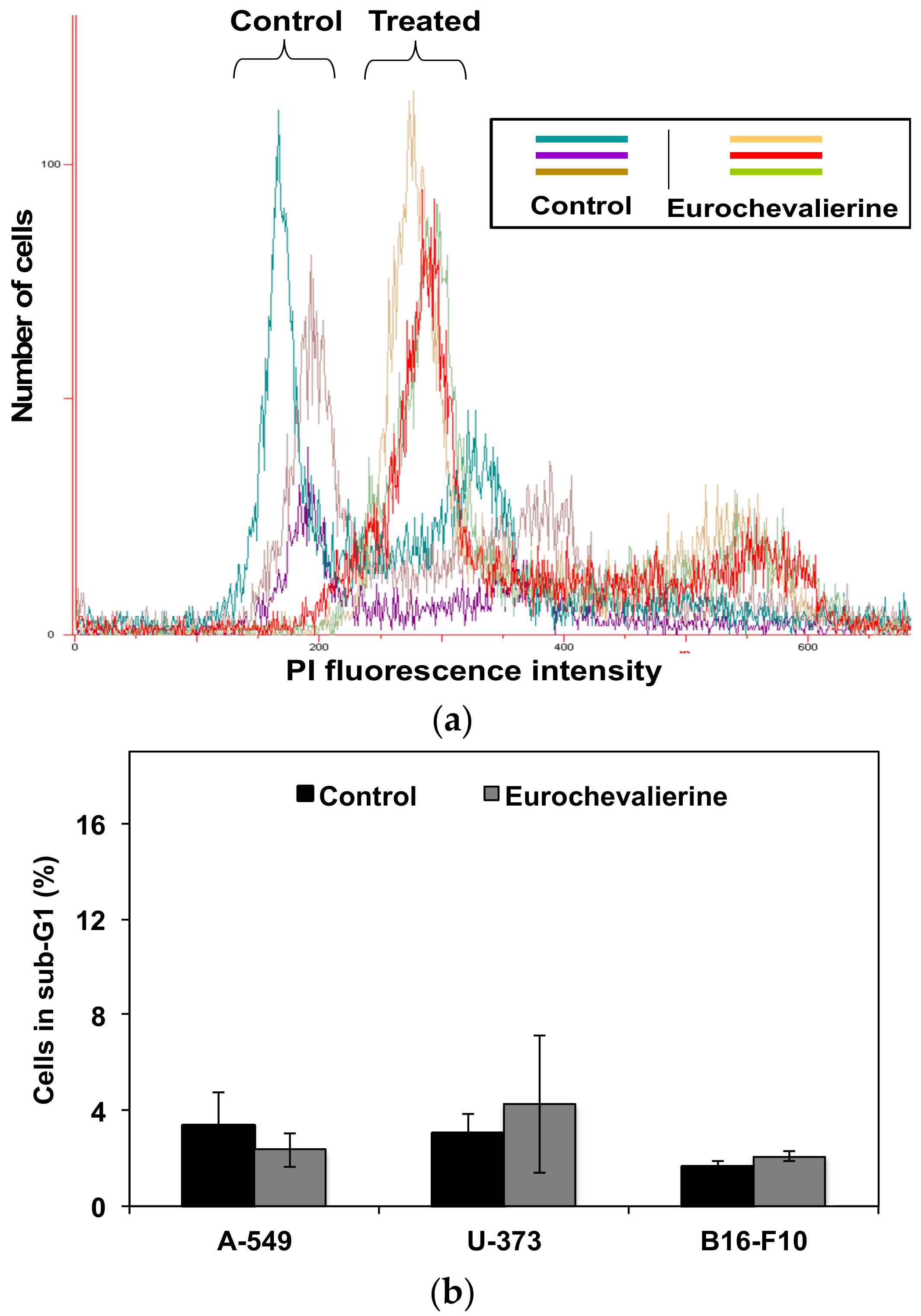
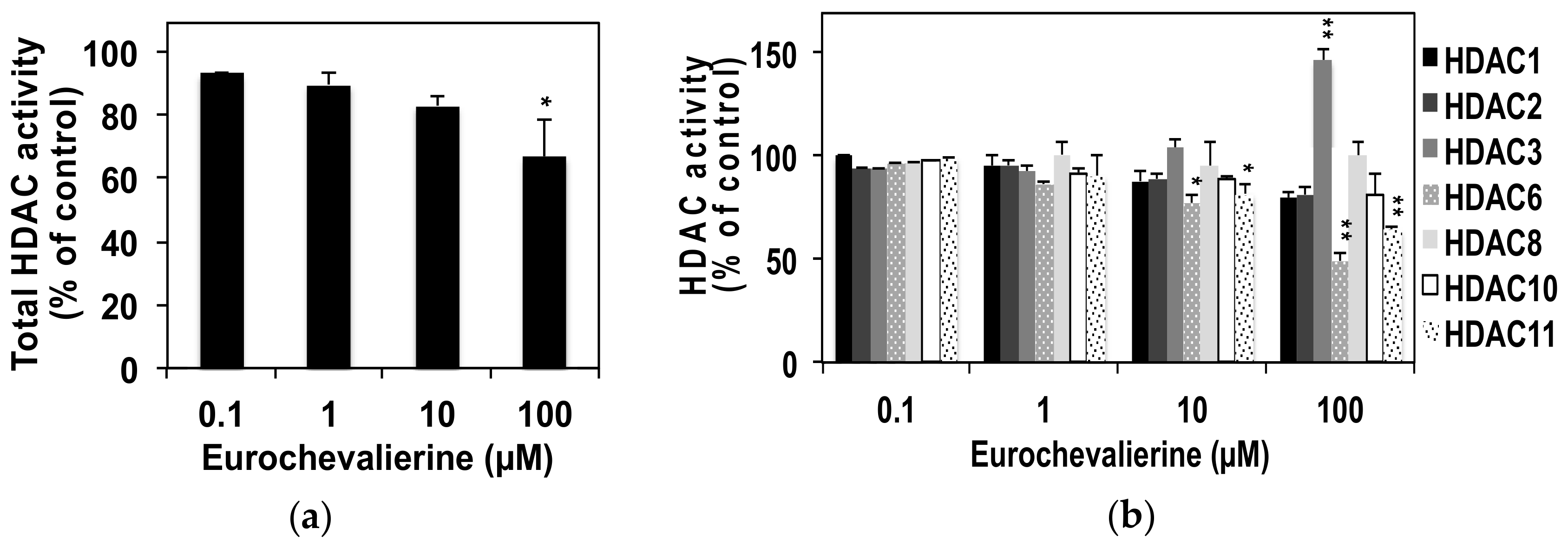
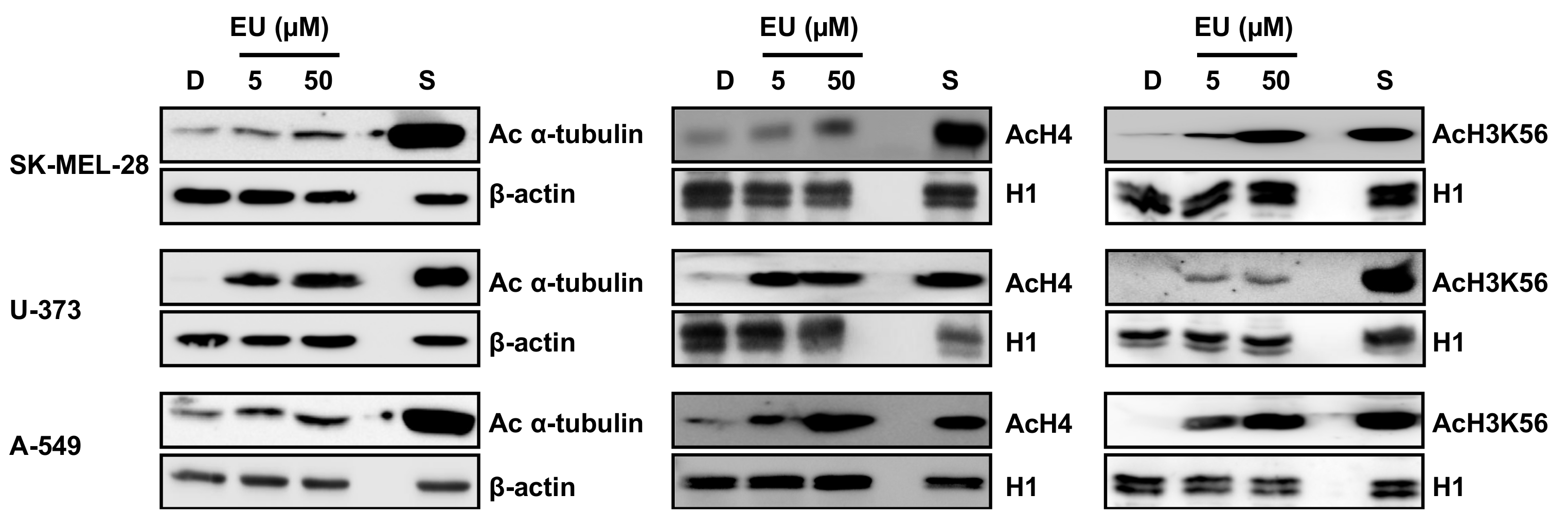
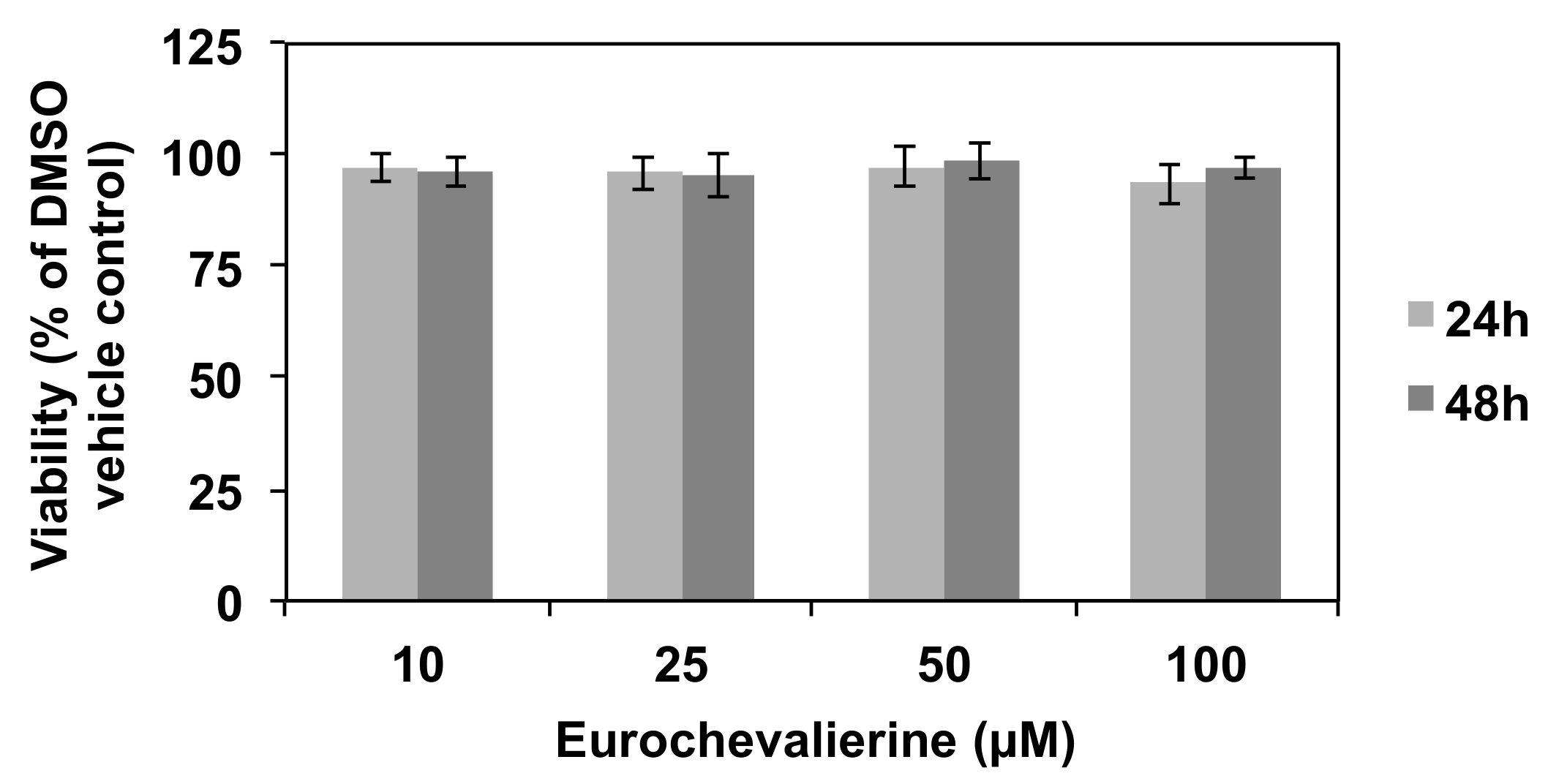
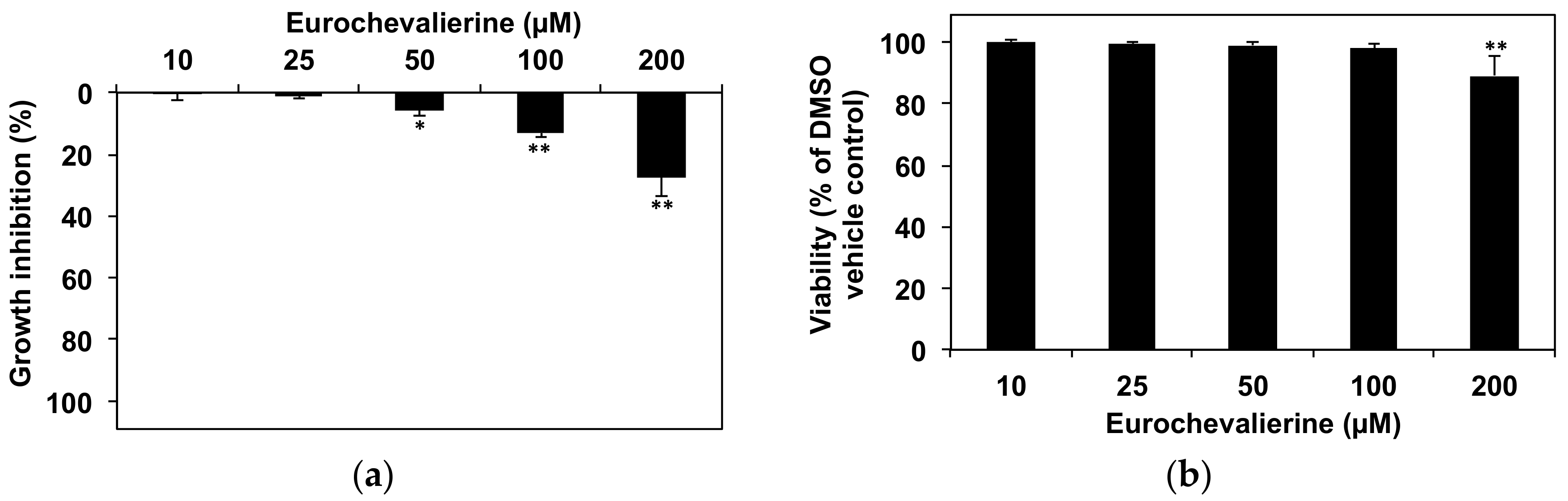
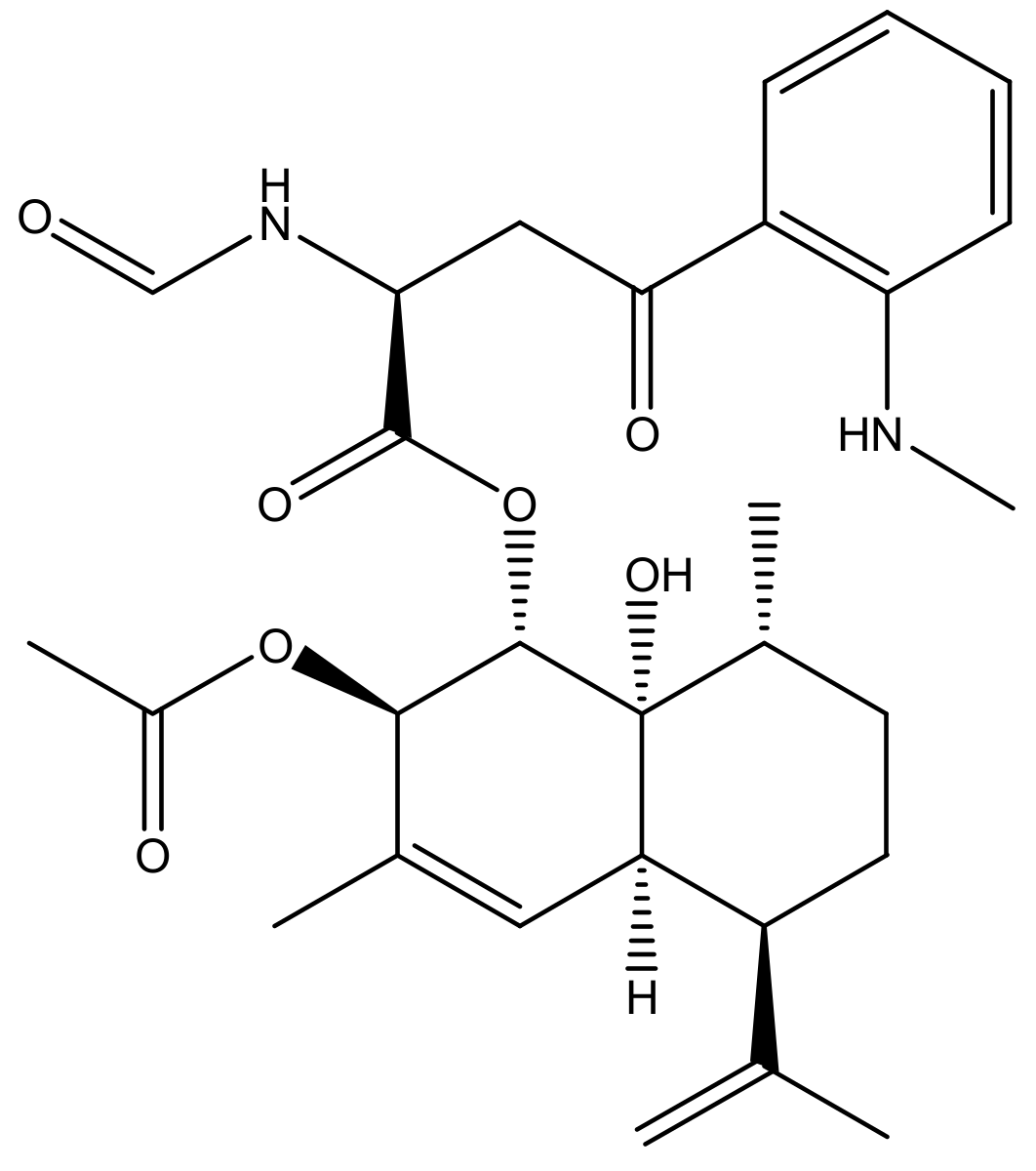
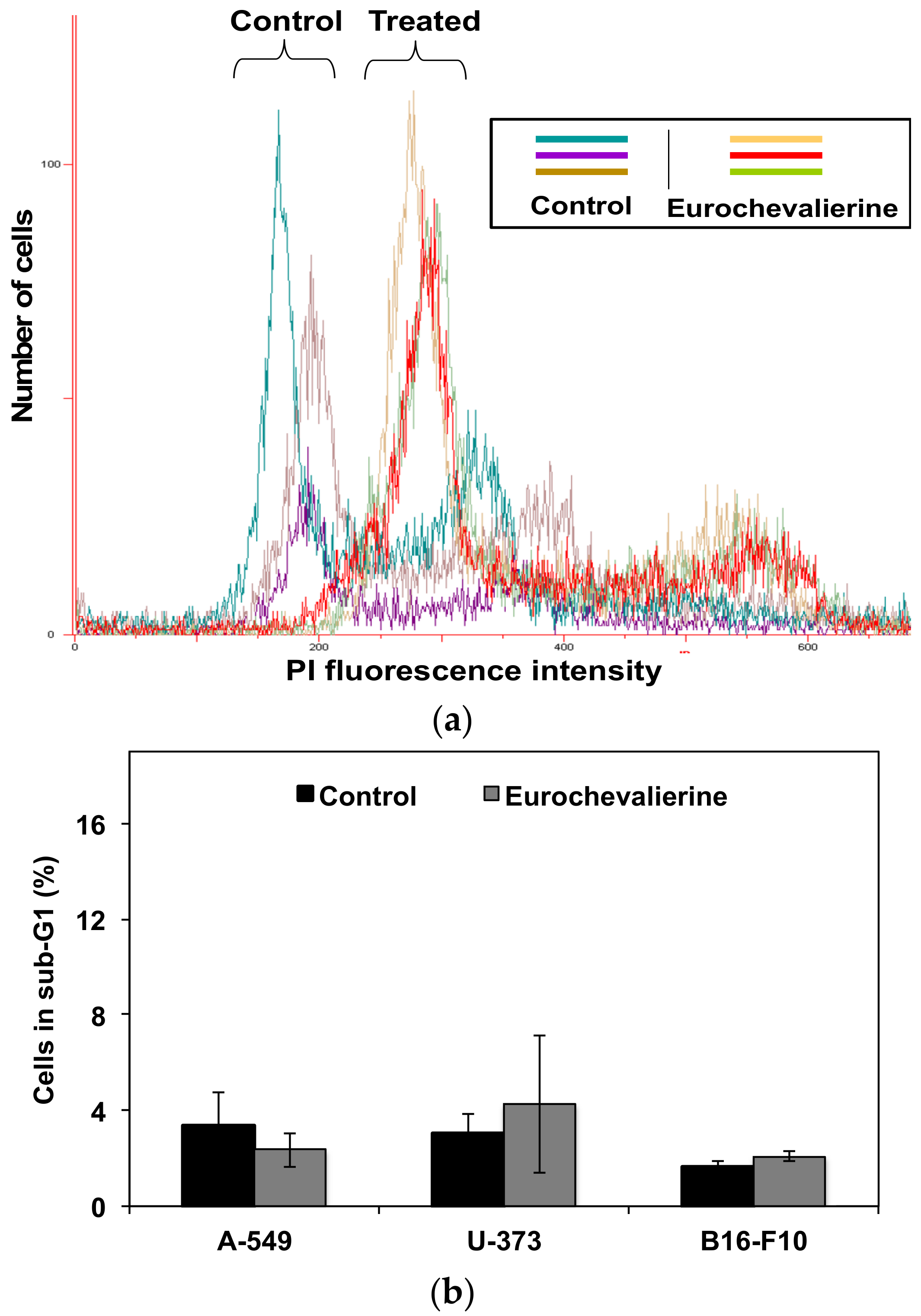
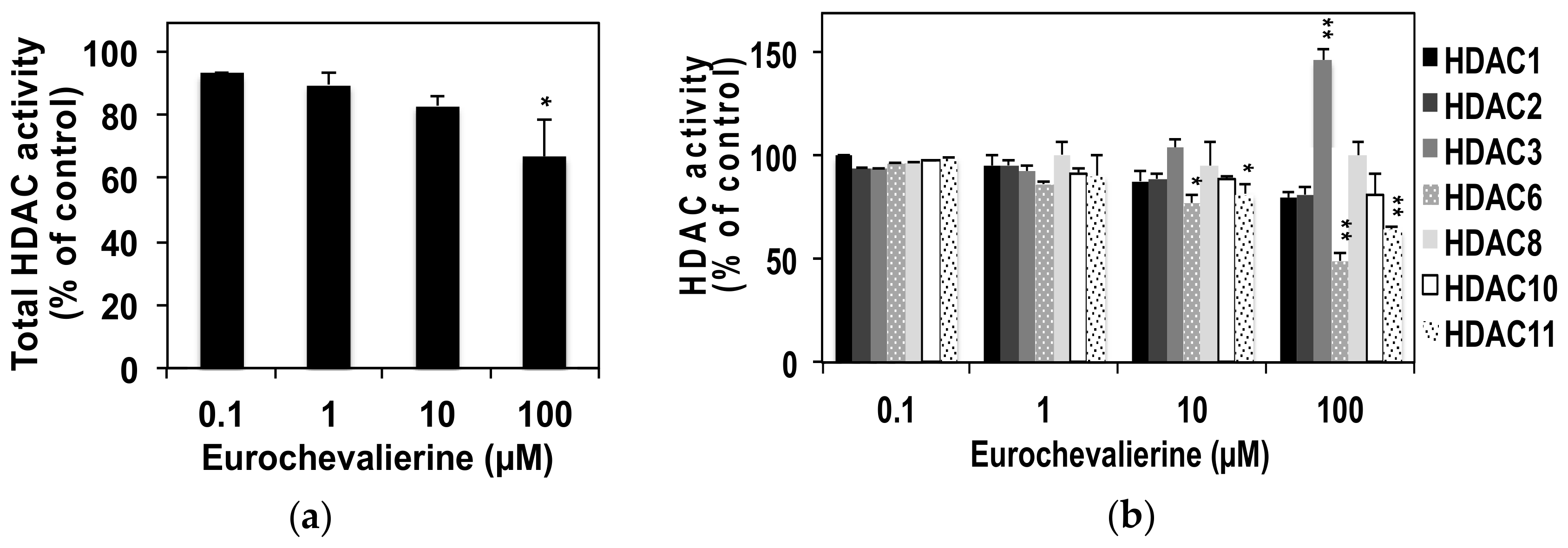
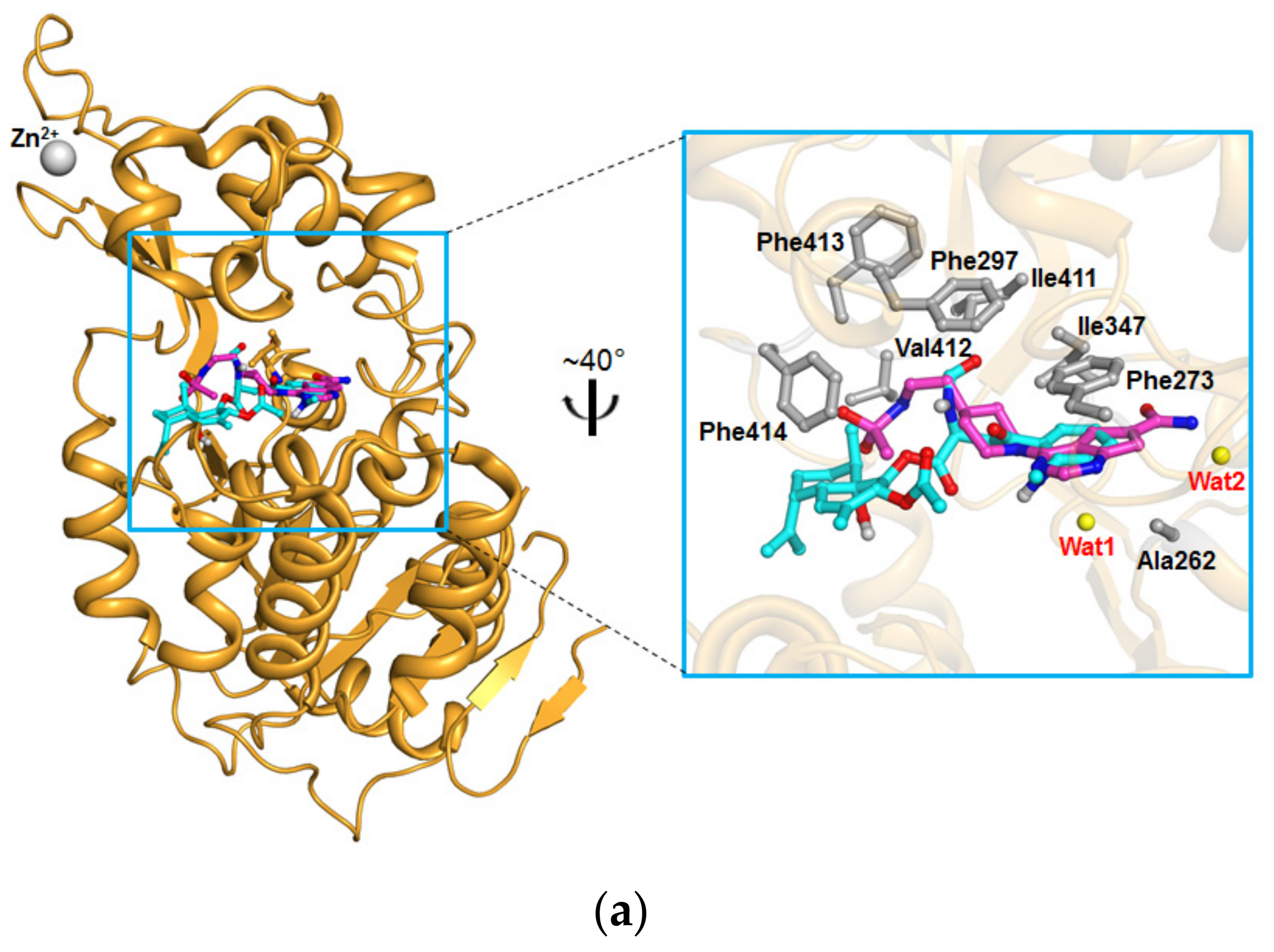
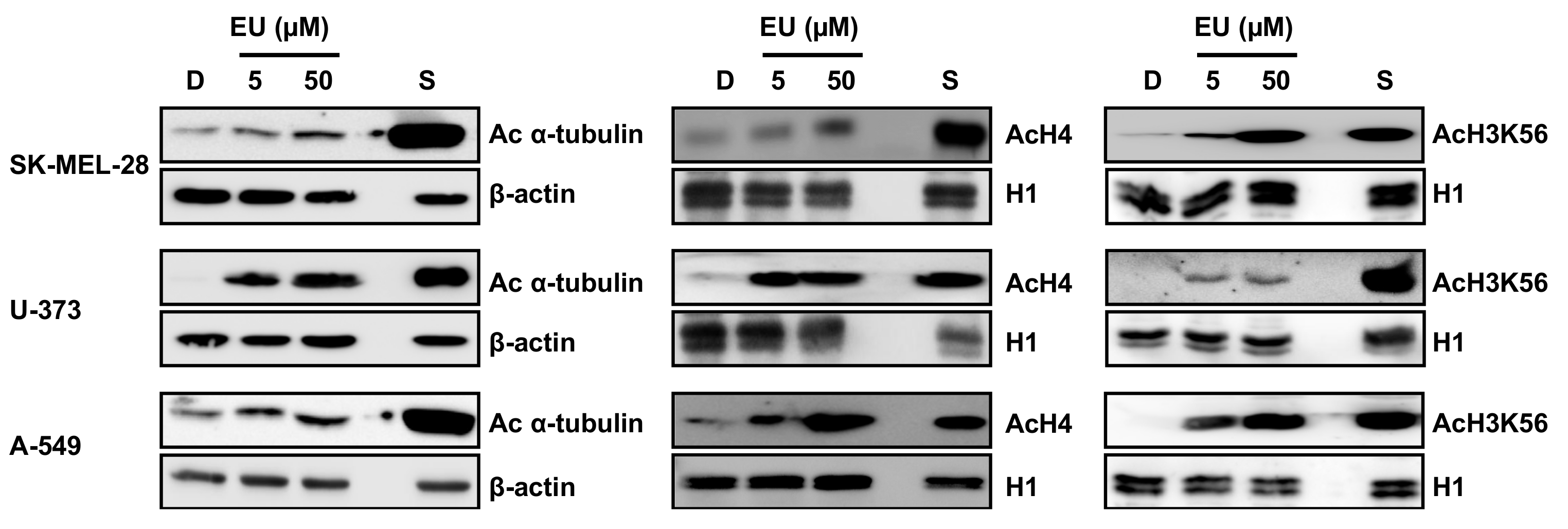
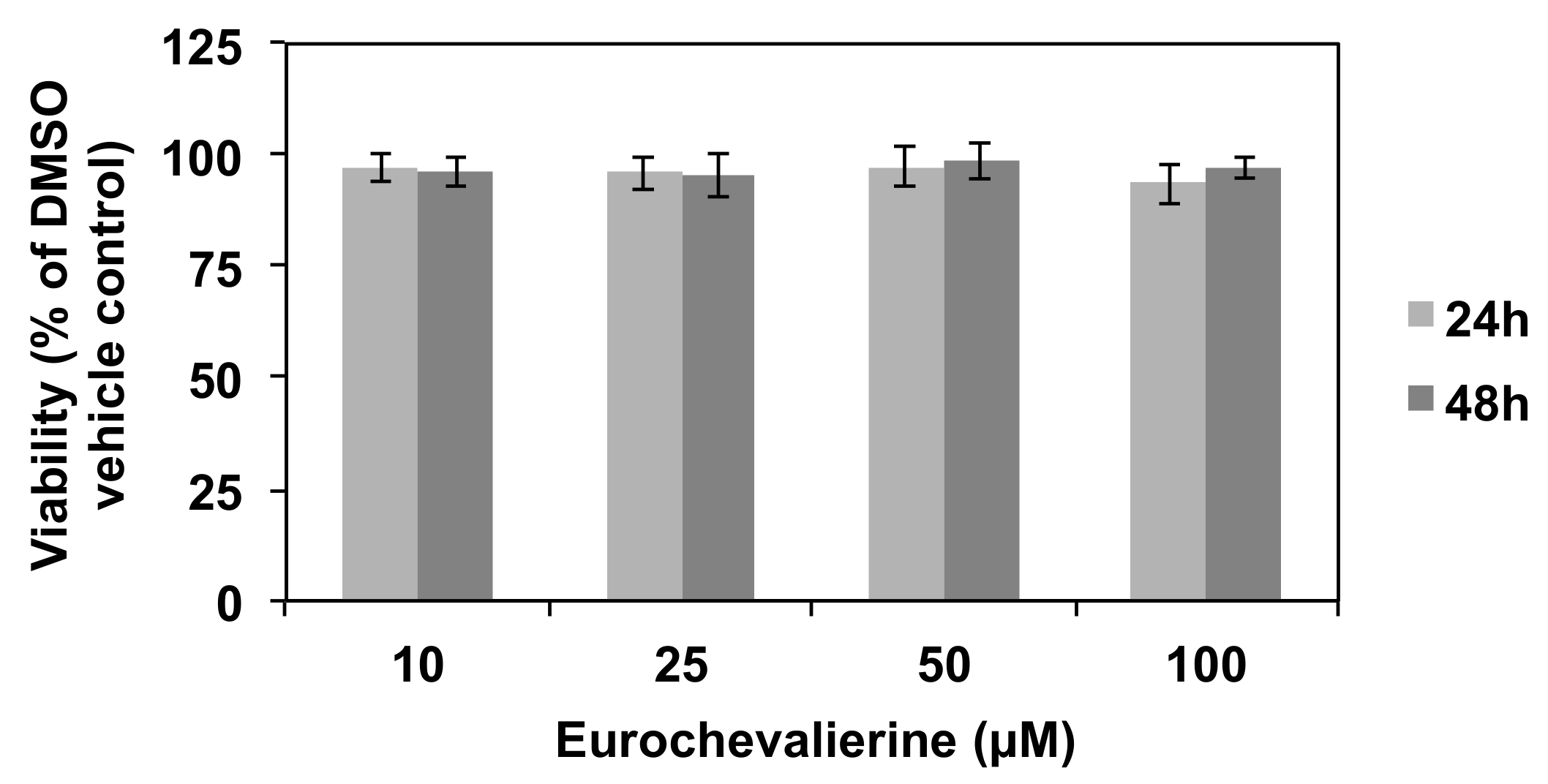
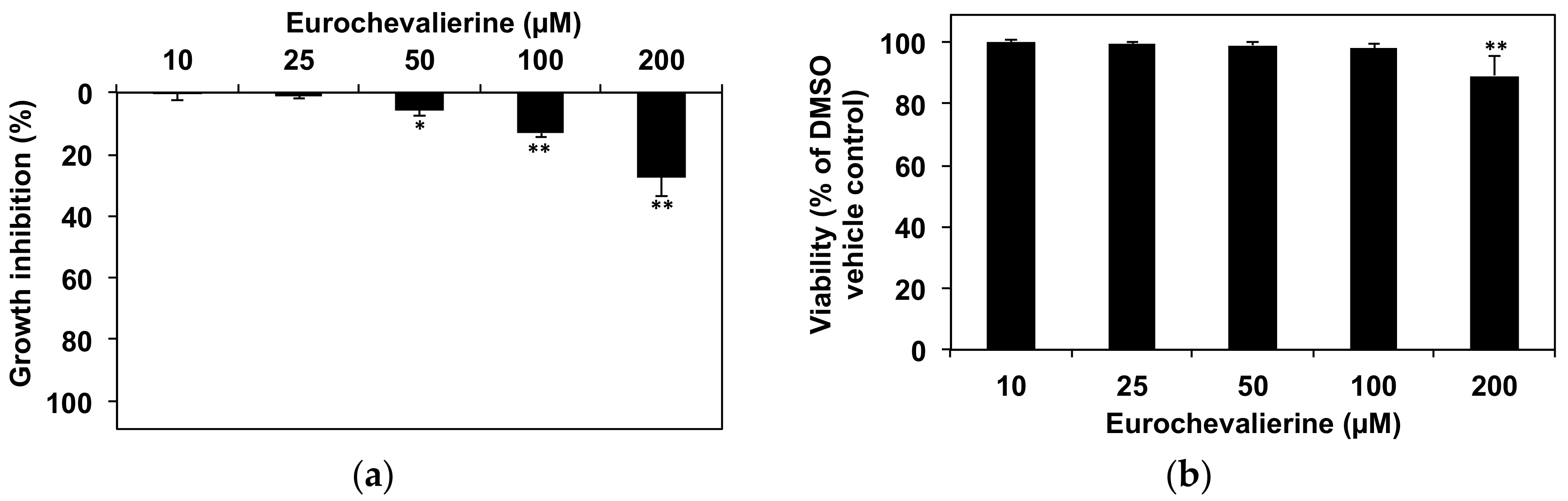
2. Results
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Cell Culture and Viability Assay
4.3. Flow Cytometry
4.4. In Vitro HDAC Activity Assay
4.5. Docking Studies
4.6. Protein Extraction and Western Blotting
4.7. Calculation of Drug-Like Properties
4.8. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Florean, C.; Schnekenburger, M.; Grandjenette, C.; Dicato, M.; Diederich, M. Epigenomics of leukemia: From mechanisms to therapeutic applications. Epigenomics 2011, 3, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Karius, T.; Schnekenburger, M.; Dicato, M.; Diederich, M. MicroRNAs in cancer management and their modulation by dietary agents. Biochem. Pharmacol. 2012, 83, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Diederich, M. Epigenetics offer new horizons for colorectal cancer prevention. Curr. Colorectal Cancer Rep. 2012, 8, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Florean, C.; Dicato, M.; Diederich, M. Epigenetic alterations as a universal feature of cancer hallmarks and a promising target for personalized treatments. Curr. Top. Med. Chem. 2016, 16, 745–776. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Florean, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Chromatin-modifying agents in anti-cancer therapy. Biochimie 2012, 94, 2264–2279. [Google Scholar] [CrossRef] [PubMed]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural compound histone deacetylase inhibitors (HDACi): Synergy with inflammatory signaling pathway modulators and clinical applications in cancer. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Folmer, F.; Orlikova, B.; Schnekenburger, M.; Dicato, M.; Diederich, M. Naturally occurring regulators of histone acetylation/deacetylation. Curr. Nutr. Food Sci. 2010, 6, 78–99. [Google Scholar] [CrossRef]
- Koprinarova, M.; Schnekenburger, M.; Diederich, M. Role of histone acetylation in cell cycle regulation. Curr. Top. Med. Chem. 2016, 16, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after Vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Tokunaga, E.; Chang, K.; Hikasa, M.; Iijima, K.; Eto, M.; Kozaki, K.; Akishita, M.; Ouchi, Y.; Kaneki, M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 2006, 25, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Goffin, E.; Lee, J.Y.; Jang, J.Y.; Mazumder, A.; Ji, S.; Rogister, B.; Bouider, N.; Lefranc, F.; Miklos, W.; et al. Discovery and characterization of R/S-N-3-Cyanophenyl-N′-(6-tert-butoxycarbonylamino-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-4-yl)urea, a new histone deacetylase class III inhibitor exerting antiproliferative activity against cancer cell lines. J. Med. Chem. 2017, 60, 4714–4733. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.; Marx-Blumel, L.; Lindig, N.; Thierbach, R.; Hoelzer, D.; Becker, S.; Wittig, S.; Lehmann, R.; Slevogt, H.; Heinzel, T.; et al. The sirtuin 1/2 inhibitor tenovin-1 induces a nonlinear apoptosis-inducing factor-dependent cell death in a p53 null Ewing’s sarcoma cell line. Investig. New Drugs 2017. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Suzuki, T.; Yoshimitsu, M.; Uchida, Y.; Kuroki, A.; Aikawa, A.; Honda, S.; Arima, N.; Soeda, S. Novel small-molecule SIRT1 inhibitors induce cell death in adult T-cell leukaemia cells. Sci. Rep. 2015, 5, 11345. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Breitenbucher, F.; Schuler, M.; Ehrenhofer-Murray, A.E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 2014, 289, 5208–5216. [Google Scholar] [CrossRef] [PubMed]
- Wilking-Busch, M.J.; Ndiaye, M.A.; Liu, X.; Ahmad, N. RNA interference-mediated knockdown of SIRT1 and/or SIRT2 in melanoma: Identification of downstream targets by large-scale proteomics analysis. J. Proteom. 2018, 170, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Cerella, C.; Teiten, M.H.; Radogna, F.; Dicato, M.; Diederich, M. From nature to bedside: Pro-survival and cell death mechanisms as therapeutic targets in cancer treatment. Biotechnol. Adv. 2014, 32, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Qurishi, Y.; Hamid, A.; Majeed, R.; Hussain, A.; Qazi, A.K.; Ahmed, M.; Zargar, M.A.; Singh, S.K.; Saxena, A.K. Interaction of natural products with cell survival and signaling pathways in the biochemical elucidation of drug targets in cancer. Future Oncol. 2011, 7, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Epigenetic modulators from “The Big Blue“: A treasure to fight against cancer. Cancer Lett. 2014, 351, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012, 7, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kanokmedhakul, K.; Kanokmedhakul, S.; Suwannatrai, R.; Soytong, K.; Prabpai, S.; Kongsaeree, P. Bioactive meroterpenoids and alkaloids from the fungus Eurotium chevalieri. Tetrahedron 2011, 67, 5461–5468. [Google Scholar] [CrossRef]
- Eamvijarn, A.; Kijjoa, A.; Bruyere, C.; Mathieu, V.; Manoch, L.; Lefranc, F.; Silva, A.; Kiss, R.; Herz, W. Secondary metabolites from a culture of the fungus Neosartorya pseudofischeri and their in vitro cytostatic activity in human cancer cells. Planta Med. 2012, 78, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Zin, W.W.; Prompanya, C.; Buttachon, S.; Kijjoa, A. Bioactive secondary metabolites from a Thai collection of soil and marine-derived fungi of the genera Neosartorya and Aspergillus. Curr. Drug Deliv. 2016, 13, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, E.; Giangare, M.C.; Bottiroli, G. Nuclease-induced DNA structural changes assessed by flow cytometry with the intercalating dye propidium iodide. Cytometry 1991, 12, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Giangare, M.C.; Prosperi, E.; Pedrali-Noy, G.; Bottiroli, G. Flow cytometric evaluation of DNA stainability with propidium iodide after histone H1 extraction. Cytometry 1989, 10, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Au, Q.; Zhang, M.; Barber, J.R.; Ng, S.C.; Zhang, B. Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem. Biophys. Res. Commun. 2009, 386, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Lin, P.; Knoll, E.; Chakrabarti, R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: Computational and experimental studies. PLoS ONE 2014, 9, e107729. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 A crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J. Med. Chem. 2013, 56, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Olah, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Olah, J.; Gerhardt, S.; Ovadi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as potent and selective Sirt2 Inhibitors: A structure-activity relationship study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcain, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Scarponi, C.; Di Felice, F.; Cesarini, E.; Avitabile, S.; Mai, A.; Mauro, M.L.; Sirri, V.; Zambruno, G.; Albanesi, C.; et al. Sirtinol treatment reduces inflammation in human dermal microvascular endothelial cells. PLoS ONE 2011, 6, e24307. [Google Scholar] [CrossRef]
- Lugrin, J.; Ciarlo, E.; Santos, A.; Grandmaison, G.; dos Santos, I.; Le Roy, D.; Roger, T. The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim. Biophys. Acta 2013, 1833, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Chen, C.Y.; Ho, K.K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.D.; Lam, E.W. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.G.; Kim, W.; Choi, M.; Kim, J.E. AK-1, a specific SIRT2 inhibitor, induces cell cycle arrest by downregulating Snail in HCT116 human colon carcinoma cells. Cancer Lett. 2015, 356, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Khanfar, M.A.; Quinti, L.; Wang, H.; Choi, S.H.; Kazantsev, A.G.; Silverman, R.B. Development and characterization of 3-(benzylsulfonamido)benzamides as potent and selective SIRT2 inhibitors. Eur. J. Med. Chem. 2014, 76, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Kim, S.A.; Ahn, S.G. Sirtuin inhibitors, EX527 and AGK2, suppress cell migration by inhibiting HSF1 protein stability. Oncol. Rep. 2016, 35, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Lernoux, M.; Schnekenburger, M.; Dicato, M.; Diederich, M. Anti-cancer effects of naturally derived compounds targeting histone deacetylase 6-related pathways. Pharmacol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Andolfi, A.; Mathieu, V.; Boari, A.; Cimmino, A.; Moreno, Y.; Banuls, L.; Vurro, M.; Kornienko, A.; Kiss, R.; et al. Fischerindoline, a pyrroloindole sesquiterpenoid isolated from Neosartorya pseudofischeri, with in vitro growth inhibitory activity in human cancer cell lines. Tetrahedron 2013, 69, 7466–7470. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Grandjenette, C.; Ghelfi, J.; Karius, T.; Foliguet, B.; Dicato, M.; Diederich, M. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem. Pharmacol. 2011, 81, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Antiproliferative and proapoptotic activities of 4-hydroxybenzoic acid-based inhibitors of histone deacetylases. Cancer Lett. 2014, 343, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.J.; Milagre, I.; Schnekenburger, M.; Gama, M.J.; Diederich, M.; Rodrigues, E. Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of CYP46A1 gene transcription. J. Neurochem. 2010, 113, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Karius, T.; Schnekenburger, M.; Ghelfi, J.; Walter, J.; Dicato, M.; Diederich, M. Reversible epigenetic fingerprint-mediated glutathione-S-transferase P1 gene silencing in human leukemia cell lines. Biochem. Pharmacol. 2011, 81, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration. Available online: http://www.molinspiration.com (accessed on 10 October 2017 and 15 November 2017).
- PreADMET v2.0. Available online: https://preadmet.bmdrc.kr/ (accessed on 10 October 2017 and 15 November 2017).
Sample Availability: Not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) 1 | ||
|---|---|---|---|
| SIRT1 | SIRT2 | SIRT3 | |
| Nicotinamide 2 | 97 ± 15 | 27 ± 3 | 67 ± 10 |
| Suramin 3 | 2.8 ± 0.3 | 13 ± 1 | >100 4 |
| Sirtinol 3 | 82.5 ± 7.1 | 47.1 ± 4.0 | ND |
| EX-527 3 | 0.10 ± 0.06 | 20.1 ± 4.2 | ND |
| AGK2 3 | 98.1 ± 2.4 | 2.8 ± 1.0 | ND |
| Eurochevalierine | 9.8 ± 2.0 | 10.2 ± 3.9 | >100 5 |
| PDB_ID | Eurochevalierine | Sirtinol | EX-527 |
|---|---|---|---|
| 4I5I | −8.0 | −9.0 | −10.3 |
| 4ZZI | −9.0 | −10.8 | −8.6 |
| 4ZZJ | −9.0 | −9.7 | −8.9 |
| Average | −8.7 | −9.8 | −9.3 |
| PDB_ID | Eurochevalierine | Sirtinol | AGK2 |
|---|---|---|---|
| 4RMG | −9.0 | −10.2 | −11.2 |
| 4RMH | −10.0 | −9.1 | −11.8 |
| 5DY4 | −9.1 | −10.4 | −10.9 |
| Average | −9.4 | −9.9 | −11.3 |
| Method | Parameter 1 | Values | ||||||
|---|---|---|---|---|---|---|---|---|
| Theoretical | Eu | Suramin | Nicotinamide | Sirtinol | EX-527 | AGK2 | ||
| Rule of 5 | n-atoms | 20 ≤ x ≤ 70 | 38 | 86 | 9 | 30 | 17 | 30 |
| MW (KDa) | 180 ≤ x ≤ 500 | 526.63 | 1297.3 | 122.13 | 394.47 | 248.71 | 434.28 | |
| miLogP | ≤5 | 4.04 | −5.72 | −0.48 | 5.67 | 2.51 | 5.73 | |
| TPSA | ≤140 | 131.03 | 483.74 | 55.99 | 61.69 | 58.88 | 78.92 | |
| n-ON | ≤10 | 9 | 29 | 3 | 4 | 3 | 5 | |
| n-OHNH | ≤5 | 3 | 12 | 2 | 2 | 3 | 1 | |
| n-rotb | ≤10 | 11 | 16 | 1 | 5 | 1 | 4 | |
| Absorption | BBBP | 0.1 ≤ MA ≤ 2 | 0.10 | 0.04 | 0.34 | 3.10 | 4.10 | 0.08 |
| IA | ≥70% | 92.6 | 65.2 | 93 | 95.8 | 90.3 | 97.3 | |
| PPB | <90% | 84.8 | 100 | 2.03 | 91.1 | 91.0 | 97.5 | |
| Toxicity | Rat | NA | Negative | Negative | Negative | Negative | Negative | Positive |
| Cardiac | NA | Ambigous | Ambigous | Medium | Ambigous | Medium | Medium | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnekenburger, M.; Mathieu, V.; Lefranc, F.; Jang, J.Y.; Masi, M.; Kijjoa, A.; Evidente, A.; Kim, H.-J.; Kiss, R.; Dicato, M.; et al. The Fungal Metabolite Eurochevalierine, a Sequiterpene Alkaloid, Displays Anti-Cancer Properties through Selective Sirtuin 1/2 Inhibition. Molecules 2018, 23, 333. https://doi.org/10.3390/molecules23020333
Schnekenburger M, Mathieu V, Lefranc F, Jang JY, Masi M, Kijjoa A, Evidente A, Kim H-J, Kiss R, Dicato M, et al. The Fungal Metabolite Eurochevalierine, a Sequiterpene Alkaloid, Displays Anti-Cancer Properties through Selective Sirtuin 1/2 Inhibition. Molecules. 2018; 23(2):333. https://doi.org/10.3390/molecules23020333
Chicago/Turabian StyleSchnekenburger, Michael, Véronique Mathieu, Florence Lefranc, Jun Young Jang, Marco Masi, Anake Kijjoa, Antonio Evidente, Hyun-Jung Kim, Robert Kiss, Mario Dicato, and et al. 2018. "The Fungal Metabolite Eurochevalierine, a Sequiterpene Alkaloid, Displays Anti-Cancer Properties through Selective Sirtuin 1/2 Inhibition" Molecules 23, no. 2: 333. https://doi.org/10.3390/molecules23020333