The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
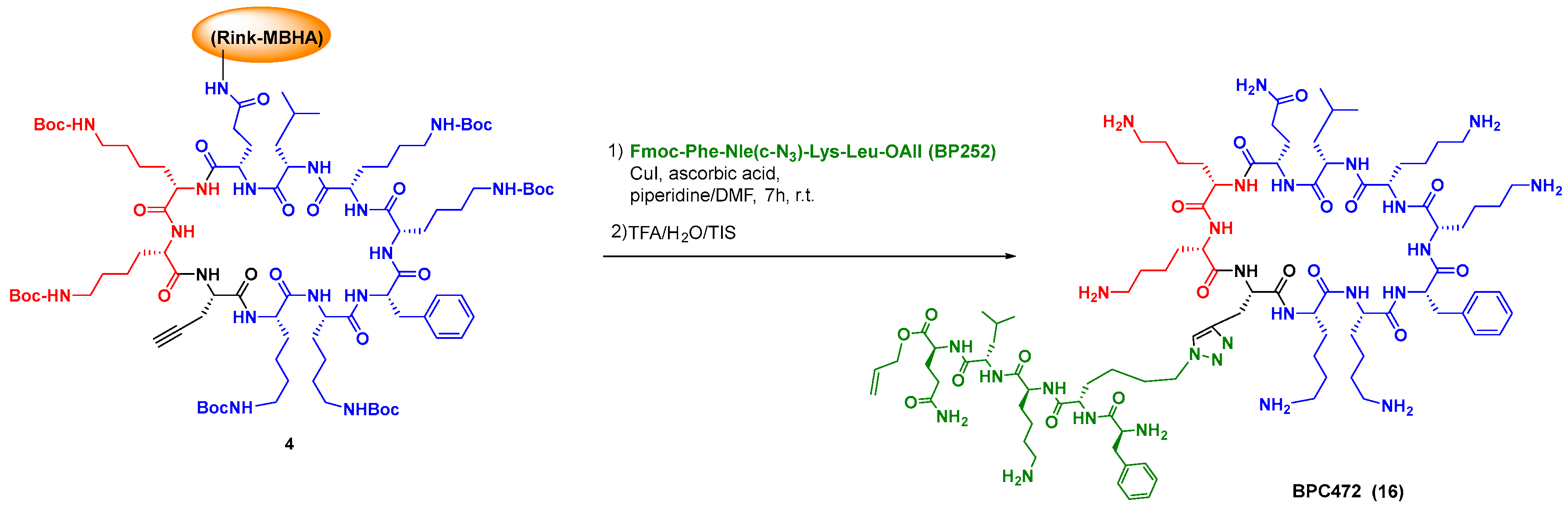{kind=link}
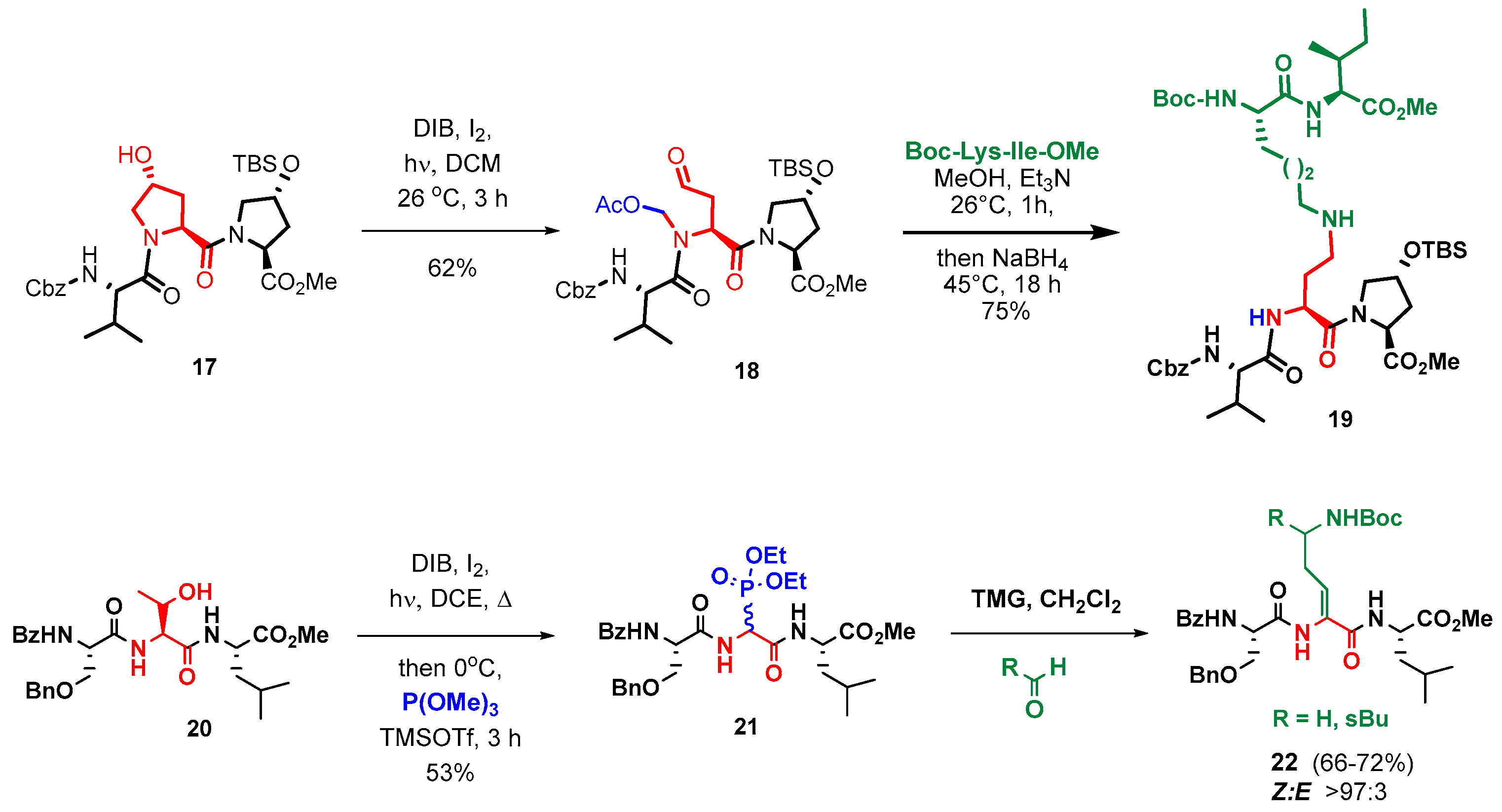{kind=link}
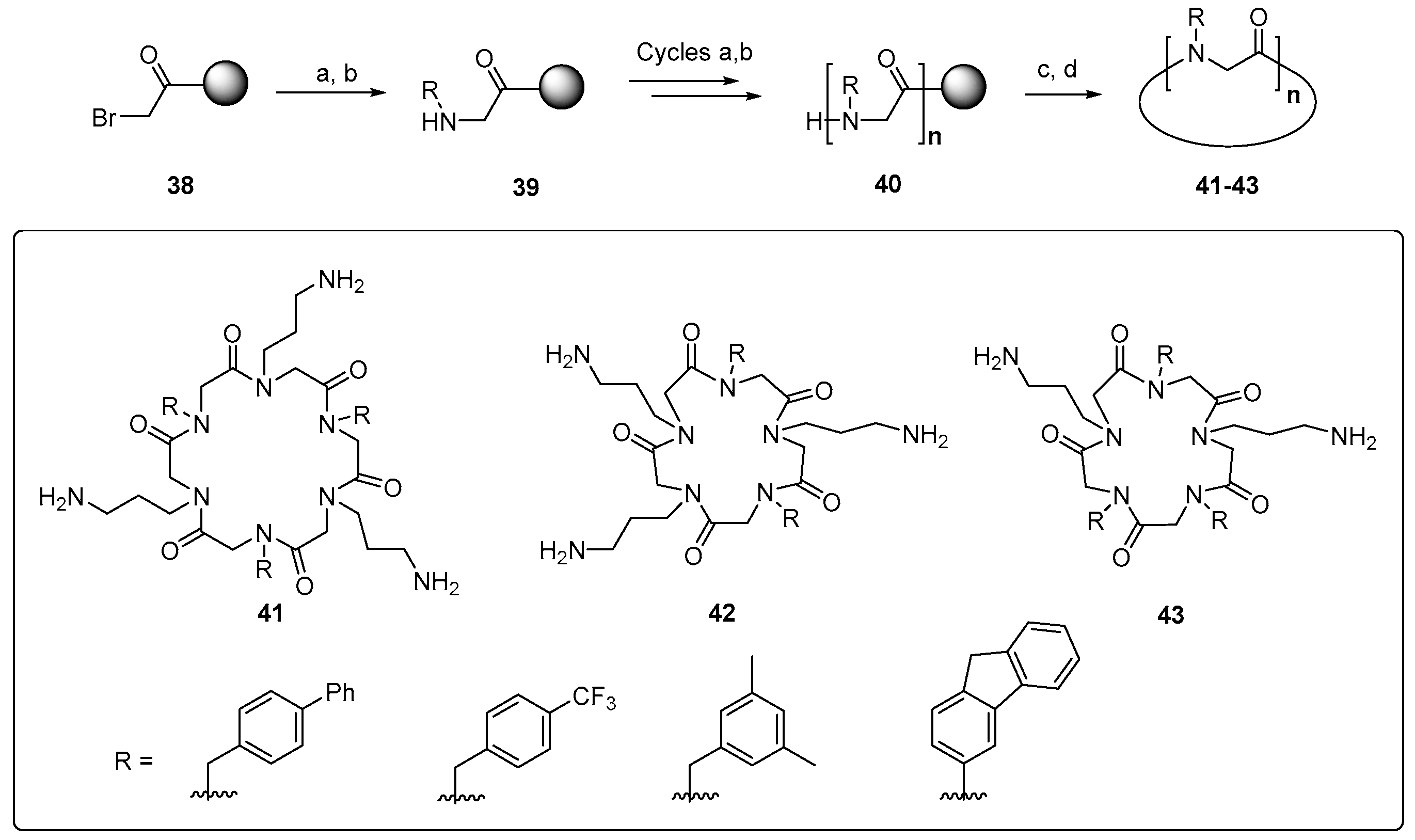{kind=link}
1. Introduction
2. Discovery of New AMPs
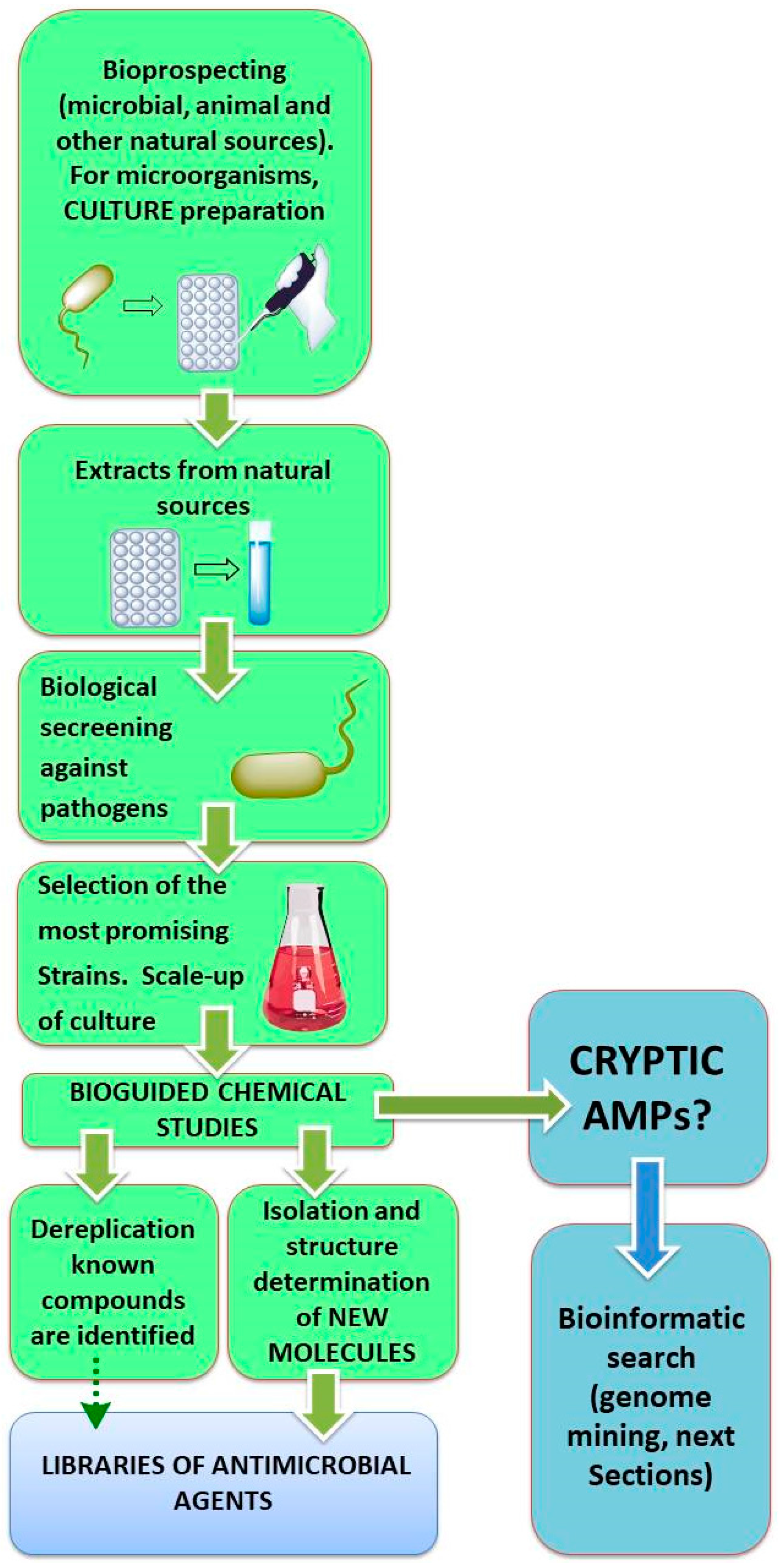
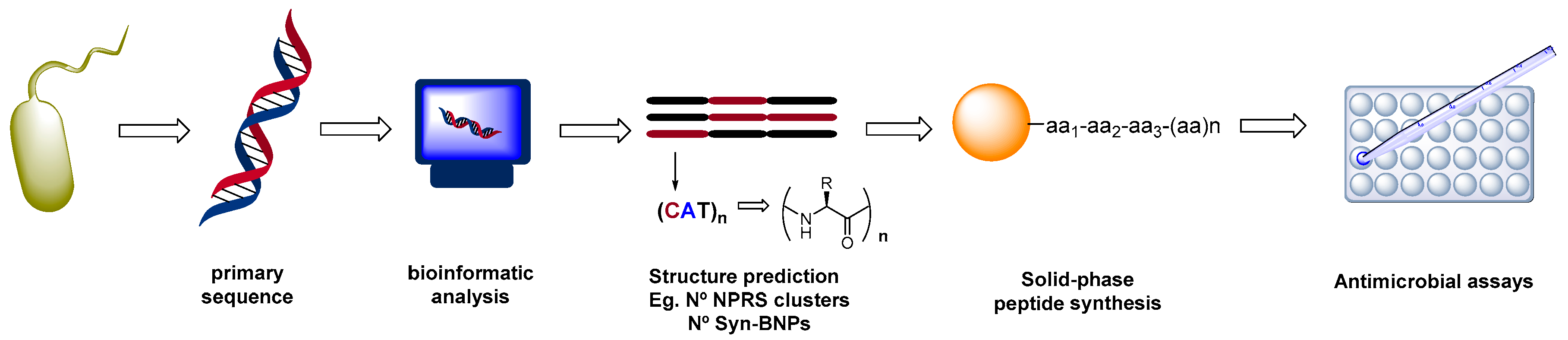
2.1. Biotechnological Discovery Strategies
2.2. Chemical Synthesis in Discovery Strategies
2.2.1. Advances in Synthetic Methodologies: Providing Scarce AMPs and Selected Analogues
2.2.2. AMP Libraries for SAR Studies
2.2.3. SAR Studies: Natural AMPs as Inspiration for Peptidomimetics
3. Delivery and Bioavailability Issues
4. Scale-Up: Overcoming Production Cost Problems
5. Perspectives and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; Wang, G., Ed.; Centre for Agriculture and Bioscience International (CABI): Wallingford, UK, 2017; ISBN 978-1-786390394 (hardback), 978-1-786390400 (e-book). [Google Scholar]
- Handbook of Biologically Active Peptides; Kastin, A.J., Ed.; Academic Press: San Diego, CA, USA, 2006. [Google Scholar]
- For Antimicrobial Peptide Databases. Antimicrobial Peptide Database-APD. Available online: http://aps.unmc.edu/AP/main.php (accessed on 16 January 2018).
- Data Repository of Antimicrobial Peptides-DRAMP. Available online: http://dramp.cpu-bioinfor.org/ (accessed on 16 January 2018).
- Defensins Knowledgebase. Available online: http://defensins.bii.a-star.edu.sg/ (accessed on 16 January 2018).
- Plant Antimicrobial Peptides-PhytAMP. Available online: http://phytamp.pfba-lab-tun.org/main.php (accessed on 16 January 2018).
- This review does not cover other antimicrobial peptides (such as valinomycin) which do not meet these structural requirements to resemble host-defense peptides.
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Wang, J.; Thompson, P.E.; Li, J. Teaching ‘Old’ Polymyxins New Tricks: New-Generation Lipopeptides Targeting Gram-Negative ‘Superbugs’. ACS Chem. Biol. 2014, 9, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Roberts, K.D.; Li, J. Rediscovering the octapeptins. Nat. Prod. Rep. 2017, 34, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.; Wu, X.; Ng, N.L.J.; Mak, C.K.; Qin, C.; Guo, Z. Synthesis of Gramicidin S and Its Analogues via an On-Resin Macrolactamization Assisted by a Predisposed Conformation of the Linear Precursors. J. Org. Chem. 2004, 69, 2681–2685. [Google Scholar] [CrossRef] [PubMed]
- Quinn, G.A.; Maloy, A.P.; McClean, S.; Carney, B.; Slater, J.W. Lipopeptide biosurfactants from Paenibacillus polymyxa inhibit single and mixed species biofilms. Biofouling 2012, 28, 1151–1166. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-D.; Won, H.-S.; Kim, J.-H.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial Peptides for Therapeutic Applications: A Review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef] [PubMed]
- Chellat, M.F.; Raguz, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem. Int. Ed. 2016, 55, 6600–6626. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Mobashery, S. Endless resistance. Endless antibiotics? MedChemComm 2016, 7, 37–49. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.K.M.G.; Hodgkinson, J.T.; Sore, H.F.; Welch, M.; Salmond, G.P.C.; Spring, D.R. Combating Multidrug-Resistant Bacteria: Current Strategies for the Discovery of Novel Antibacterials. Angew. Chem. Int. Ed. 2013, 52, 10706–10733. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Hughes, D.; Devocelle, M.; Humphreys, H. Beyond conventional antibiotics for the future treatment of methicillin-resistant Staphylococcus aureus infections: Two novel alternatives. FEMS Immunol. Med. Microbiol. 2012, 65, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Leng, Q.; Begum, M.; Woodle, M.; Scaria, P.; Chou, S.; Mixson, A. Peptide-based antifungal therapies against emerging infections. Drugs Future 2010, 35, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Vriens, K.; Cammue, B.P.A.; Thevissen, K. Antifungal Plant Defensins: Mechanisms of Action and Production. Molecules 2014, 19, 12280–12303. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Lee, M.W.; Fulan, B.M.; Ferguson, A.L.; Wong, G.C.L. What can machine learning do for antimicrobial peptides, and what can antimicrobial peptides do for machine learning? Interface Focus 2017, 7, 20160153. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.-A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.D.; Tieleman, D.P. The Importance of Membrane Defects: Lessons from Simulations. Acc. Chem. Res. 2014, 47, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Bürck, J.; Wadhwani, P.; Fanghänel, S.; Ulrich, A.S. Oriented Circular Dichroism: A Method to Characterize Membrane-Active Peptides in Oriented Lipid Bilayers. Acc. Chem. Res. 2016, 49, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Pushpanathan, M.; Pooja, S.; Gunasekaran, P.; Rajendhran, J. Critical Evaluation and Compilation of Physicochemical Determinants and Membrane Interactions of MMGP1 Antifungal Peptide. Mol. Pharm. 2016, 13, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Chairatana, P.; Nolan, E.M. Human α-Defensin 6: A Small Peptide That Self-Assembles and Protects the Host by Entangling Microbes. Acc. Chem. Res. 2017, 50, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Heruka De Zoysa, G.; Cameron, A.J.; Hegde, V.V.; Raghothama, S.; Sarojini, V. Antimicrobial Peptides with Potential for Biofilm Eradication: Synthesis and Structure Activity Relationship Studies of Battacin Peptides. J. Med. Chem. 2015, 58, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Golla, R.M.; Lau, K.; Lushnikova, T.; Wang, G. Anti-Staphylococcal Biofilm Effects of Human Cathelicidin Peptides. ACS Med. Chem. Lett. 2016, 7, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Gomes, M.S. Immuno-Stimulatory Peptides as a Potential Adjunct Therapy against Intra-Macrophagic Pathogens. Molecules 2017, 22, 1297. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Silva, O.N.; Lu, T.K.; Franco, O.L. Antimicrobial peptides: Role in human disease and potential as immunotherapies. Pharmacol. Ther. 2017, 178, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-A.; Xiang, Y.; Wang, Y.-J.; Liu, J.; Lee, W.-H.; Zhang, Y. Naturally Occurring Antimicrobial Peptide OH-CATH30 Selectively Regulates the Innate Immune Response to Protect against Sepsis. J. Med. Chem. 2013, 56, 9136–9145. [Google Scholar] [CrossRef] [PubMed]
- Rudilla, H.; Fusté, E.; Cajal, Y.; Rabanal, F.; Vinuesa, T.; Viñas, M. Synergistic Antipseudomonal Effects of Synthetic Peptide AMP38 and Carbapenems. Molecules 2016, 21, 1223. [Google Scholar] [CrossRef] [PubMed]
- For More Data about Approved Drugs or Candidates in Clinical Trials. Available online: https://clinicaltrials.gov/ (Update: https://www.opm.gov/); (accessed on 16 January 2018).
- FDA Page. Available online: https://www.fda.gov/Drugs/default.htm (accessed on 31 January 2018).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 16 January 2018).
- Gottler, L.M.; Ramamoorthy, A. Structure, Membrane Orientation, Mechanism, and Function of Pexiganan—A Highly Potent Antimicrobial Peptide Designed from Magainin. Biochim. Biophys. Acta 2009, 1788, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- For On-Line Information on Gramicidins. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/gramicidin (accessed on 16 January 2018).
- Gramicidin, S. Available online:. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Gramicidin_S (accessed on 16 January 2018).
- Falagas, M.E.; Grammatikos, A.P.; Michalopoulos, A. Potential of old-generation antibiotics to address current need for new antibiotics. Expert Rev. Anti-Infect. Ther. 2008, 6, 593–600. [Google Scholar] [CrossRef] [PubMed]
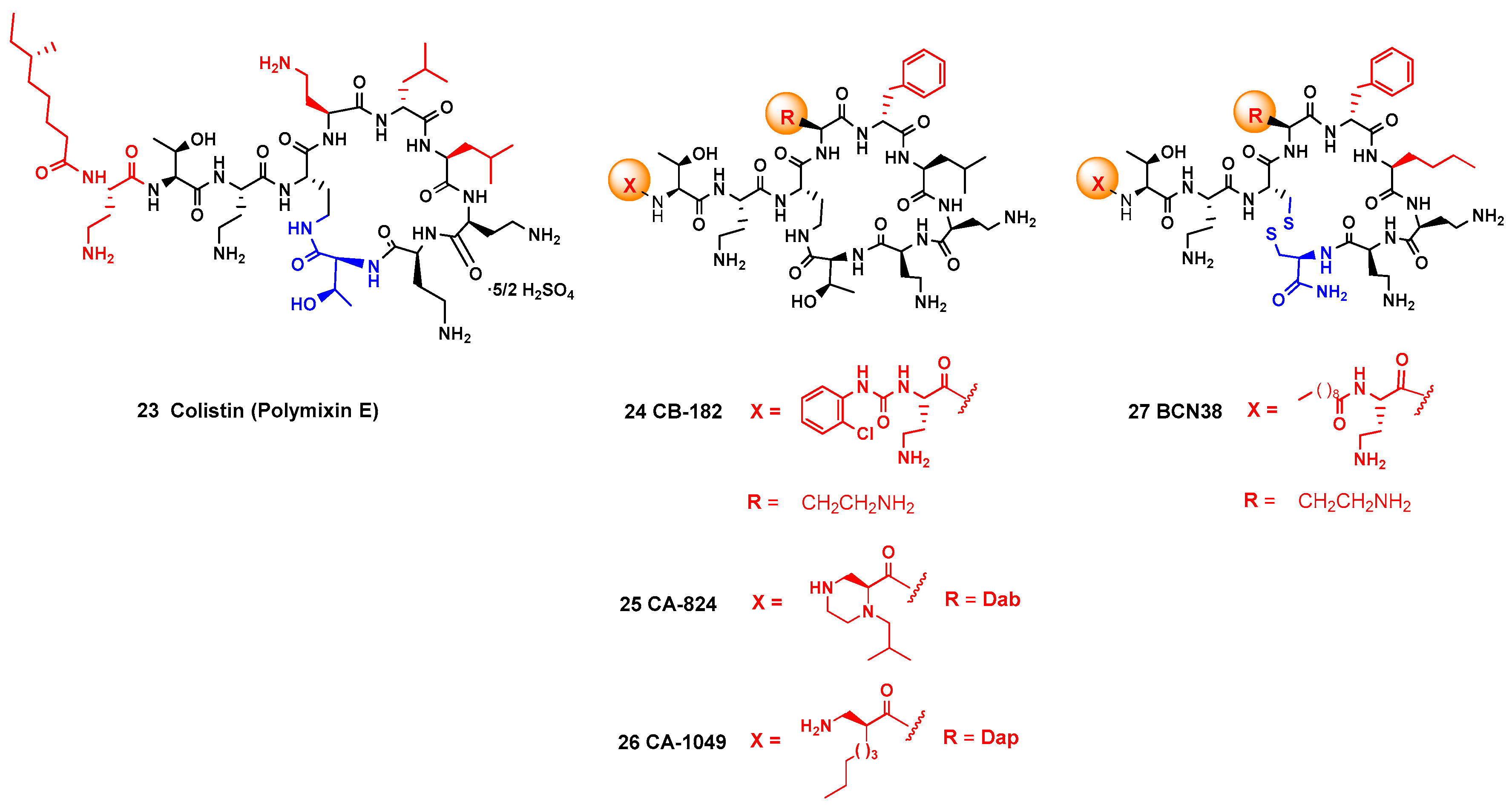
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L.; Perry, C.M. Caspofungin: A review of its use in the treatment of fungal infections. Drugs 2005, 65, 2049–2068. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W. Echinocandins: A new class of antifungal. J. Antimicrob. Chemother. 2002, 49, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Redman, R.; Yazji, S.; Bellm, L. Iseganan HCl: A novel antimicrobial agent. Expert Opin. Investig. Drugs 2002, 11, 1161–1170. [Google Scholar] [PubMed]
- Elad, S.; Epstein, J.B.; Raber-Durlacher, J.; Donnelly, P.; Strahilevitz, J. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: Analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J. Oral Pathol. Med. 2012, 41, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Fritschel, T.R.; Rhomberg, P.R.; Sader, H.S.; Jones, R.N. Antimicrobial Activity of Omiganan Pentahydrochloride against Contemporary Fungal Pathogens Responsible for Catheter-Associated Infections. Antimicrob. Agents Chemother. 2008, 52, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- A Study to Evaluate the Safety and Efficacy of Omiganan (CLS001) Topical Gel Versus Vehicle in Female Subjects with Moderate to Severe Acne Vulgaris. Available online: https://clinicaltrials.gov/ct2/show/NCT02571998 (accessed on 16 January 2018).
- Brilacidin: For On-Line Drug Information. Available online: http://www.ipharminc.com/brilacidin-1/, https://pubchem.ncbi.nlm.nih.gov/compound/Brilacidin; (accessed on 16 January /2018).
- Mensa, B.; Howell, G.L.; Scott, R.; DeGrado, W.F. Comparative Mechanistic Studies of Brilacidin, Daptomycin, and the Antimicrobial Peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- For on-Line Information on Novexatin®. Available online: https://www.novabiotics.co.uk/pipeline/novexatin-np213 (accessed on 16 January 2018).
- Nibbering, P.H.; Breij, A.D.E.; Cordfunke, R.A.; Zaat, S.A.J.; Drijfhout, J.W. Antimicrobial Peptide and Uses Thereof. U.S. Patent WO 2015088344 A1, 18 June 2015. [Google Scholar]
- Riool, M.; de Breij, A.; Drijfhout, J.W.; Nibbering, P.H.; Zaat, S.A.J. Antimicrobial Peptides in Biomedical Device Manufacturing. Front. Chem. 2017, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Van der Does, A.M.; Hensbergen, P.J.; Bogaards, S.J.; Cansoy, M.; Deelder, A.M.; van Leeuwen, H.C.; Drijfhout, J.W.; van Dissel, J.T.; Nibbering, P.H. The human lactoferrin-derived peptide hLF1-11 exerts immunomodulatory effects by specific inhibition of myeloperoxidase activity. J. Immunol. 2012, 188, 5012–5019. [Google Scholar] [CrossRef] [PubMed]
- Safety of a Single Dose of 5 mg of hLF1-11 Given to Autologous Haematopoietic Stem Cell Transplant Recipients. Clinical Trials. Available online: https://clinicaltrials.gov/show/NCT00509938 (accessed on 16 January 2018).
- Cheng, D.J.; Oppenheim, F.G.; Helmerhorst, E.J. Antifungal Formulation and Method of Preparation. U.S. Patent WO 2009005798 A2, 8 January 2009. [Google Scholar]
- For on-Line Information about PAC-113. Available online: https://www.drugbank.ca/drugs/DB05756, http://bciq.biocentury.com/products/pac-113 (accessed on 16 January 2018).
- For On-Line Information on the Stage of Development. Available online: www.pharmaasia.com/2016/08/pacgen-announces-progress-pac-113-license/ (accessed on 16 January 2018).
- Scott, M.G.; Dullaghan, E.; Mookherjee, N.; Glavas, N.; Walsbrook, M.; Thompson, A.; Wang, A.; Lee, K.; Doria, S.; Hamill, P.; et al. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 2007, 25, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.B.; Kielczewska, A.; Rozek, A.; Takenaka, S.; Li, Y.; Thorson, L.; Hancock, R.E.; Guarna, M.M.; North, J.R.; Foster, L.J.; et al. Sequestosome-1/p62 is the key intracellular target of innate defense regulator peptide. J. Biol. Chem. 2009, 284, 36007–36011. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Madera, L.; Ma, S.; Waldbrook, M.; Elliott, M.R.; Easton, D.M.; Mayer, M.L.; Mullaly, S.C.; Kindrachuk, J.; Jenssen, H.; et al. Synthetic Cationic Peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhances leukocyte recruitment. J. Immunol. 2010, 184, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Tokumoto, K.; Kaji, T.; Paudel, A.; Panthee, S.; Hamamoto, H.; Sekimizu, K.; Inoue, M. Total Synthesis and Biological Mode of Action of WAP-8294A2: A Menaquinone-Targeting Antibiotic. J. Org. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Novamycin NP339. Available online: https://www.novabiotics.co.uk/pipeline/novamycin-np339 (accessed on 16 January 2018).
- Novamycin NP432. Available online: https://www.novabiotics.co.uk/pipeline/novarifyn-np432 (accessed on 16 January 2018).
- Farha, M.A.; Brown, E.D. Strategies for target identification of antimicrobial natural products. Nat. Prod. Rep. 2016, 33, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, D.; Wang, L.; Lin, C.; Ma, C.; Xi, X.; Chen, T.; Shaw, C.; Zhou, M. Dermaseptin-PH: A Novel Peptide with Antimicrobial and Anticancer Activities from the Skin Secretion of the South American Orange-Legged Leaf Frog, Pithecopus (Phyllomedusa) hypochondrialis. Molecules 2017, 22, 1805. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Gao, Y.; Wang, L.; Xi, X.; Wu, Y.; Zhou, M.; Zhang, Y.; Ma, C.; Chen, T.; Shaw, C. A Combined Molecular Cloning and Mass Spectrometric Method to Identify, Characterize, and Design Frenatin Peptides from the Skin Secretion of Litoria infrafrenata. Molecules 2016, 21, 1429. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pan, J.; Wu, Y.; Xi, X.; Ma, C.; Wang, L.; Zhou, M.; Chen, T. PSN-PC: A Novel Antimicrobial and Anti-Biofilm Peptide from the Skin Secretion of Phyllomedusa-camba with Cytotoxicity on Human Lung Cancer Cell. Molecules 2017, 22, 1896. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, D.; Xi, X.; Wu, Y.; Ma, C.; Zhou, M.; Wang, L.; Yang, M.; Chen, T.; Shaw, C. Identification and Characterisation of the Antimicrobial Peptide, Phylloseptin-PT, from the Skin Secretion of Phyllomedusa tarsius, and Comparison of Activity with Designed, Cationicity-Enhanced Analogues and Diastereomers. Molecules 2016, 21, 1667. [Google Scholar] [CrossRef] [PubMed]
- Onaka, H. Novel antibiotic screening methods to awaken silent or cryptic secondary metabolic pathways in actinomycetes. J. Antibiot. 2017, 70, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Hertweck, C. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009, 7, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Zarins-Tutt, J.S.; Triscari-Barbieri, T.; Gao, H.; Mearns-Spragg, A.; Zhang, L.; Newman, D.J.; Goss, R.J.M. Prospecting for new bacterial metabolites: A glossary of approaches for inducing, activating and upregulating the biosynthesis of bacterial cryptic or silent natural products. Nat. Prod. Rep. 2016, 33, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Vila-Farres, X.; Chu, J.; Inoyama, D.; Ternei, M.A.; Lemetre, C.; Cohen, L.J.; Cho, W.; Reddy, B.V.; Zebroski, H.A.; Freundlich, J.S.; Perlin, D.S.; Brady, S.F. Antimicrobials Inspired by Nonribosomal Peptide Synthetase Gene Clusters. J. Am. Chem. Soc. 2017, 139, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Lastra, J.M.; Borges, A.A.; Boto, A. Antimicrobial Peptides, Compositions Containing Them and Uses. ES Patent P201630875, 26 June 2016. [Google Scholar]
- Bals, R.; Wilson, J.M. Cathelicidins—A family of multifunctional antimicrobial peptides. Cell. Mol. Life Sci. 2003, 60, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Cathelicidins: A family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 2002, 9, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Genebank Public Databases. Available online: http://www.ncbi.nlm.nih.gov/genome (accessed on 16 January 2018).
- Nucleotide BLAST. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 16 January 2018).
- GENSCAN. Available online: http://genes.mit.edu/GENSCAN.html (accessed on 16 January 2018).
- GeneMark. Available online: http://exon.gatech.edu/GeneMark/ (accessed on 16 January 2018).
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- ADP3 Tool. Available online: http://aps.unmc.edu/AP/prediction/prediction_main.php (accessed on 16 January 2018).
- Torrent, M.; Di Tommaso, P.; Pulido, D.; Nogués, M.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An automated web server for prediction of protein antimicrobial regions. Bioinformatics 2012, 28, 130–131. [Google Scholar] [CrossRef] [PubMed]
- AMPA Tool. Available online: http://tcoffee.crg.cat/apps/ampa/do (accessed on 16 January 2018).
- PSIPred Tool. Available online: http://bioinf.cs.ucl.ac.uk/psipred/ (accessed on 16 January 2018).
- Wang, P.; Ge, R.; Liu, L.; Xiao, X.; Li, Y.; Cai, Y. Multi-label Learning for Predicting the Activities of Antimicrobial Peptides. Sci. Rep. 2017, 7, 2202. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.; Villalobos, A.; Gustafsson, C.; Minshull, J. You’re one in a googol: Optimizing genes for protein expression. J. R. Soc. Interface 2009, 6 (Suppl. 4), S467–S476. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
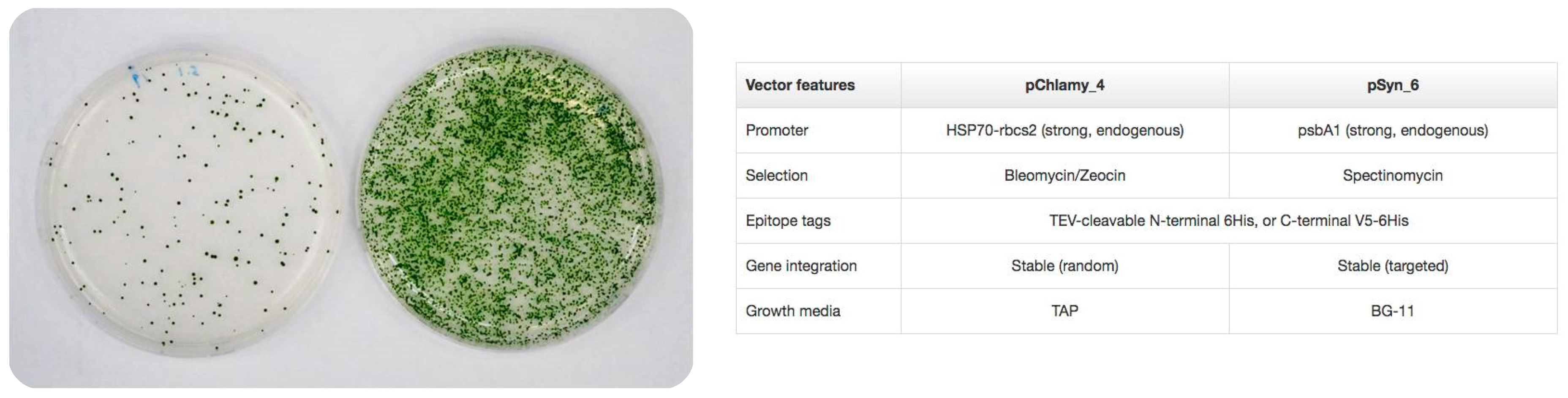
- Potvin, G.; Zhang, Z. Strategies for high-level recombinant protein expression in transgenic microalgae: A review. Biotechnol. Adv. 2010, 28, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Wijffels, R.H.; Kruse, O.; Hellingwerf, K.J. Potential of industrial biotechnology with cyanobacteria and eukaryotic microalgae. Curr. Opin. Biotechnol. 2013, 24, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Rasala, B.A.; Lee, P.A.; Shen, Z.; Briggs, S.P.; Mendez, M.; Mayfield, S.P. Robust expression and secretion of Xylanase1 in Chlamydomonas reinhardtii by fusion to a selection gene and processing with the FMDV 2A peptide. PLoS ONE 2012, 7, e43349. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Sewald, N.; Jakubke, H.D. Peptides: Chemistry and Biology; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef] [PubMed]
- Brand, G.D.; Santos, R.C.; Arake, L.M.; Silva, V.G.; Veras, L.M.C.; Costa, V.; Costa, C.H.N.; Kuckelhaus, S.S.; Alexandre, J.G.; Feio, M.J.; et al. The Skin Secretion of the Amphibian Phyllomedusa nordestina: A Source of Antimicrobial and Antiprotozoal Peptides. Molecules 2013, 18, 7058–7070. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Dahiya, R.; Khokra, S.L.; Mourya, R.; Chennupati, S.V.; Maharaj, S. Total Synthesis and Pharmacological Investigation of Cordyheptapeptide A. Molecules 2017, 22, 682. [Google Scholar] [CrossRef] [PubMed]
- Riahifard, N.; Tavakoli, K.; Yamaki, J.; Parang, K.; Tiwari, R. Synthesis and Evaluation of Antimicrobial Activity of [R4W4K]-Levofloxacin and [R4W4K]-Levofloxacin-Q Conjugates. Molecules 2017, 22, 957. [Google Scholar] [CrossRef] [PubMed]
- Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of ‘‘difficult peptides’’. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef] [PubMed]
- Martí-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S.V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, A.; Trinh, T.B.; Pei, D. Global Analysis of Peptide Cyclization Efficiency. ACS Comb. Sci. 2013, 15, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-F.; Zhang, X.-H.; Ding, Y.-J.; Yang, Y.-S.; Bi, X.-B.; Liu, C.F. Facile Synthesis of Peptidyl Salicylaldehyde Esters and Its Use in Cyclic Peptide Synthesis. Org. Lett. 2013, 15, 5182–5185. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; McIntosh, J.; Hathaway, B.J.; Schmidt, E.W. Using Marine Natural Products to Discover a Protease that Catalyzes Peptide Macrocyclization of Diverse Substrates. J. Am. Chem. Soc. 2009, 131, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Güell, I.; Vilà, S.; Micaló, L.; Badosa, E.; Montesinos, E.; Planas, M.; Feliu, L. Synthesis of Cyclic Peptidotriazoles with Activity Against Phytopathogenic Bacteria. Eur. J. Org. Chem. 2013, 2013, 4933–4943. [Google Scholar] [CrossRef]
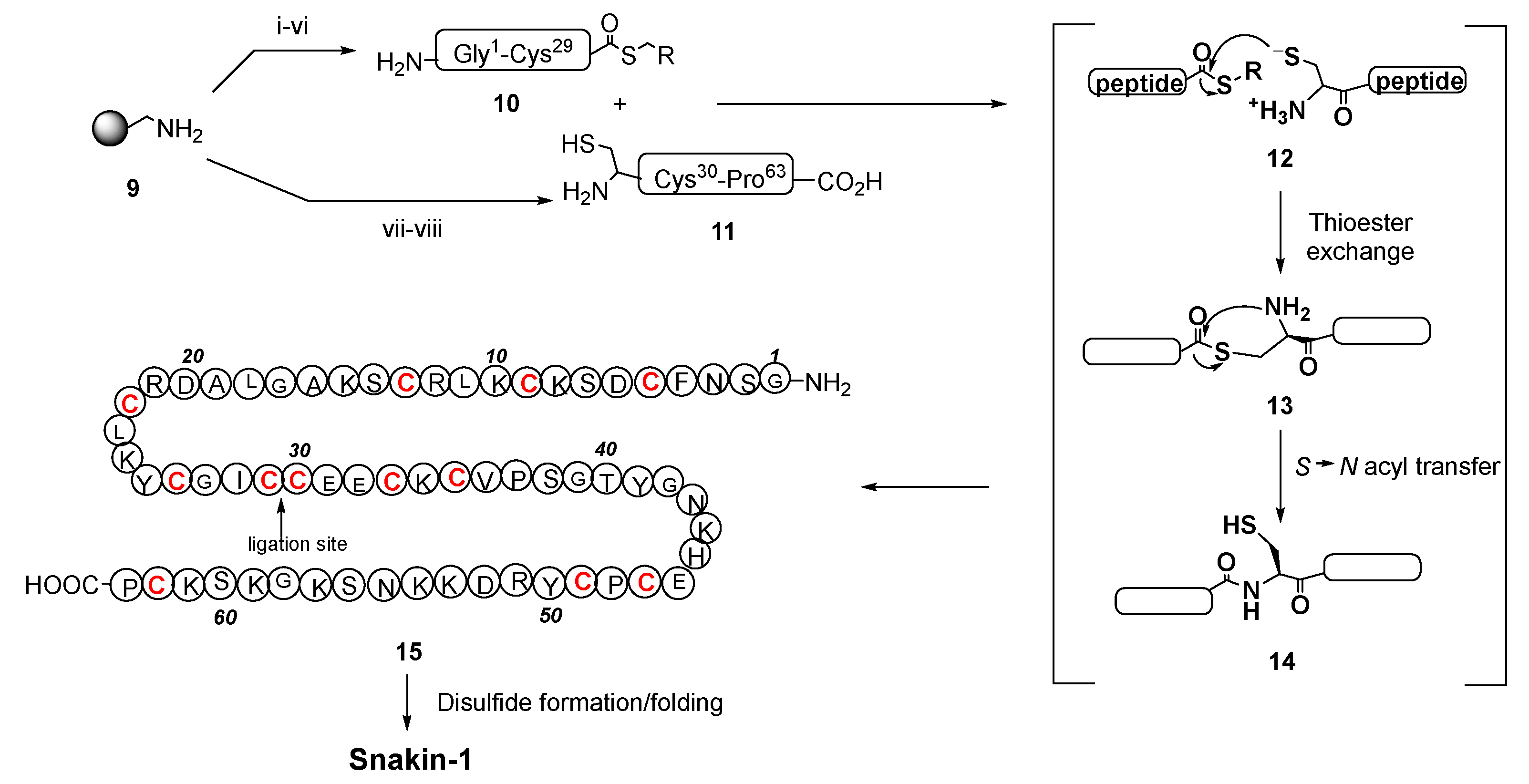
- Harris, P.W.R.; Yang, S.-H.; Molina, A.; López, G.; Middleditch, M.; Brimble, M.A. Plant Antimicrobial Peptides Snakin-1 and Snakin-2: Chemical Synthesis and Insights into the Disulfide Connectivity. Chem. Eur. J. 2014, 20, 5102–5110. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Craik, D.J. The Chemistry and Biology of Theta Defensins. Angew. Chem. Int. Ed. 2014, 53, 10612–10623. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.-K.; Guo, Y.; He, Y.; Wang, F.-L.; Chang, H.-N.; Wang, Y.-J.; Wu, F.-M.; Tian, C.-L.; Liu, L. Diaminodiacid-Based Solid-Phase Synthesis of Peptide Disulfide Bond Mimics. Angew. Chem. Int. Ed. 2013, 52, 9558–9562. [Google Scholar] [CrossRef] [PubMed]
- Magee, T.V.; Brown, M.F.; Starr, J.T.; Ackley, D.C.; Abramite, J.A.; Aubrecht, J.; Butler, A.; Crandon, J.L.; Dib-Hajj, F.; Flanagan, M.E.; et al. Discovery of Dap-3 Polymyxin Analogues for the Treatment of Multidrug-Resistant Gram-Negative Nosocomial Infections. J. Med. Chem. 2013, 56, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Romero-Estudillo, I.; Boto, A. Domino Process Achieves Site-Selective Peptide Modification with High Optical Purity. Applications to Chain Diversification and Peptide Ligation. J. Org. Chem. 2015, 80, 9379–9391. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.M.; Bernardes, G.J.L.; Davis, B.G. A “Tag-and-Modify” Approach to Site-Selective Protein Modification. Acc. Chem. Res. 2011, 44, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Vilà, S.; Badosa, E.; Montesinos, E.; Feliu, L.; Planas, M. Solid-Phase Synthesis of Peptide Conjugates Derived from the Antimicrobial Cyclic Decapeptide BPC194. Eur. J. Org. Chem. 2015, 1117–1129. [Google Scholar] [CrossRef]
- Vilà, S.; Camó, C.; Figueras, E.; Badosa, E.; Montesinos, E.; Planas, M.; Feliu, L. Solid-Phase Synthesis of Cyclic Lipopeptidotriazoles. Eur. J. Org. Chem. 2014, 4785–4794. [Google Scholar] [CrossRef]
- Saavedra, C.J.; Hernández, D.; Boto, A. Metal-Free, Site-Selective Peptide Modification by Conversion of “Customizable” Units into β-substituted Dehydroamino Acids. Chem. Eur. J. 2018, 24, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Hernández, D.; Boto, A.; Guzmán, D.; Alvarez, E. Metal-free, direct conversion of α-amino acids into α-keto γ-amino esters for the synthesis of α,γ-peptides. Org. Biomol. Chem. 2017, 15, 7736–7742. [Google Scholar] [CrossRef] [PubMed]
- Romero-Estudillo, I.; Saavedra, C.J.; Boto, A.; Alvarez, E. Site-Selective Modification of Peptides: From “Customizable Units” to Novel α-Aryl and α-Alkyl Glycine Derivatives, and Components of Branched Peptides. Biopolymers 2015, 104, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, C.J.; Boto, A.; Hernández, R. Synthesis of α,γ-Peptide Hybrids by Selective Conversion of Glutamic Acid Units. Org. Lett. 2012, 14, 3542–3545. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, C.J.; Boto, A.; Hernández, R.; Miranda, J.I.; Aizpurua, J.M. Conformation and Chiral Effects in α,β,α-Tripeptides. J. Org. Chem. 2012, 77, 5907–5913. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Kaji, T.; Kuranaga, T.; Hamamoto, H.; Sekimizu, K.; Inoue, M. Total Synthesis and Biological Evaluation of the Antibiotic Lysocin E and Its Enantiomeric, Epimeric, and N-Demethylated Analogues. Angew. Chem. Int. Ed. 2015, 54, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Kaji, T.; Murai, M.; Itoh, H.; Yasukawa, J.; Hamamoto, H.; Sekimizu, K.; Inoue, M. Total Synthesis and Functional Evaluation of Fourteen Derivatives of Lysocin E: Importance of Cationic, Hydrophobic, and Aromatic Moieties for Antibacterial Activity. Chem. Eur. J. 2016, 22, 16912–16919. [Google Scholar] [CrossRef] [PubMed]
- Malgieri, G.; Avitabile, C.; Palmieri, M.; D’Andrea, L.D.; Isernia, C.; Romanelli, A.; Fattorusso, R. Structural Basis of a Temporin 1b Analogue Antimicrobial Activity against Gram Negative Bacteria Determined by CD and NMR Techniques in Cellular Environment. ACS Chem. Biol. 2015, 10, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Nell, M.J.; Tjabringa, G.S.; Wafelman, A.R.; Verrijk, R.; Hiemstra, P.S.; Drijfhout, J.W.; Grote, J.J. Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides 2006, 27, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, E.; Bardají, E. Synthetic Antimicrobial Peptides as Agricultural Pesticides for Plant-Disease Control. Chem. Biodivers. 2008, 5, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Kumar, S.; Shekhar, S.; Dhawan, B.; Dey, S. Synthesis and Biological Evaluation of Novel Peptide BF2 as an Antibacterial Agent against Clinical Isolates of Vancomycin-Resistant Enterococci. J. Med. Chem. 2014, 57, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- De Jesús-Huertas, N.; Vargas-Casanova, Y.; Gómez-Chimbi, A.K.; Hernández, E.; Leal-Castro, A.L.; Melo-Díaz, J.M.; Rivera-Monroy, Z.J.; García-Castañeda, J.E. Synthetic Peptides Derived from Bovine Lactoferricin Exhibit Antimicrobial Activity against E. coli ATCC 11775, S. maltophilia ATCC 13636 and S. enteritidis ATCC 13076. Molecules 2017, 22, 452. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Guo, Y.; Qiao, X.; Shang, X.; Niu, W.; Jin, M. Design, Recombinant Fusion Expression and Biological Evaluation of Vasoactive Intestinal Peptide Analogue as Novel Antimicrobial Agent. Molecules 2017, 22, 1963. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, J.-S.; Lee, Y.-S.; Sim, D.-W.; Lee, S.-H.; Bahk, Y.-Y.; Lee, K.-H.; Kim, E.-H.; Park, S.-J.; Lee, B.-J.; et al. Structural Characterization of de Novo Designed L5K5W Model Peptide Isomers with Potent Antimicrobial and Varied Hemolytic Activities. Molecules 2013, 18, 859–876. [Google Scholar] [CrossRef] [PubMed]
- De Jesús-Huertas, N.; Rivera-Monroy, Z.J.; Fierro-Medina, R.; García-Castañeda, J.E. Antimicrobial Activity of Truncated and Polyvalent Peptides Derived from the FKCRRQWQWRMKKGLA Sequence against Escherichia coli ATCC 25922 and Staphylococcus aureus ATCC 25923. Molecules 2017, 22, 987. [Google Scholar] [CrossRef] [PubMed]
- Laverty, G.; McCloskey, A.P.; Gilmore, B.F.; Jones, D.S.; Zhou, J.; Xu, B. Ultrashort Cationic Naphthalene-Derived Self-Assembled Peptides as Antimicrobial Nanomaterials. Biomacromolecules 2014, 15, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Slootweg, J.C.; Prochnow, P.; Bobersky, S.; Bandow, J.E.; Metzler-Nolte, N. Exploring Structure—Activity Relationships in Synthetic Antimicrobial Peptides (synAMPs) by a Ferrocene Scan. Eur. J. Inorg. Chem. 2017, 2017, 360–367. [Google Scholar] [CrossRef]
- Karstad, R.; Isaksen, G.; Wynendaele, E.; Guttormsen, Y.; De Spiegeleer, B.; Brandsdal, B.-O.; Svendsen, J.S.; Svenson, J. Targeting the S1 and S3 subsite of trypsin with unnatural cationic amino acids generates antimicrobial peptides with potential for oral administration. J. Med. Chem. 2012, 55, 6294–6305. [Google Scholar] [CrossRef] [PubMed]
- Bojsen, R.; Torbensen, R.; Larsen, C.E.; Folkesson, A.; Regenberg, B. The synthetic amphipathic peptidomimetic LTX109 is a potent fungicide that disturbs plasma membrane integrity in a sphingolipid dependent manner. PLoS ONE 2013, 8, e69483. [Google Scholar] [CrossRef] [PubMed]
- Stensen, W.; Turner, R.; Brown, M.; Kondori, N.; Svendsen, J.S.; Svenson, J. Short Cationic Antimicrobial Peptides Display Superior Antifungal Activities toward Candidiasis and Onychomycosis in Comparison with Terbinafine and Amorolfine. Mol. Pharm. 2016, 13, 3595–3600. [Google Scholar] [CrossRef] [PubMed]
- Stromstedt, A.A.; Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob. Agents Chemother. 2009, 53, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Hayouka, Z.; Bella, A.; Stern, T.; Ray, S.; Jiang, H.; Grovenor, C.R.M.; Ryadnov, M.G. Binary Encoding of Random Peptide Sequences for Selective and Differential Antimicrobial Mechanisms. Angew. Chem. Int. Ed. 2017, 56, 8099–8103. [Google Scholar] [CrossRef] [PubMed]
- Zerbe, K.; Moehle, K.; Robinson, J.A. Protein Epitope Mimetics: From New Antibiotics to Supramolecular Synthetic Vaccines. Acc. Chem. Res. 2017, 50, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic Antibiotics Target Outer-Membrane Biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Patora-Komisarska, K.; Moehle, K.; Obrecht, D.; Robinson, J.A. Structural studies of β-hairpin peptidomimetic antibiotics that target LptD in Pseudomonas sp. Bioorg. Med. Chem. 2013, 21, 5806–5810. [Google Scholar] [CrossRef] [PubMed]
- Urfer, M.; Bogdanovic, J.; Lo Monte, F.; Moehle, K.; Zerbe, K.; Omasits, U.; Ahrens, C.H.; Pessi, G.; Eberl, L.; Robinson, J.A.A. Peptidomimetic Antibiotic Targets Outer Membrane Proteins and Disrupts Selectively the Outer Membrane in Escherichia coli. J. Biol. Chem. 2016, 291, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Hartgerink, J.D.; Ghadiri, M.R. Oriented Self-Assembly of Cyclic Peptide Nanotubes in Lipid Membranes. J. Am. Chem. Soc. 1998, 120, 4417–4424. [Google Scholar] [CrossRef]
- Fernandez-Lopez, S.; Kim, H.S.; Choi, E.C.; Delgado, M.; Granja, J.R.; Khasanov, A.; Kraehenbuehl, K.; Long, G.; Weinberger, D.A.; Wilcoxen, K.M.; et al. Antibacterial agents based on the cyclic d,l-alpha-peptide architecture. Nature 2001, 412, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Jelokhani-Niaraki, M.; Kondejewski, L.H.; Wheaton, L.C.; Hodges, R.S. Effect of Ring Size on Conformation and Biological Activity of Cyclic Cationic Antimicrobial Peptides. J. Med. Chem. 2009, 52, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Oddo, A.; Thomsen, T.T.; Britt, H.M.; Løbner-Olesen, A.; Thulstrup, P.W.; Sanderson, J.M.; Hansen, P.R. Modulation of Backbone Flexibility for Effective Dissociation of Antibacterial and Hemolytic Activity in Cyclic Peptides. ACS Med. Chem. Lett. 2016, 7, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Sarig, H.; Rotem, S.; Ziserman, L.; Danino, D.; Mor, A. Impact of Self-Assembly Properties on Antibacterial Activity of Short Acyl-Lysine Oligomers. Antimicrob. Agents Chemother. 2008, 52, 4308–4314. [Google Scholar] [CrossRef] [PubMed]
- Radzishevsky, I.S.; Rotem, S.; Bourdetsky, D.; Navon-Venezia, S.; Carmeli, Y.; Mor, A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 2007, 25, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Sarig, H.; Goldfeder, Y.; Rotem, S.; Mor, A. Mechanisms Mediating Bactericidal Properties and Conditions That Enhance the Potency of a Broad-Spectrum Oligo-Acyl-Lysyl. Antimicrob. Agents Chemother. 2011, 55, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Benson, M.A.; Shin, S.B.Y.; Torres, V.J.; Kirshenbaum, K. Amphiphilic Cyclic Peptoids That Exhibit Antimicrobial Activity by Disrupting Staphylococcus aureus Membranes. Eur. J. Org. Chem. 2013, 2013, 3560–3566. [Google Scholar] [CrossRef]
- Jahnsen, R.D.; Sandberg-Schaal, A.; Juul Vissing, K.; Mørck Nielsen, H.; Frimodt-Møller, N.; Franzyk, H. Tailoring Cytotoxicity of Antimicrobial Peptidomimetics with High Activity against Multidrug-Resistant Escherichia coli. J. Med. Chem. 2014, 57, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Smith, C.; Wu, H.; Padhee, S.; Manoj, N.; Cardiello, J.; Qiao, Q.; Cao, C.; Yin, H.; Cai, J. Lipidated Cyclic γ-AApeptides Display Both Antimicrobial and Antiinflammatory Activity. ACS Chem. Biol. 2014, 9, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, J.; Hernandez-Gordillo, V.; Nepal, M.; Brezden, A.; Pozzi, V.; Seleem, M.N.; Chmielewski, J. Targeting Intracellular Pathogenic Bacteria with Unnatural Proline-Rich Peptides: Coupling Antibacterial Activity with Macrophage Penetration. Angew. Chem. Int. Ed. 2013, 52, 9664–9667. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Pohane, A.A.; Bansal, S.; Bajaj, A.; Jain, V.; Srivastava, A. Cell Penetrating Synthetic Antimicrobial Peptides (SAMPs) Exhibiting Potent and Selective Killing of Mycobacterium by Targeting Its DNA. Chem. Eur. J. 2015, 21, 3540–3545. [Google Scholar] [CrossRef] [PubMed]
- Albada, H.B.; Prochnow, P.; Bobersky, S.; Langklotz, S.; Schriek, P.; Bandow, J.E.; Metzler-Nolte, N. Tuning the Activity of a Short Arg-Trp Antimicrobial Peptide by Lipidation of a C- or N-Terminal Lysine Side-Chain. ACS Med. Chem. Lett. 2012, 3, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, L. Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, P.I.; Ryder, N.S.; Weiss, H.M.; Hook, D.F.; Escalante, J.; Seebach, D. Exploring the Antibacterial and Hemolytic Activity of Shorter- and Longer-Chain β-, a,β-, and g-Peptides, and of β-Peptides from β2-3-Aza- and β3-2-Methylidene-amino Acids Bearing Proteinogenic Side Chain—A Survey. Chem. Biodivers. 2005, 2, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.P.; Gellman, S.H.; De Grado, W.F. β-Peptides: From Structure to Function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Gellman, S.H. Foldamers with Heterogeneous Backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A. β-Peptoid ‘‘Foldamers’’—Why the Additional Methylene Unit? Biopolymers 2011, 96, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, J.; Shankaramma, S.C.; Balaram, P. Design of Folded Peptides. Chem. Rev. 2001, 101, 3131–3152. [Google Scholar] [CrossRef] [PubMed]
- Claudon, P.; Violette, A.; Lamour, K.; Decossas, M.; Fournel, S.; Heurtault, B.; Godet, J.; Mély, Y.; Jamart-Grégoire, B.; Averlant-Petit, M.C.; et al. Consequences of Isostructural Main-Chain Modifications for the Design of Antimicrobial Foldamers: Helical Mimics of Host-Defense Peptides Based on a Heterogeneous Amide/Urea Backbone. Angew. Chem. Int. Ed. 2010, 49, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.L.H.; Hjørringgaard, C.U.; Vad, B.S.; Otzen, D.; Skrydstrup, T. Incorporation of β-Silicon-β3-Amino Acids in the Antimicrobial Peptide Alamethicin Provides a 20-Fold Increase in Membrane Permeabilization. Chem. Eur. J. 2016, 22, 8358–8367. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Teng, P.; Bai, G.; Lin, X.; Zuo, X.; Cao, C.; Cai, J. Helical Antimicrobial Sulfono-γ-AApeptides. J. Med. Chem. 2015, 58, 4802–4811. [Google Scholar] [CrossRef] [PubMed]
- Sgolastra, F.; Deronde, B.M.; Sarapas, J.M.; Som, A.; Tew, G.N. Designing mimics of membrane active proteins. Acc. Chem. Res. 2013, 46, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, U.; Sobczak, M.; Oledzka, E. Current state of a dual behaviour of antimicrobial peptides-Therapeutic agents and promising delivery vectors. Chem. Biol. Drug Des. 2017, 90, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Bray, B.L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Rotem, S.; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 2009, 1788, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Ingham, A.B.; Moore, R.J. Recombinant production of antimicrobial peptides in heterologous microbial systems. Biotechnol. Appl. Biochem. 2007, 47, 1–9. [Google Scholar] [PubMed]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sönksen, C.P.; Ludvigsen, S.; Raventós, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Nguyen, L.T.; Gopal, R.; Aizawa, T.; Vogel, H.J. Overexpression of Antimicrobial, Anticancer, and Transmembrane Peptides in Escherichia coli through a Calmodulin-Peptide Fusion System. J. Am. Chem. Soc. 2016, 138, 11318–11326. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.E.; Hancock, R.E.; Kalman, D. Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 2010, 31, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Herbel, V.; Schäfer, H.; Wink, M. Recombinant Production of Snakin-2 (an Antimicrobial Peptide from Tomato) in E. coli and Analysis of Its Bioactivity. Molecules 2015, 20, 14889–14901. [Google Scholar] [CrossRef] [PubMed]
- Fahimirad, S.; Abtahi, H.; Razavi, S.H.; Alizadeh, H.; Ghorbanpour, M. Production of Recombinant Antimicrobial Polymeric Protein Beta Casein-E 50-52 and Its Antimicrobial Synergistic Effects Assessment with Thymol. Molecules 2017, 22, 822. [Google Scholar] [CrossRef] [PubMed]
- Bundó, M.; Montesinos, L.; Izquierdo, E.; Campo, S.; Mieulet, D.; Guiderdoni, E.; Rossignol, M.; Badosa, E.; Montesinos, E.; San Segundo, B.; et al. Production of cecropin A antimicrobial peptide in rice seed endosperm. BMC Plant Biol. 2014, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, L.; Bundó, M.; Izquierdo, E.; Campo, S.; Badosa, E.; Rossignol, M.; Montesinos, E.; San Segundo, B.; Coca, M. Production of Biologically Active Cecropin A Peptide in Rice Seed Oil Bodies. PLoS ONE 2016, 11, e0146919. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Suga, H. Discovering functional, non-proteinogenic amino acid containing, peptides using genetic code reprogramming. Org. Biomol. Chem. 2015, 13, 9353–9363. [Google Scholar] [CrossRef] [PubMed]

















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boto, A.; Pérez de la Lastra, J.M.; González, C.C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23, 311. https://doi.org/10.3390/molecules23020311
Boto A, Pérez de la Lastra JM, González CC. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules. 2018; 23(2):311. https://doi.org/10.3390/molecules23020311
Chicago/Turabian StyleBoto, Alicia, Jose Manuel Pérez de la Lastra, and Concepción C. González. 2018. "The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs" Molecules 23, no. 2: 311. https://doi.org/10.3390/molecules23020311
APA StyleBoto, A., Pérez de la Lastra, J. M., & González, C. C. (2018). The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules, 23(2), 311. https://doi.org/10.3390/molecules23020311