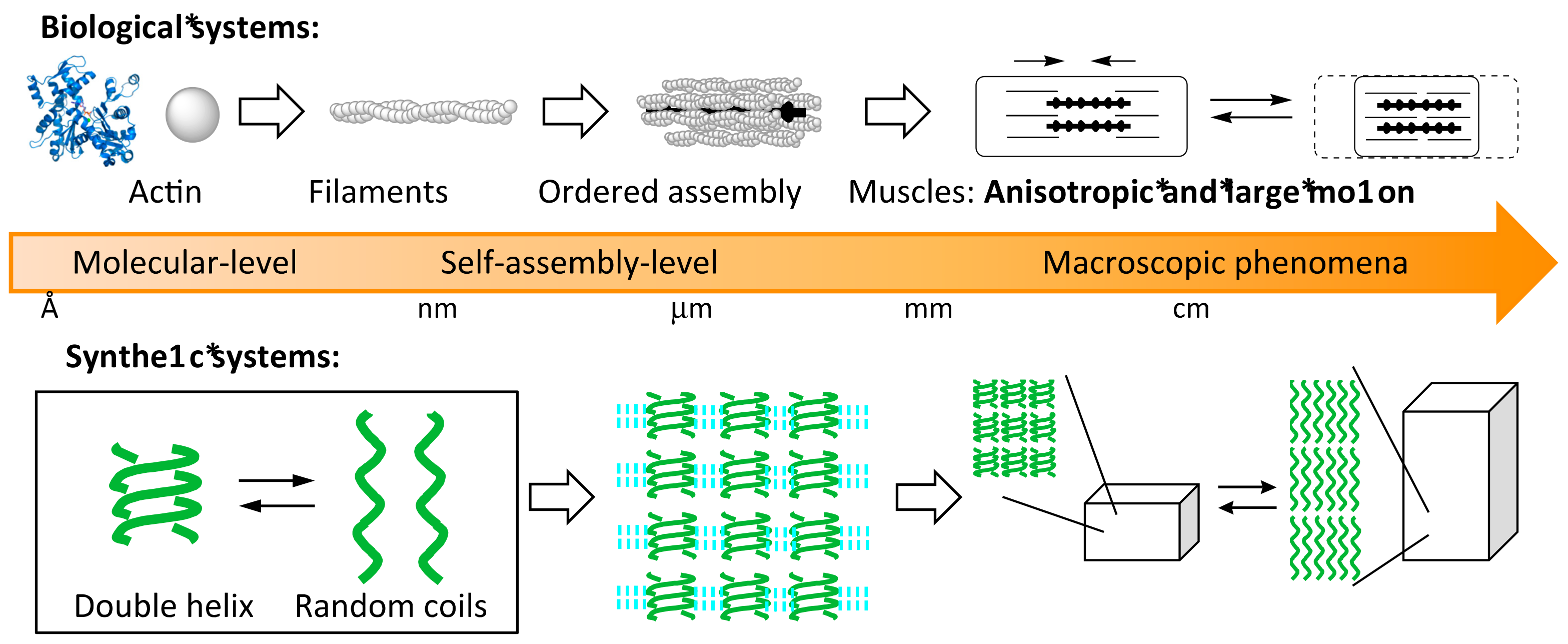
Synthesis and Self-Assembly of Chiral Cylindrical Molecular Complexes: Functional Heterogeneous Liquid-Solid Materials Formed by Helicene Oligomers

Abstract
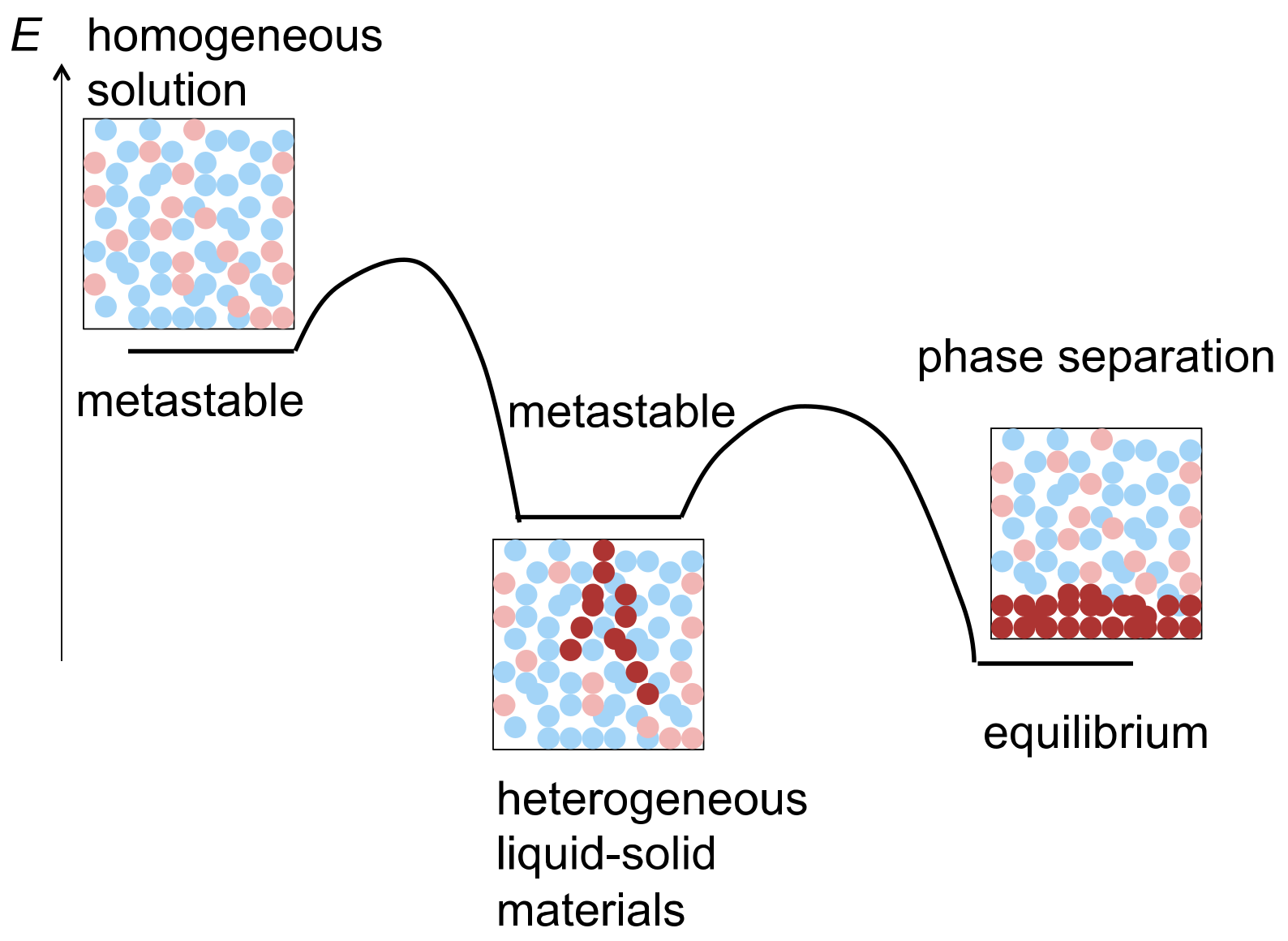
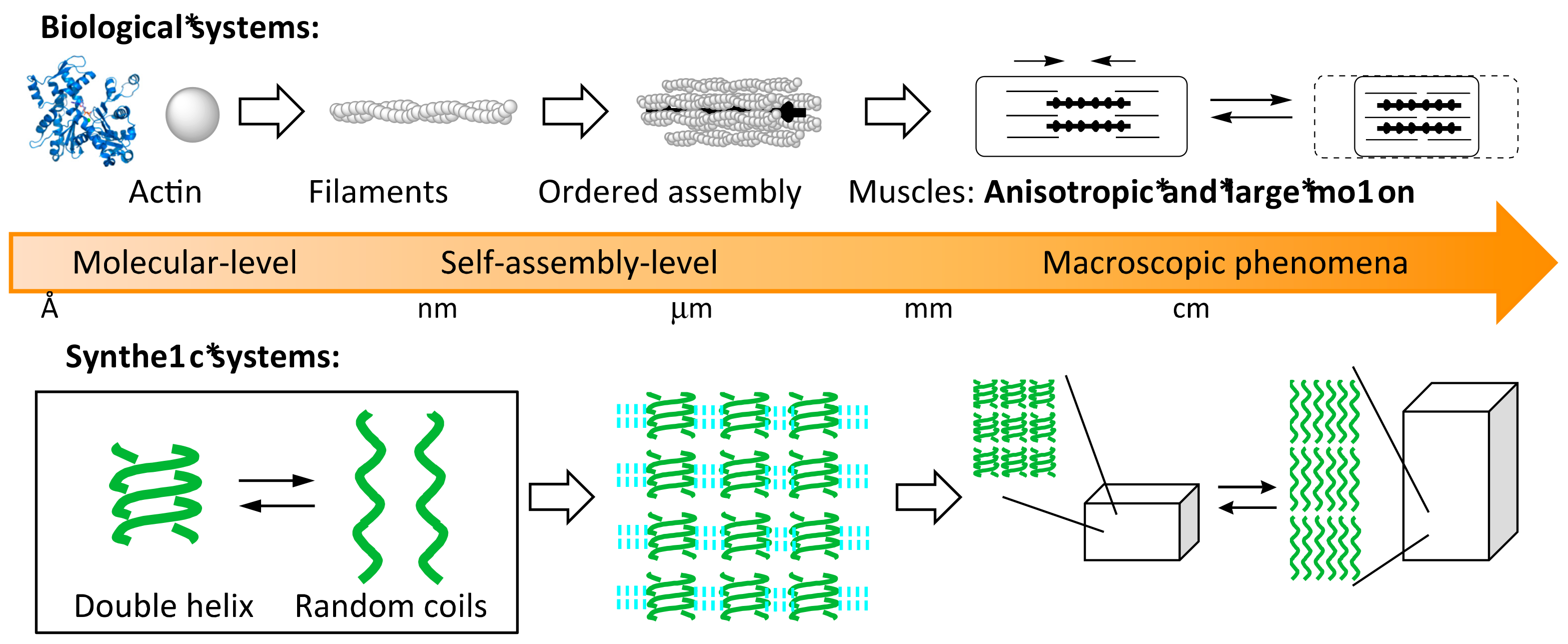
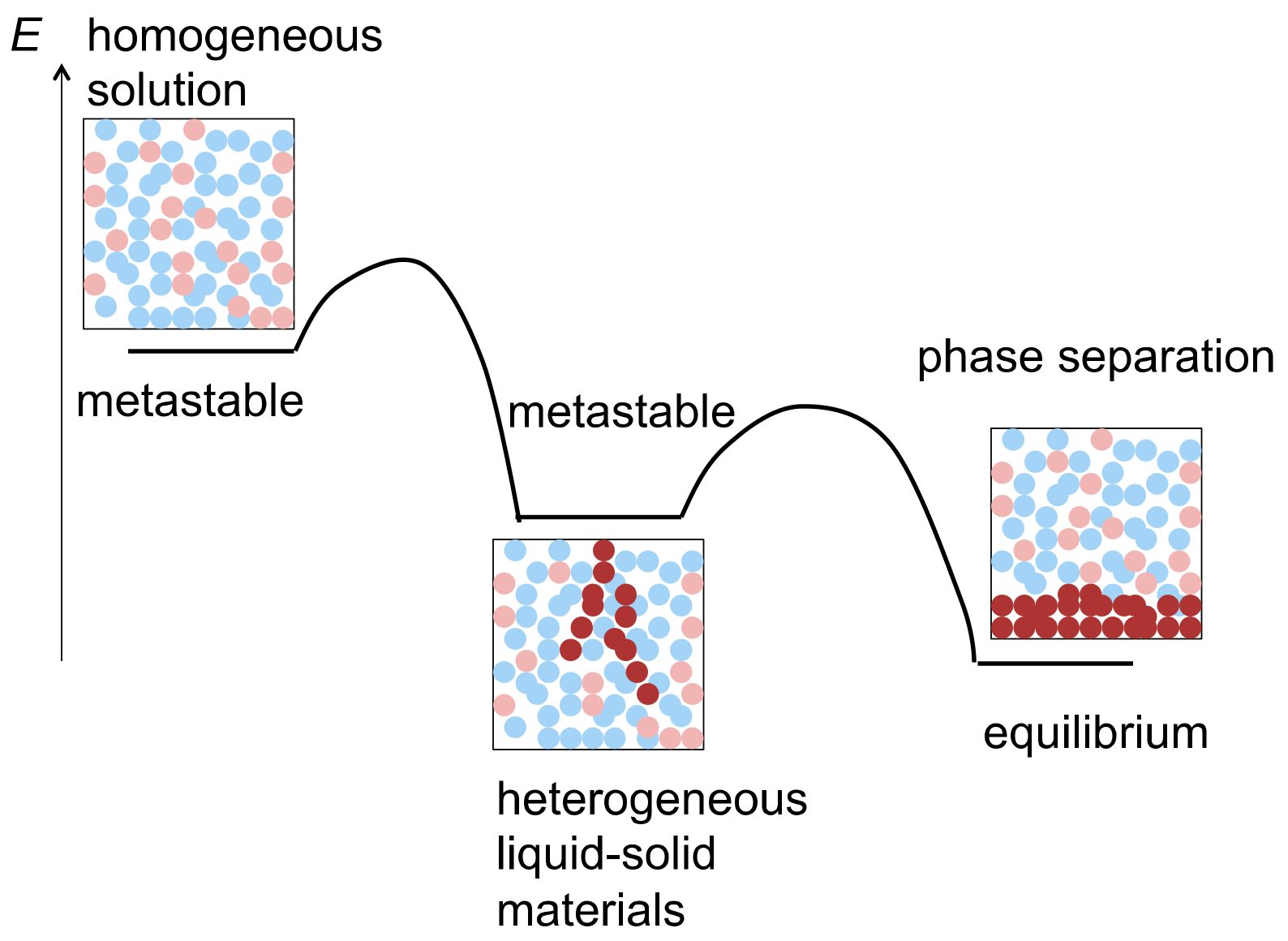
:1. Heterogeneous Liquid-Solid Materials
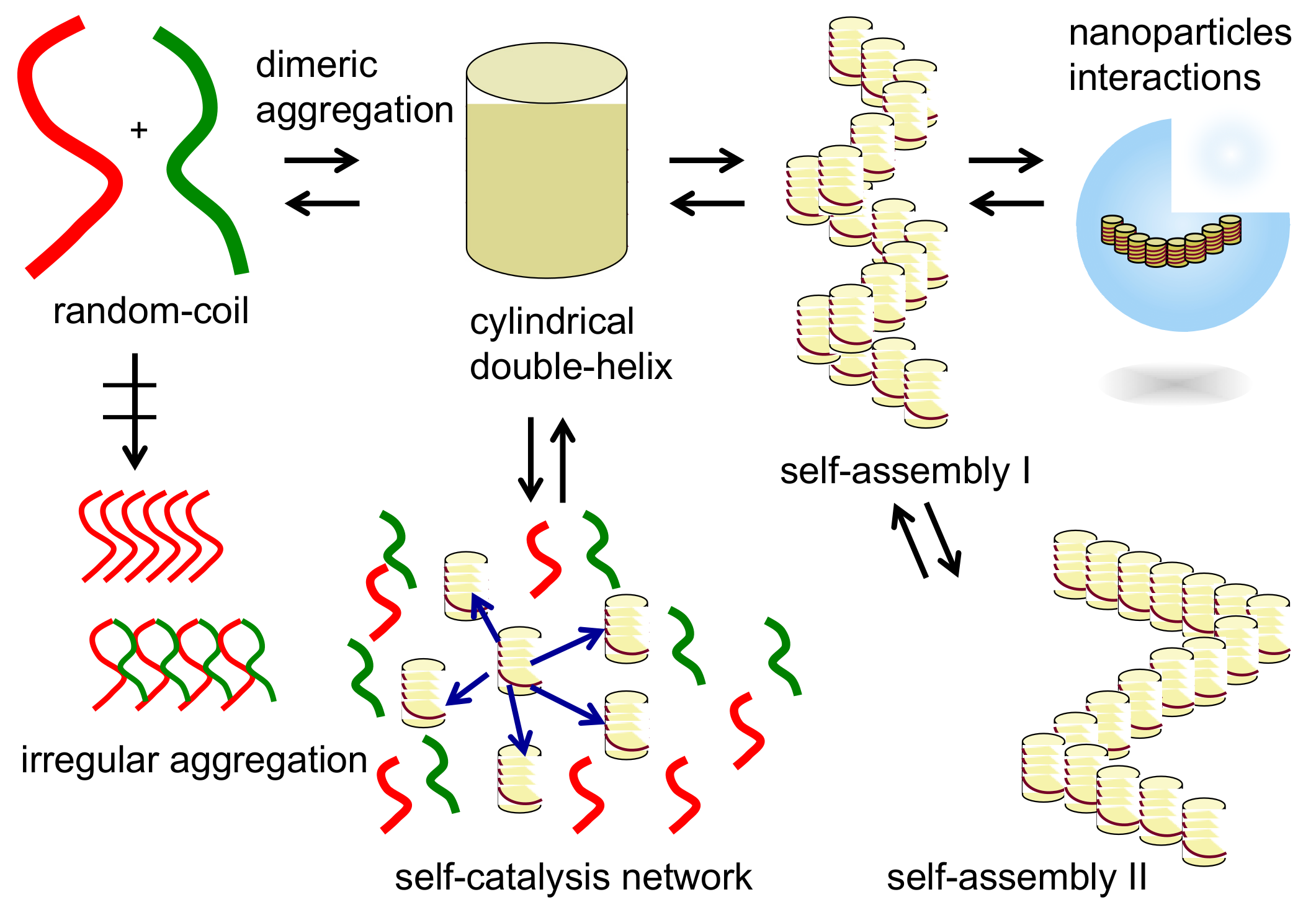
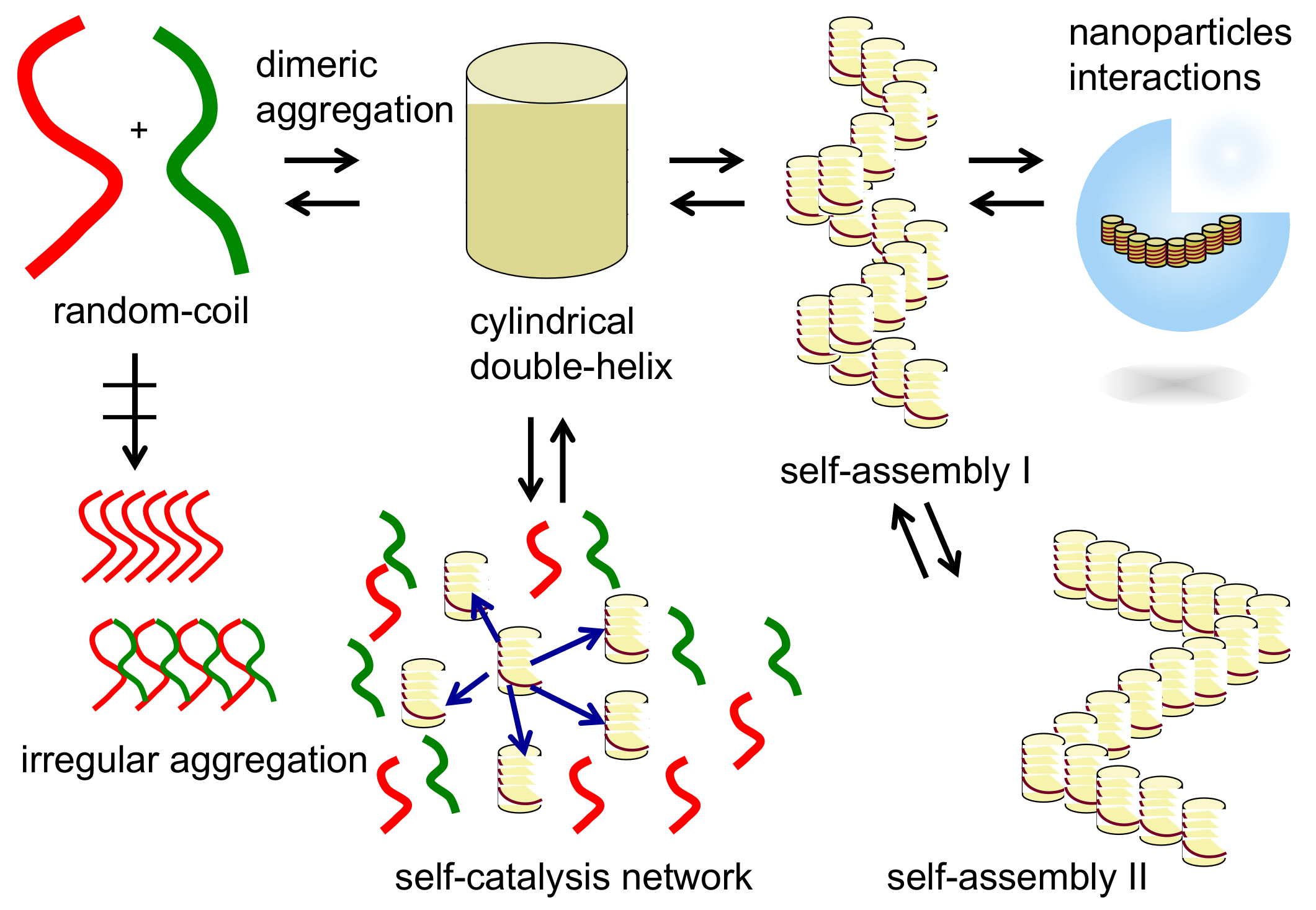
2. Self-Assembly of Chiral Cylindrical Molecular Complexes
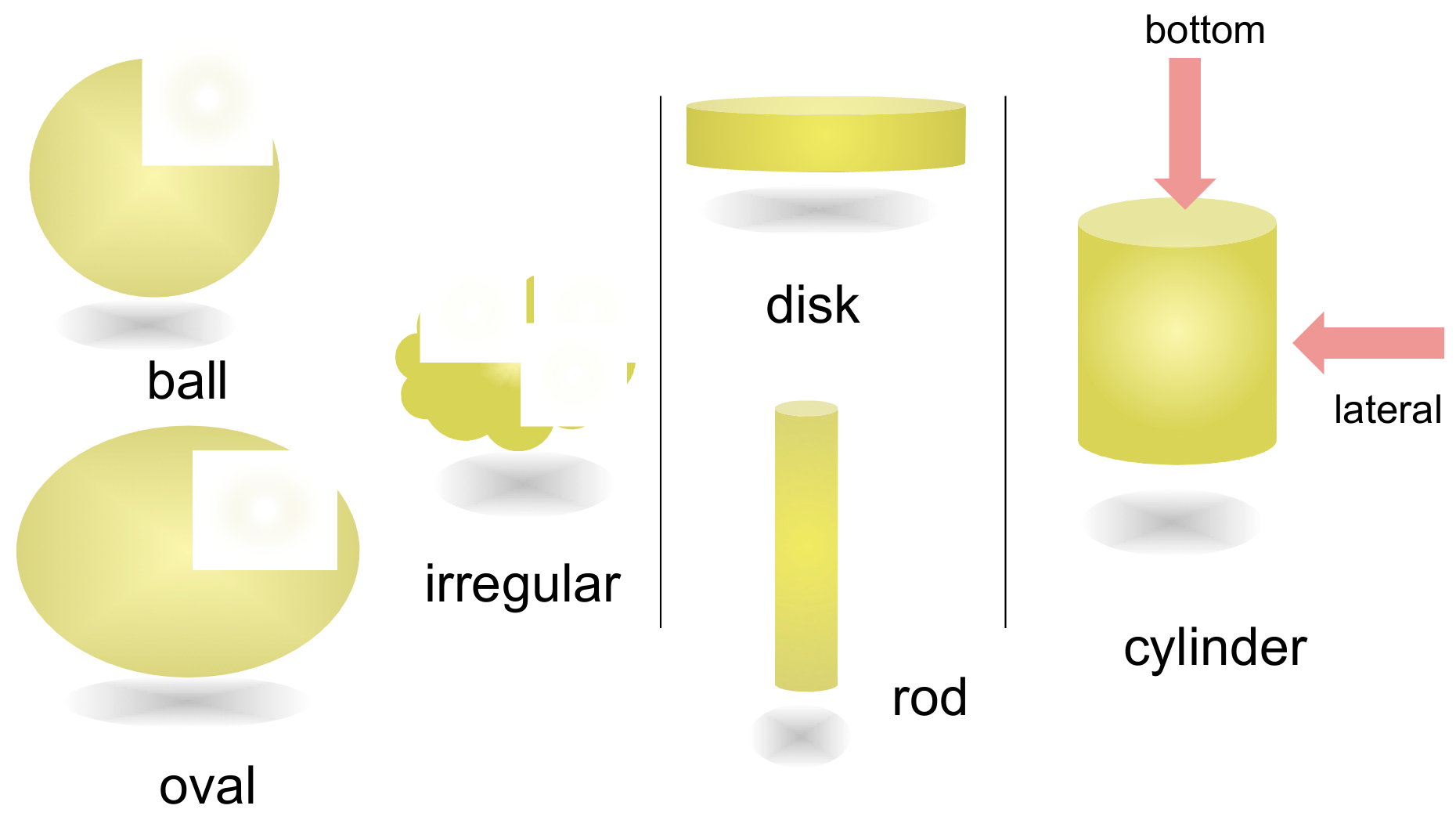
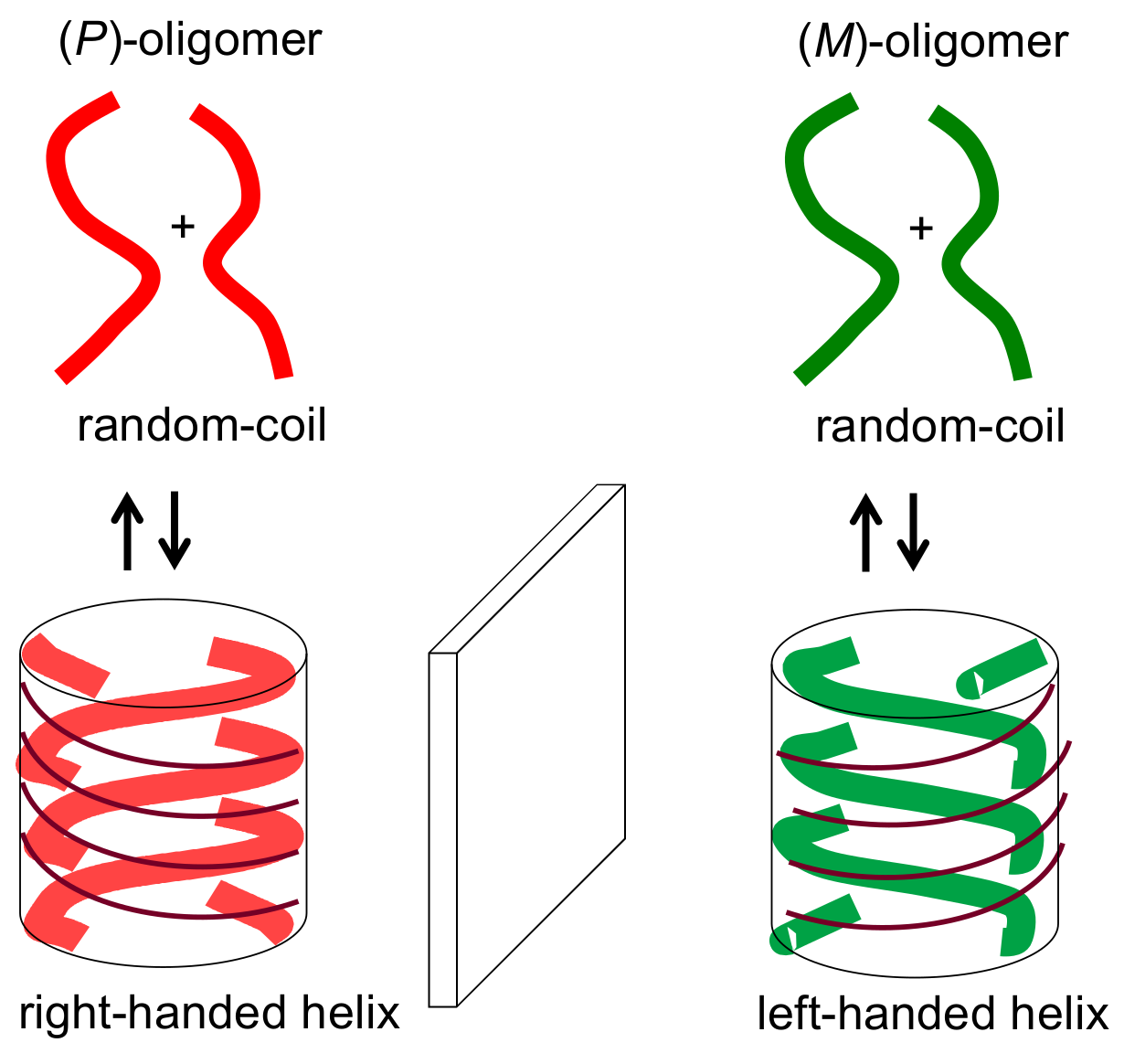
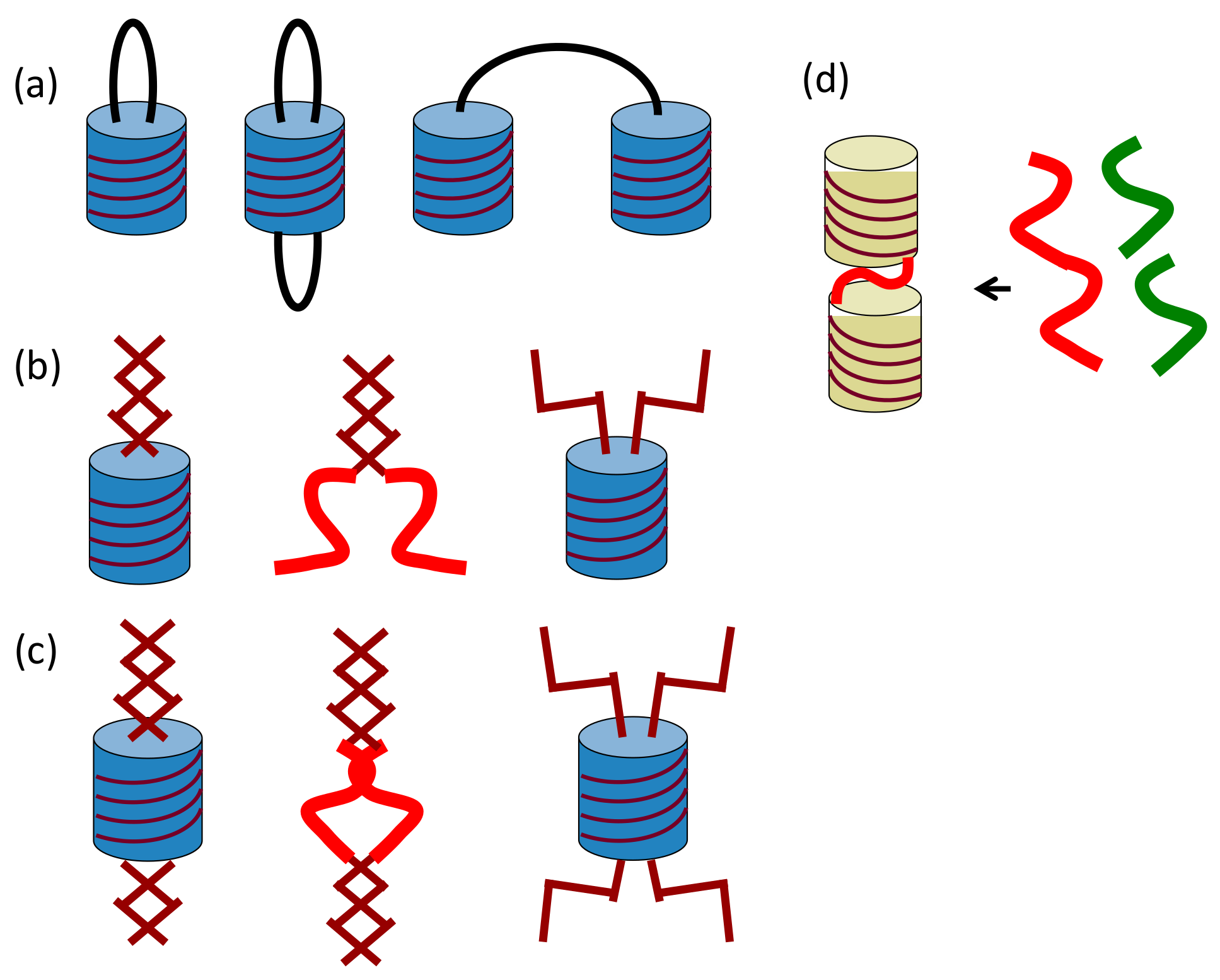
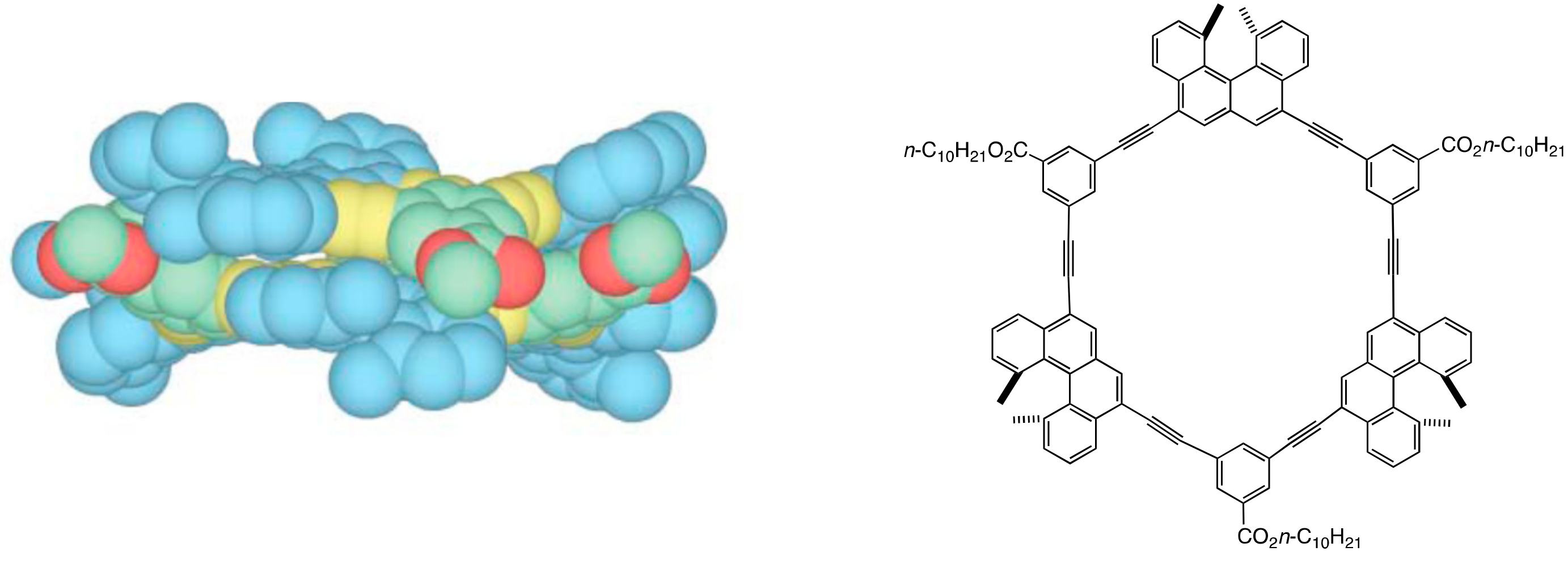
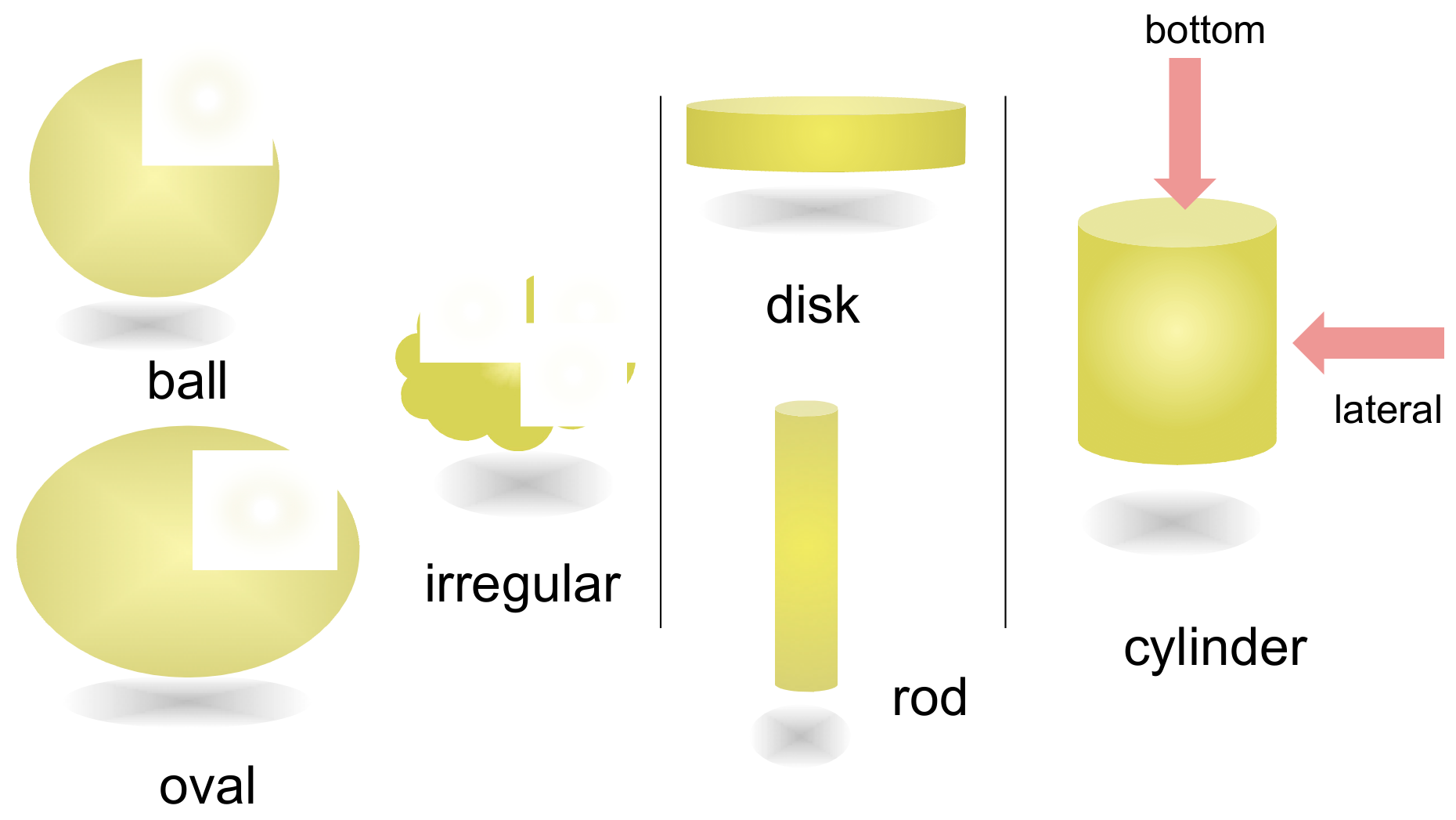
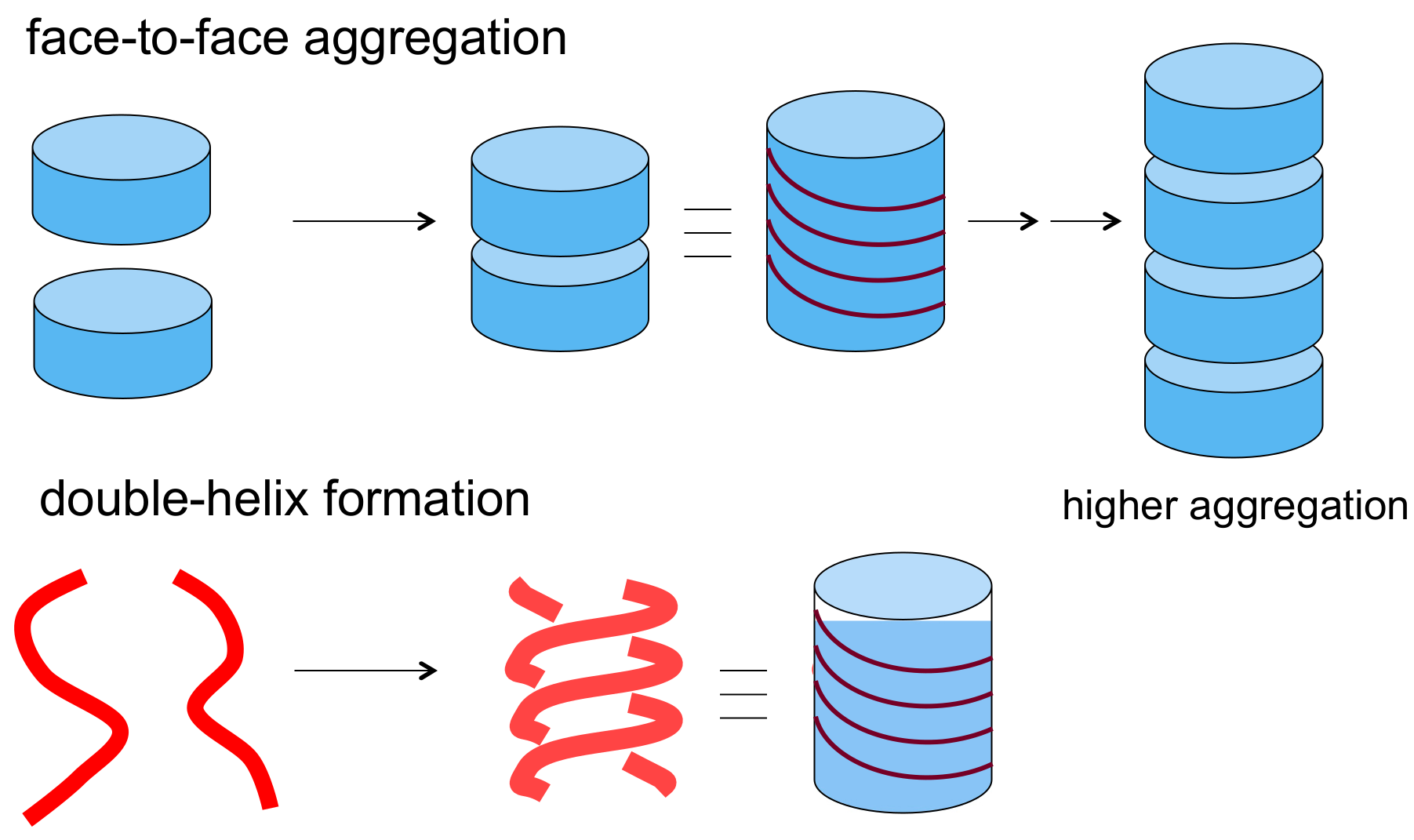
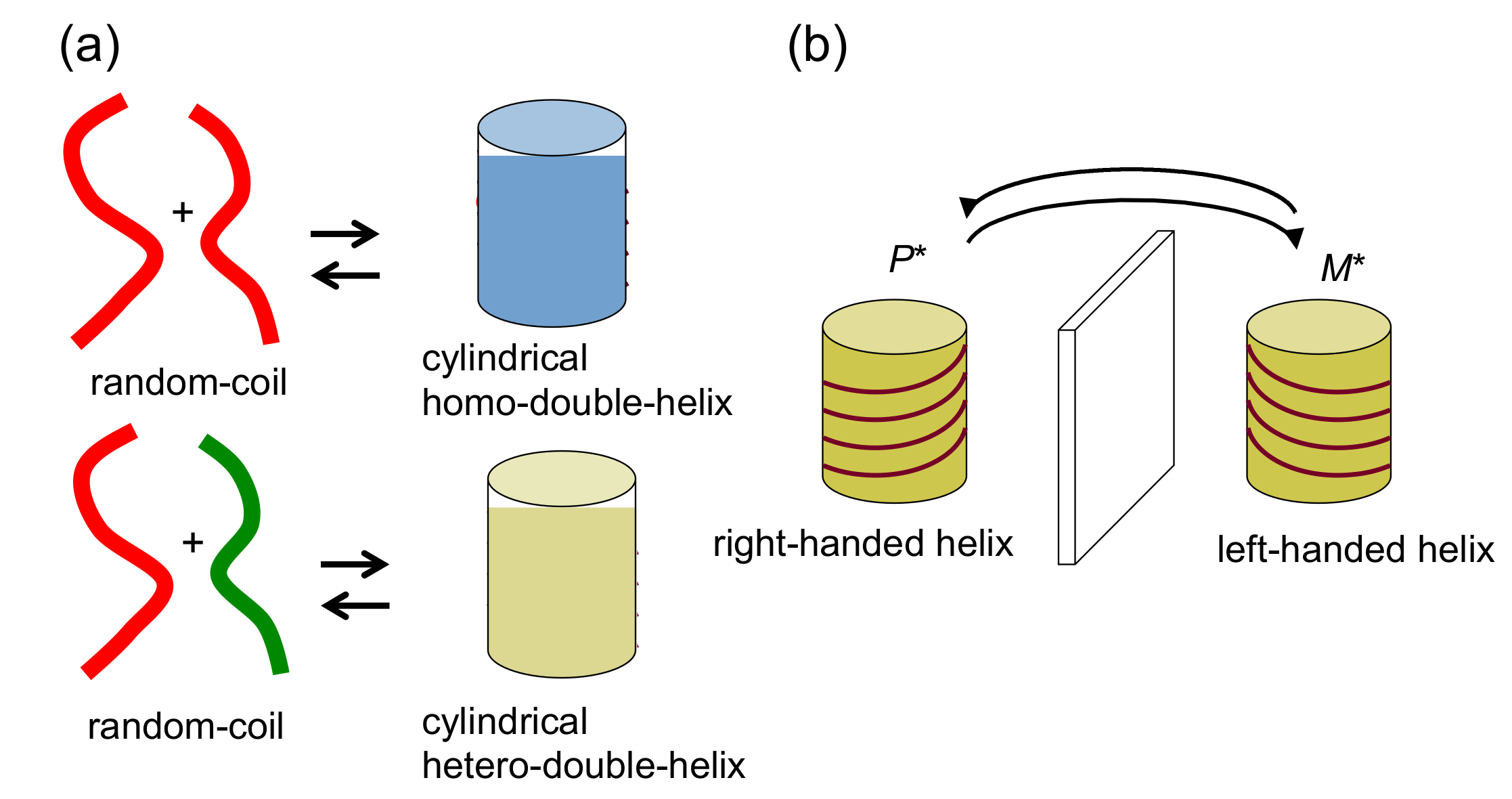
2.1. Cylindrical Molecular Complexes
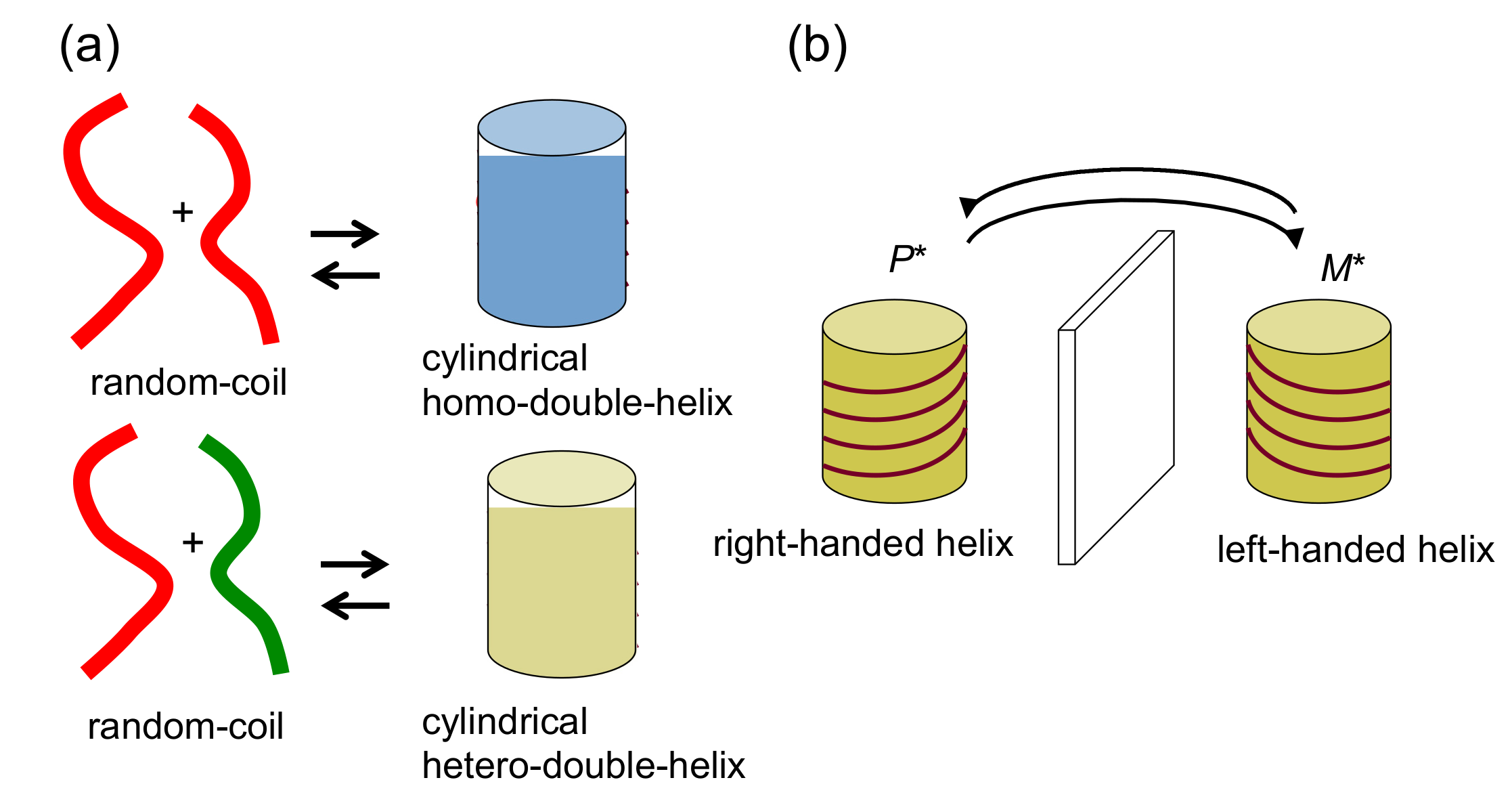
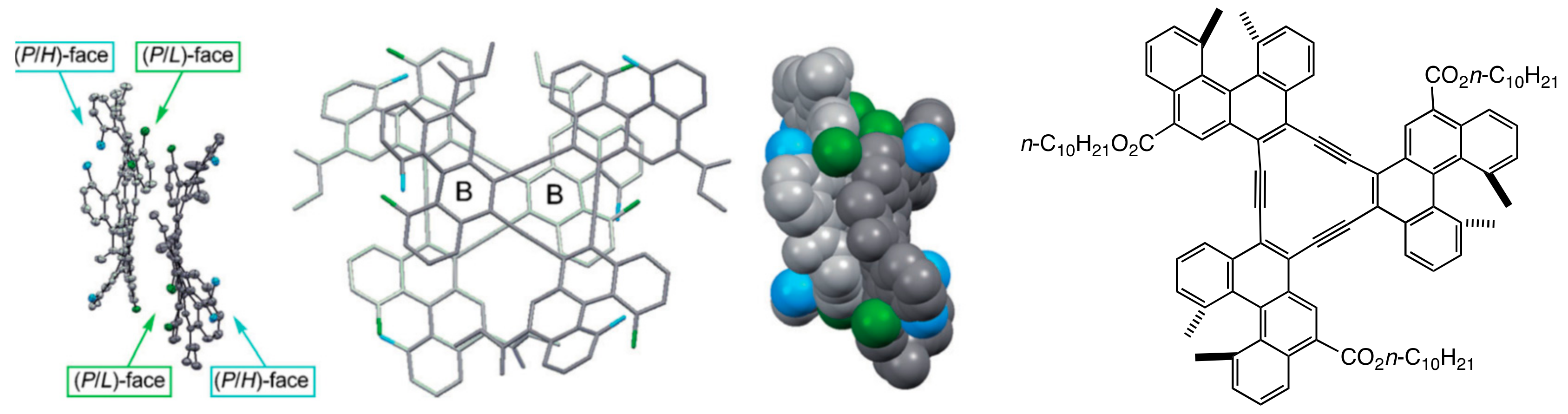
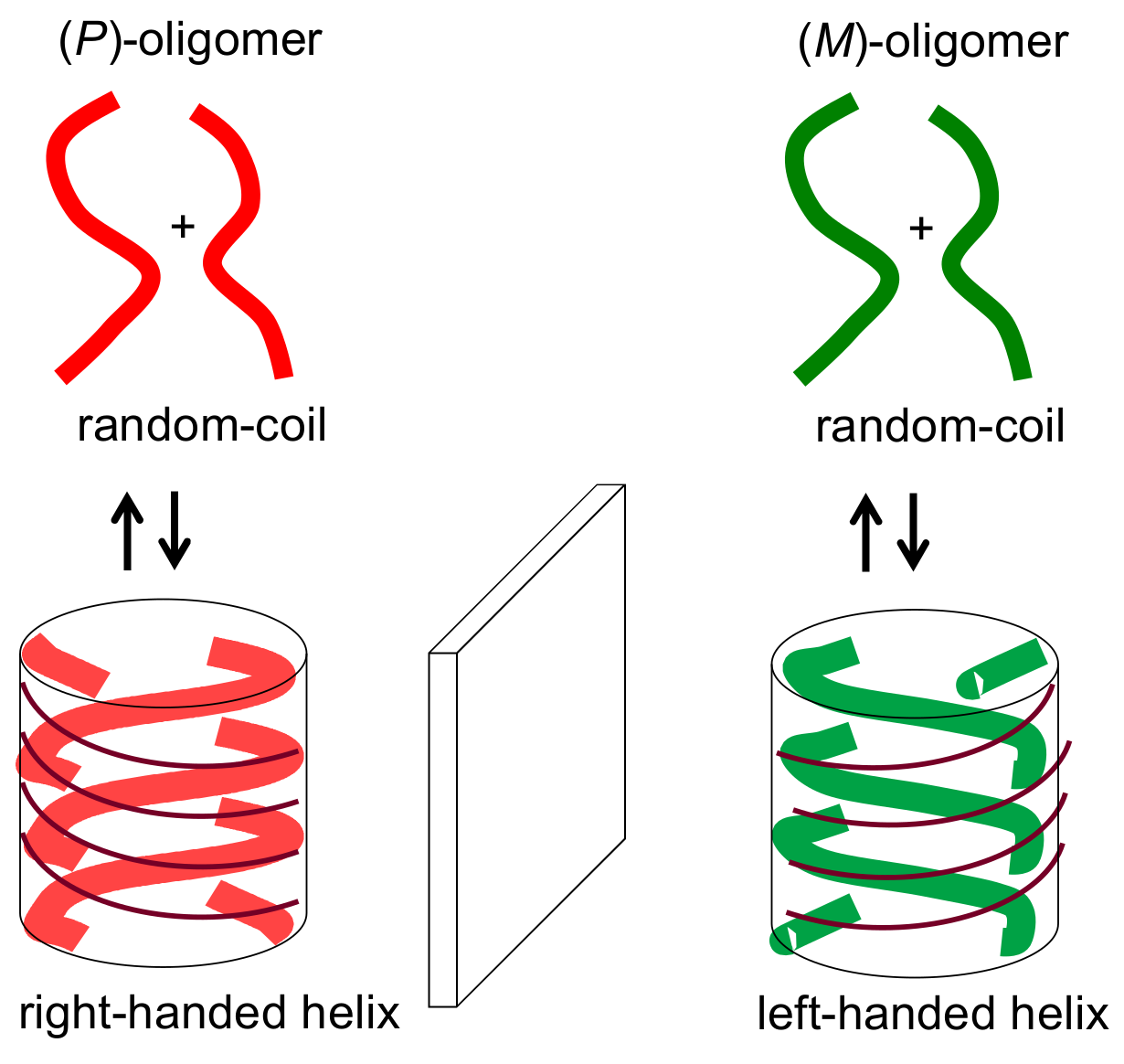
2.2. Double-Helix Chiral Cylindrical Molecular Complex
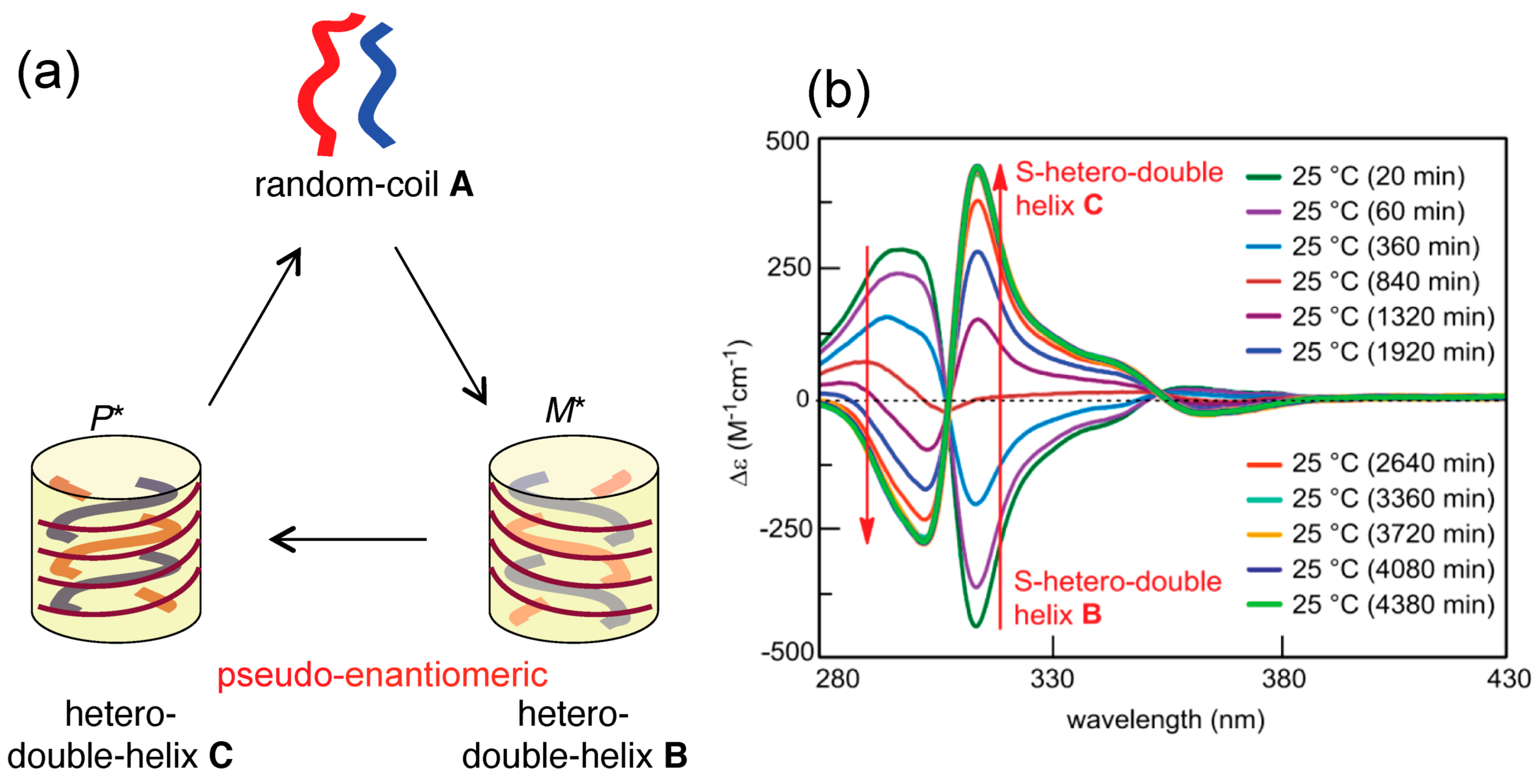
2.3. Dynamics of Double-Helix Chiral Cylindrical Molecular Complex Formation
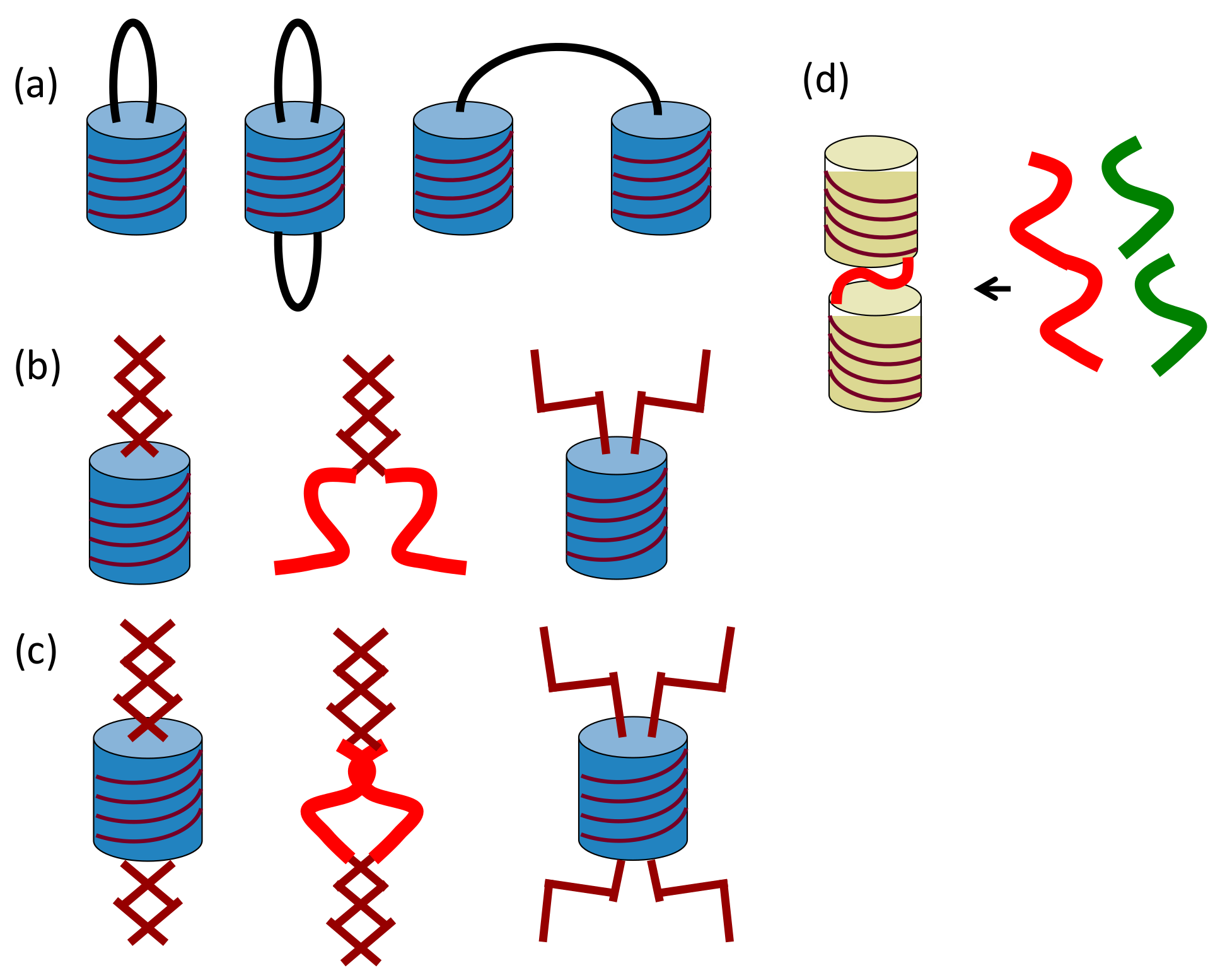
2.4. Self-Assembly of Double-Helix Chiral Cylindrical Molecular Complexes
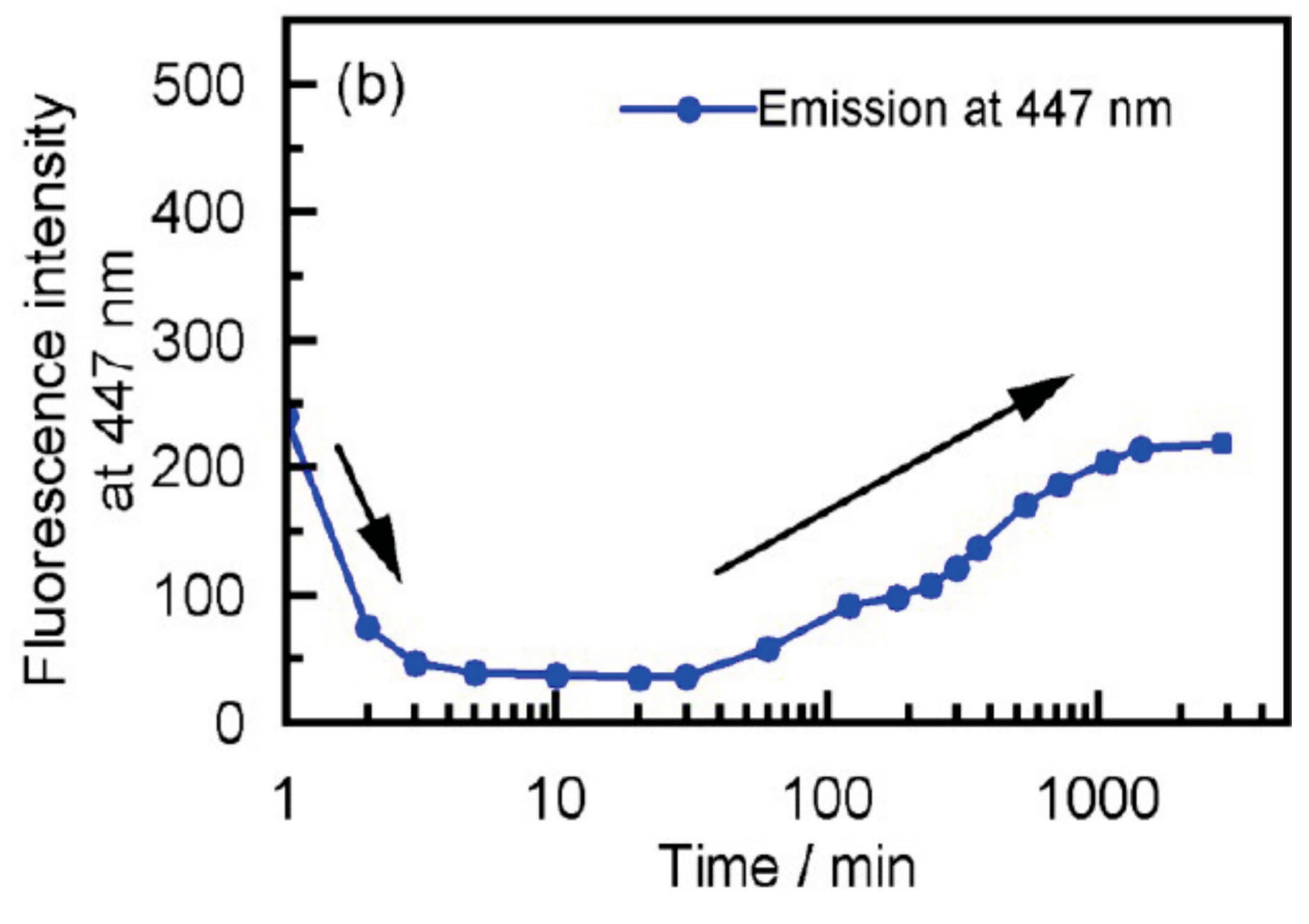
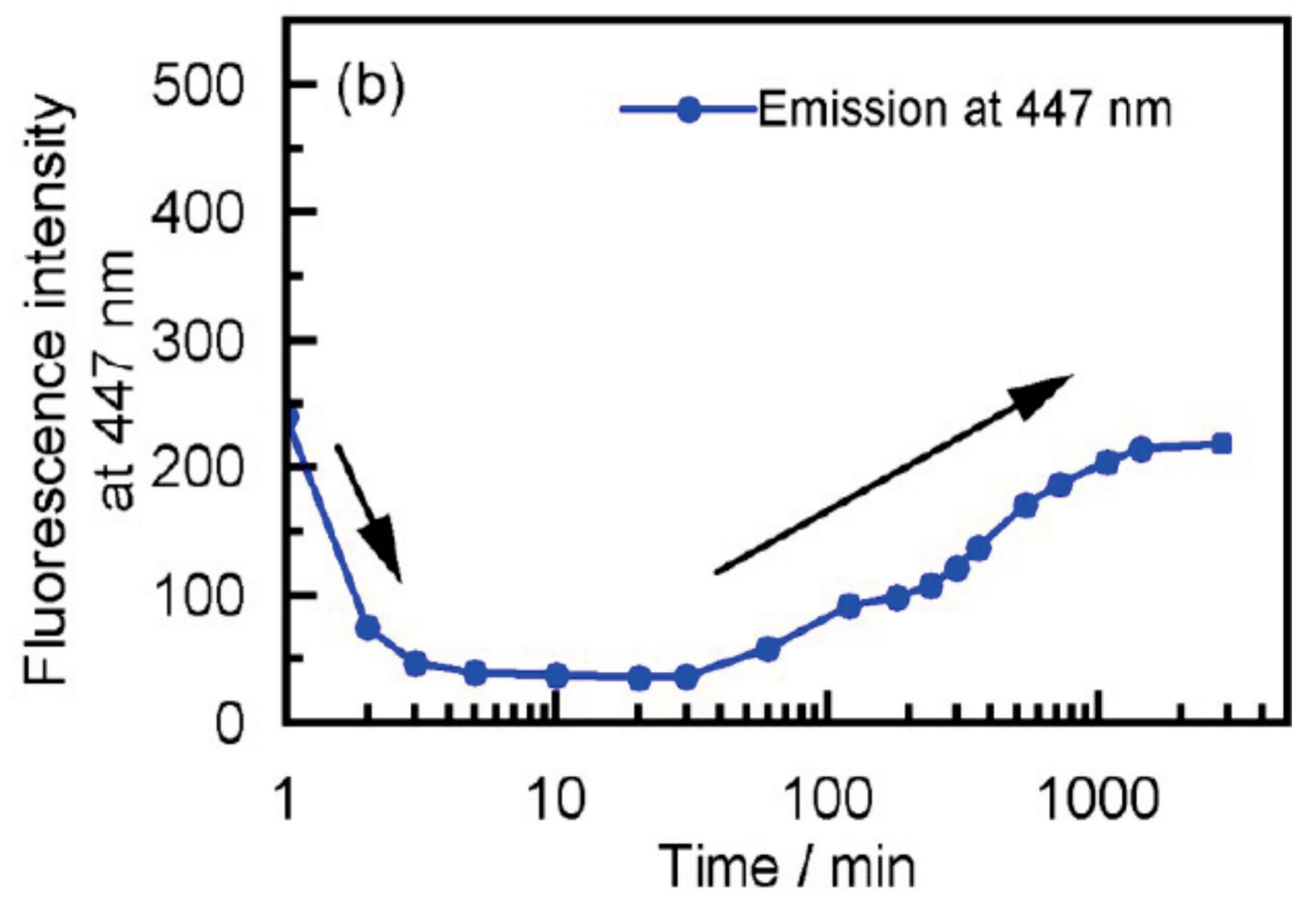
2.5. Dynamics in Self-Assembly of Double-Helix Chiral Cylindrical Molecular Complexes
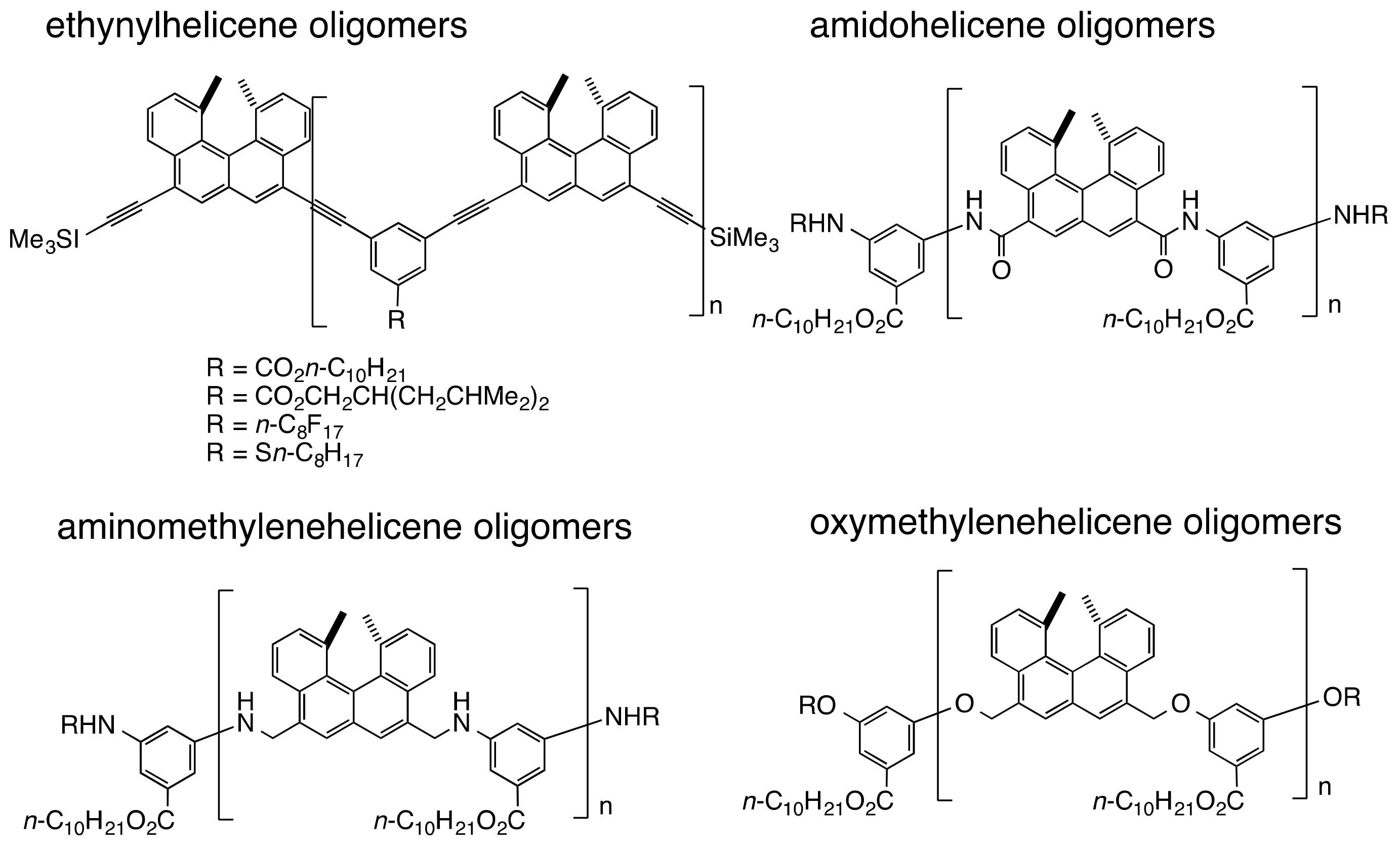
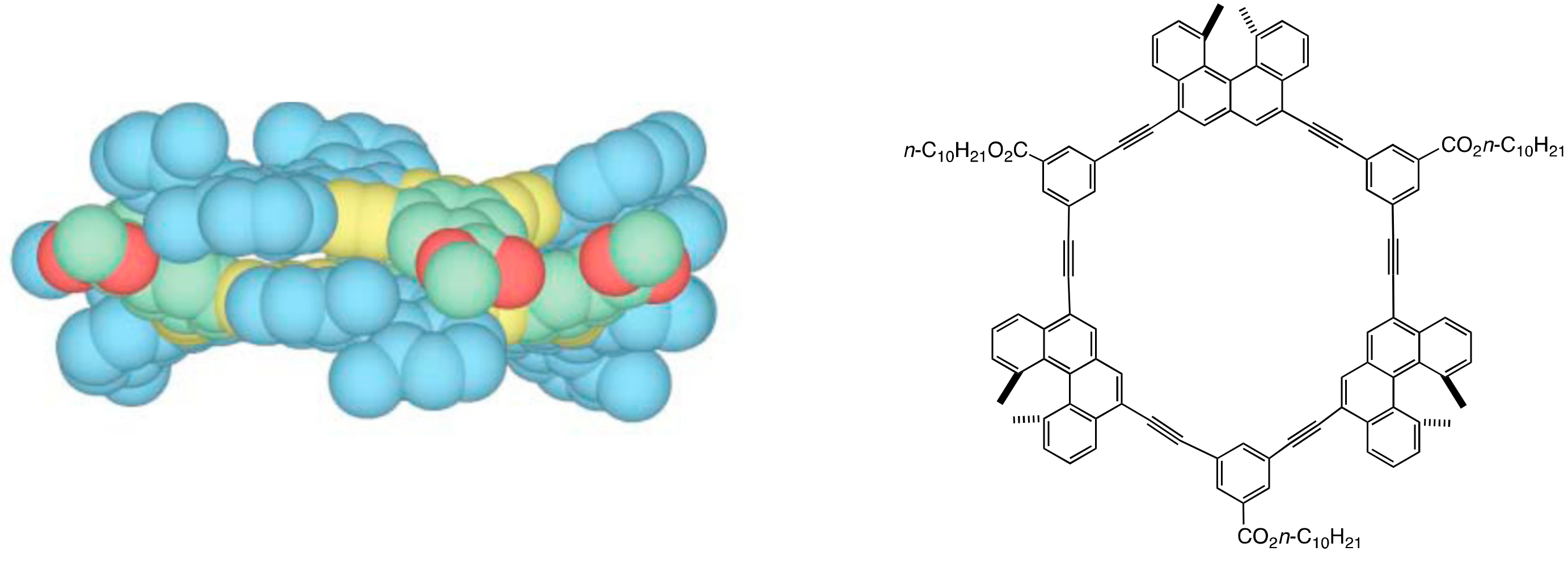
3. Double-Helix Chiral Cylindrical Molecular Complexes
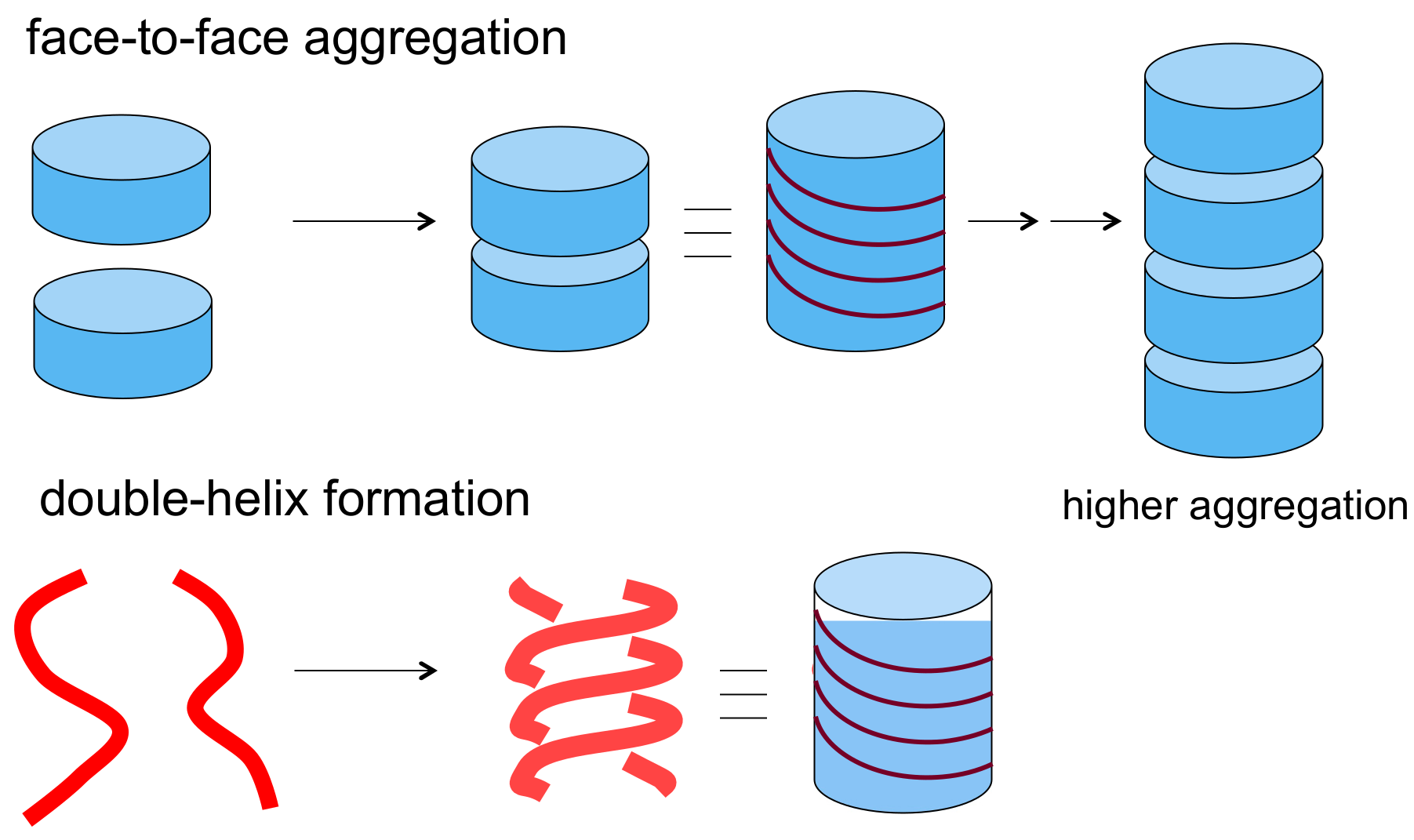
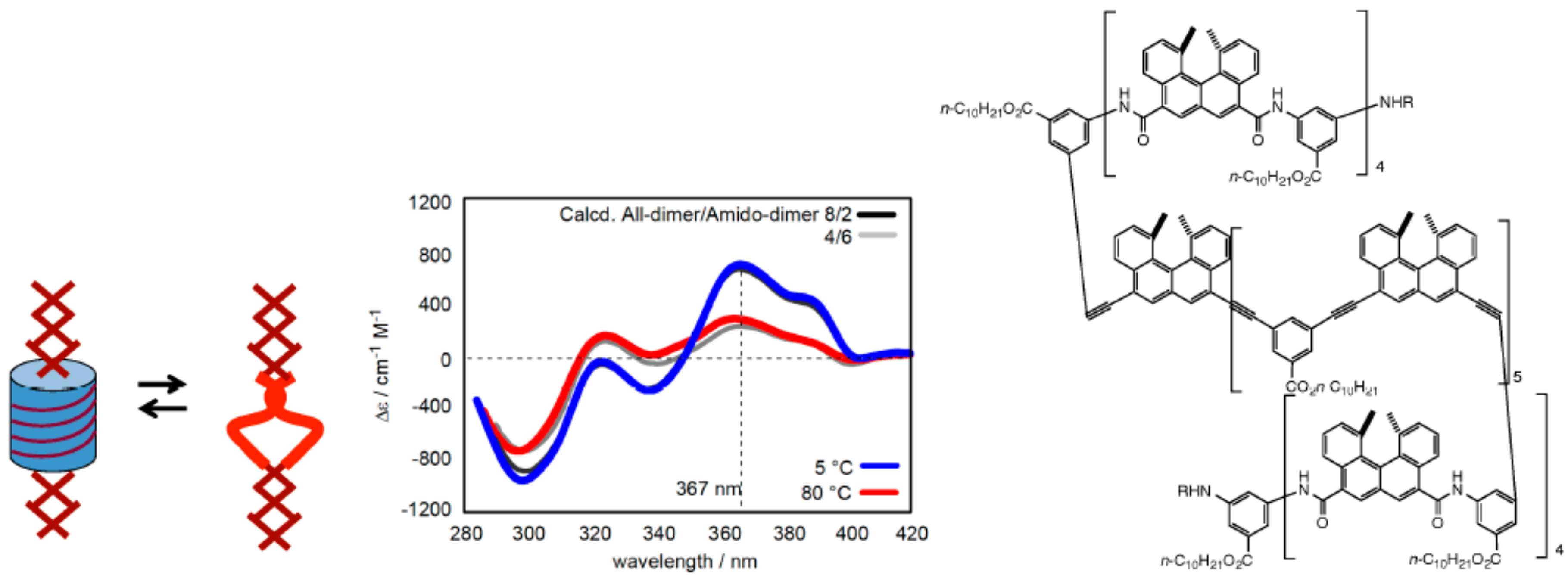
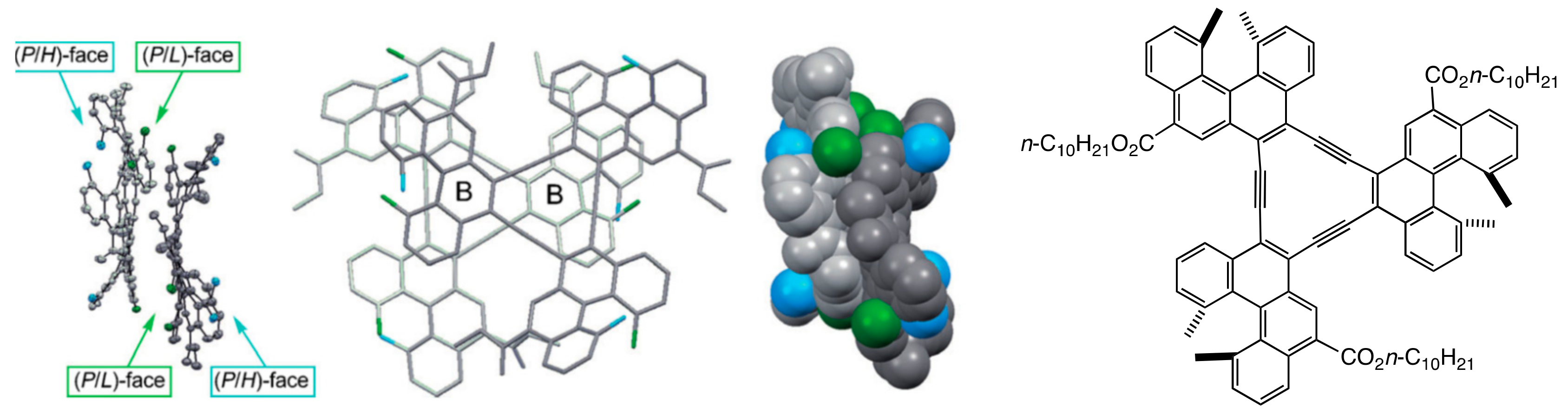
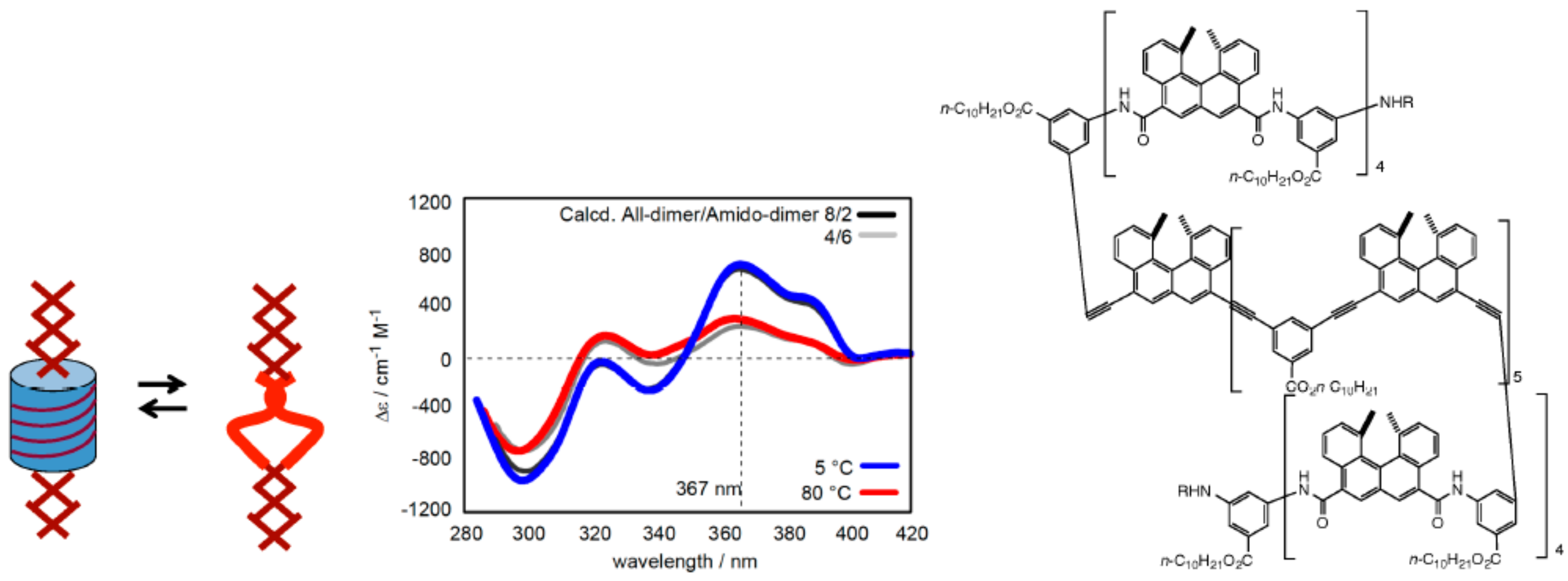
3.1. Face-to-Face Dimeric Aggregation of Cyclic Ethynylhelicene Oligomers
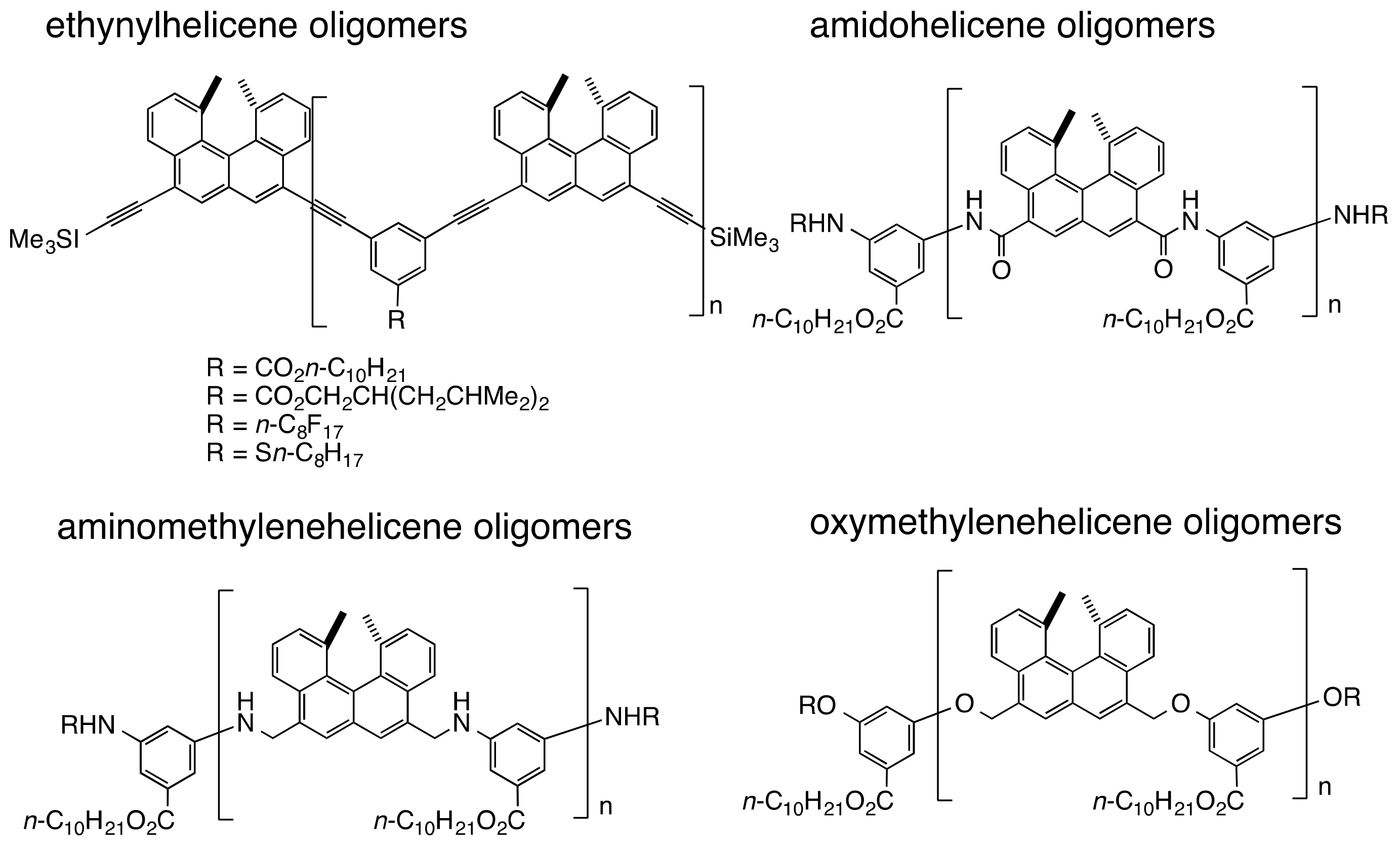
3.2. Double-Helix Formation by Ethynylhelicene Oligomers
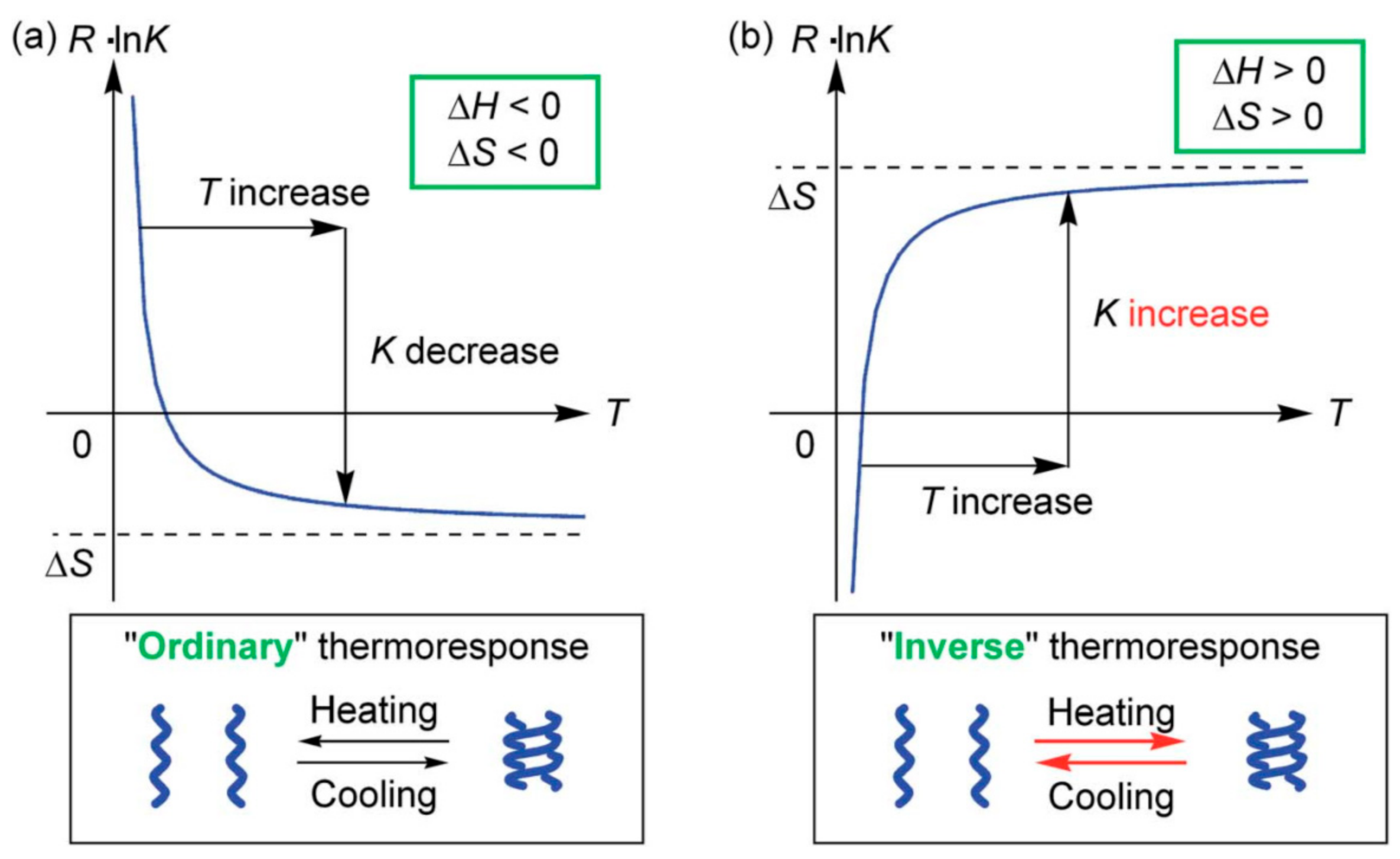
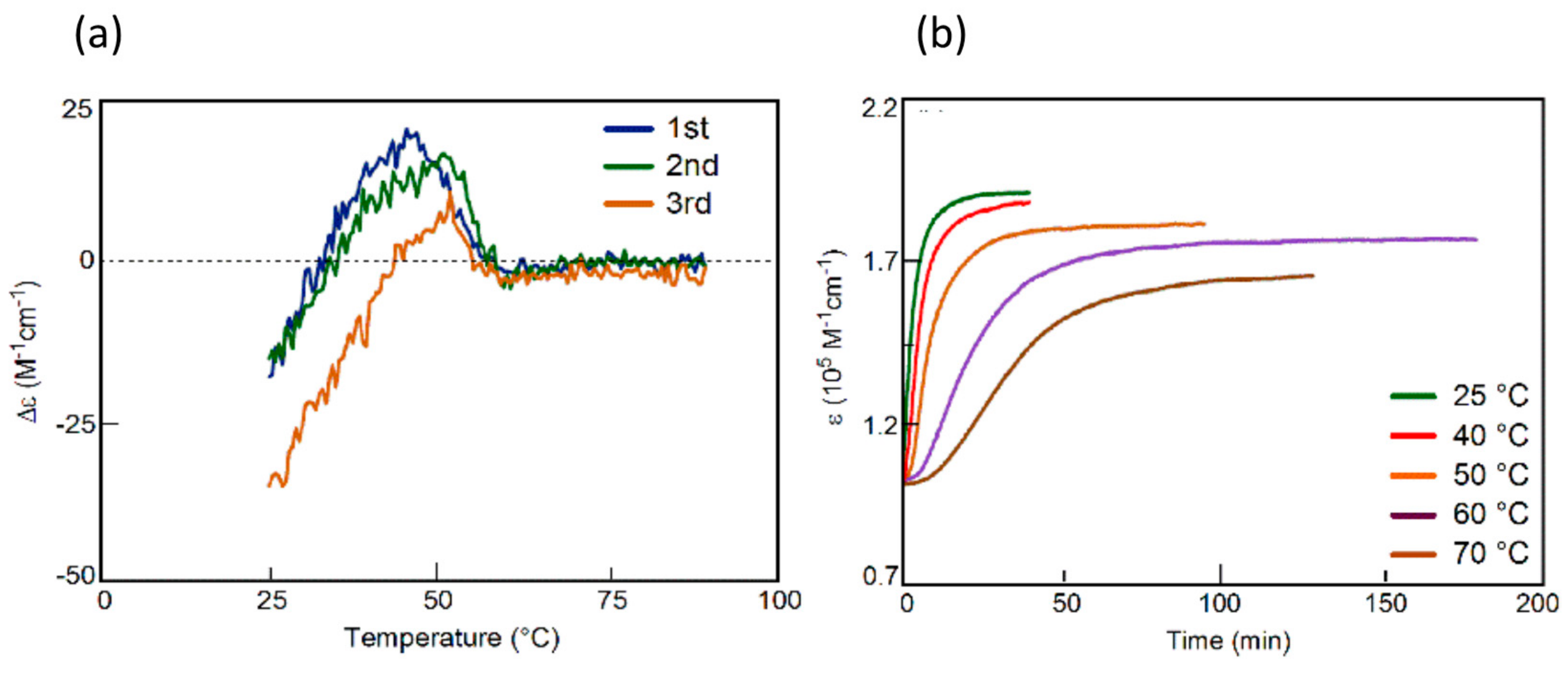
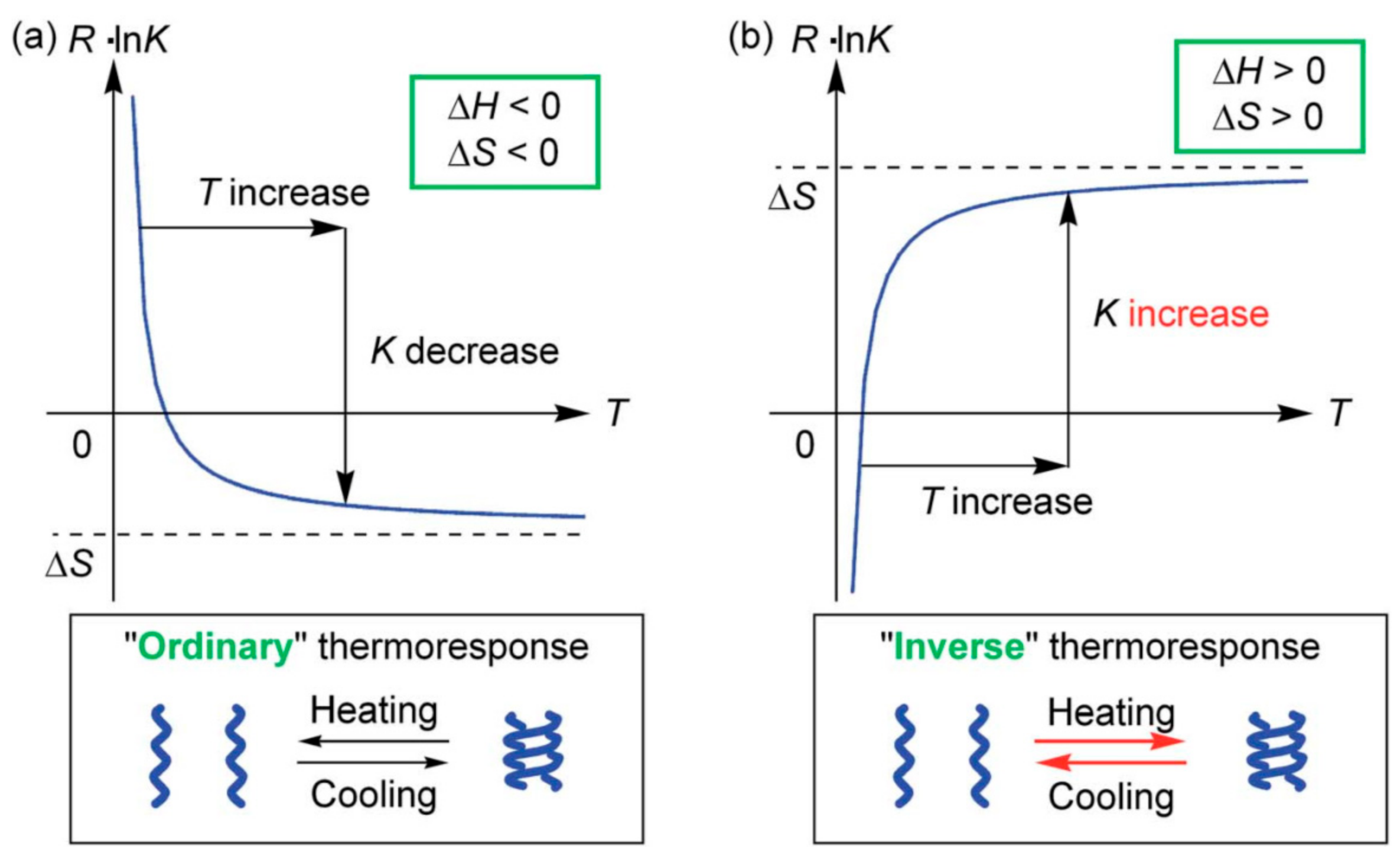
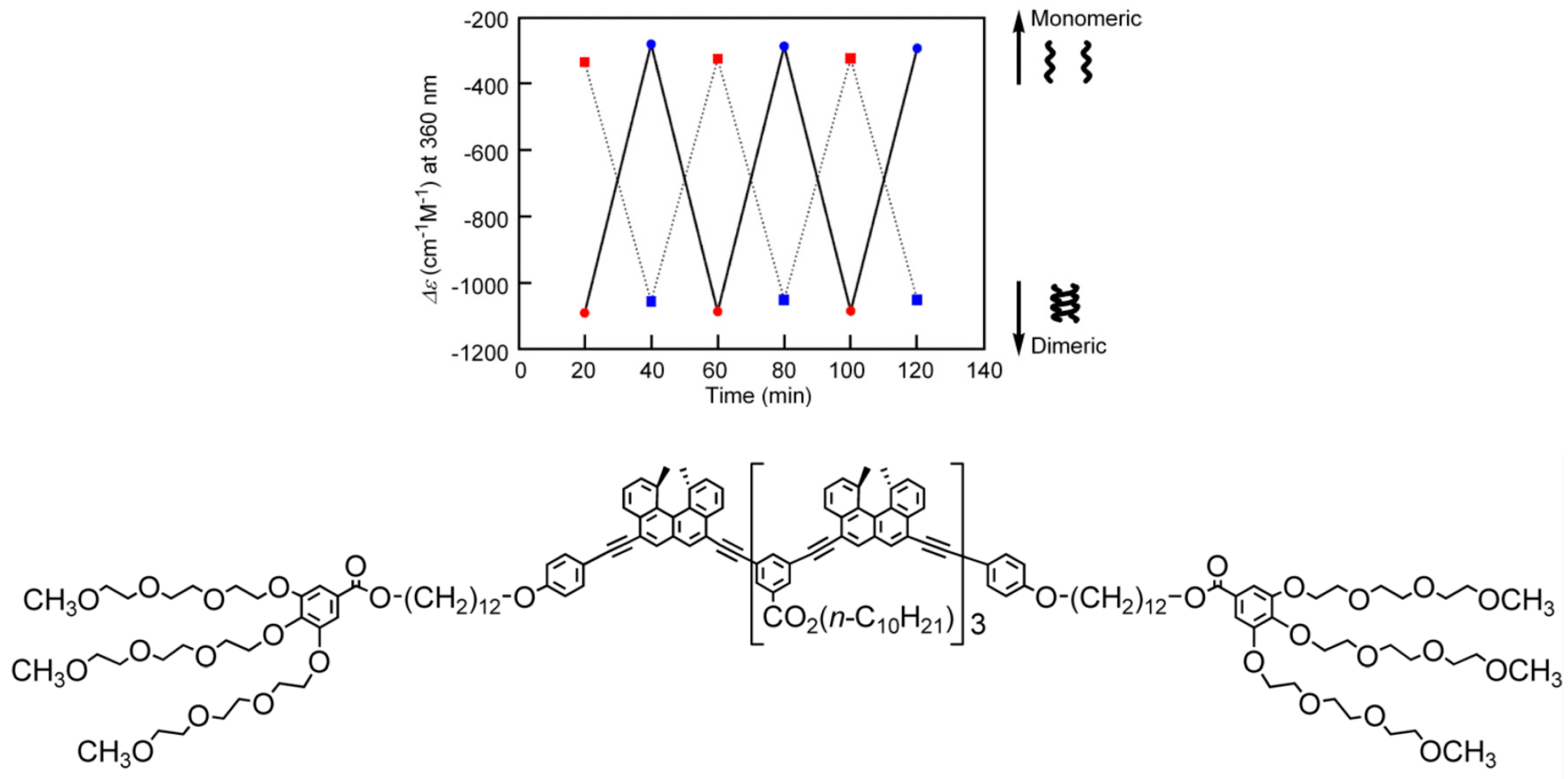
3.3. Thermo Response of Ethynylhelicene Oligomers
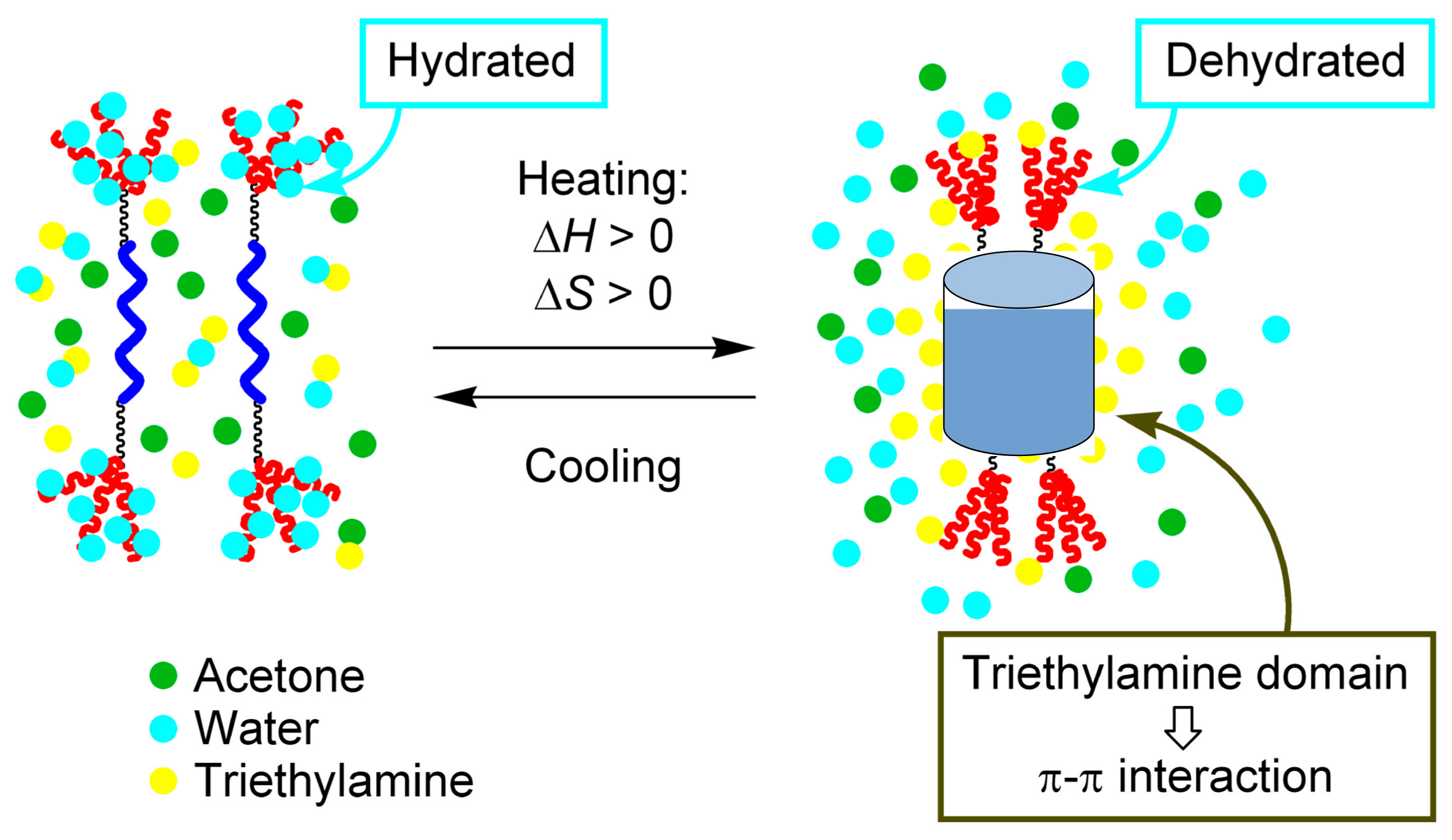
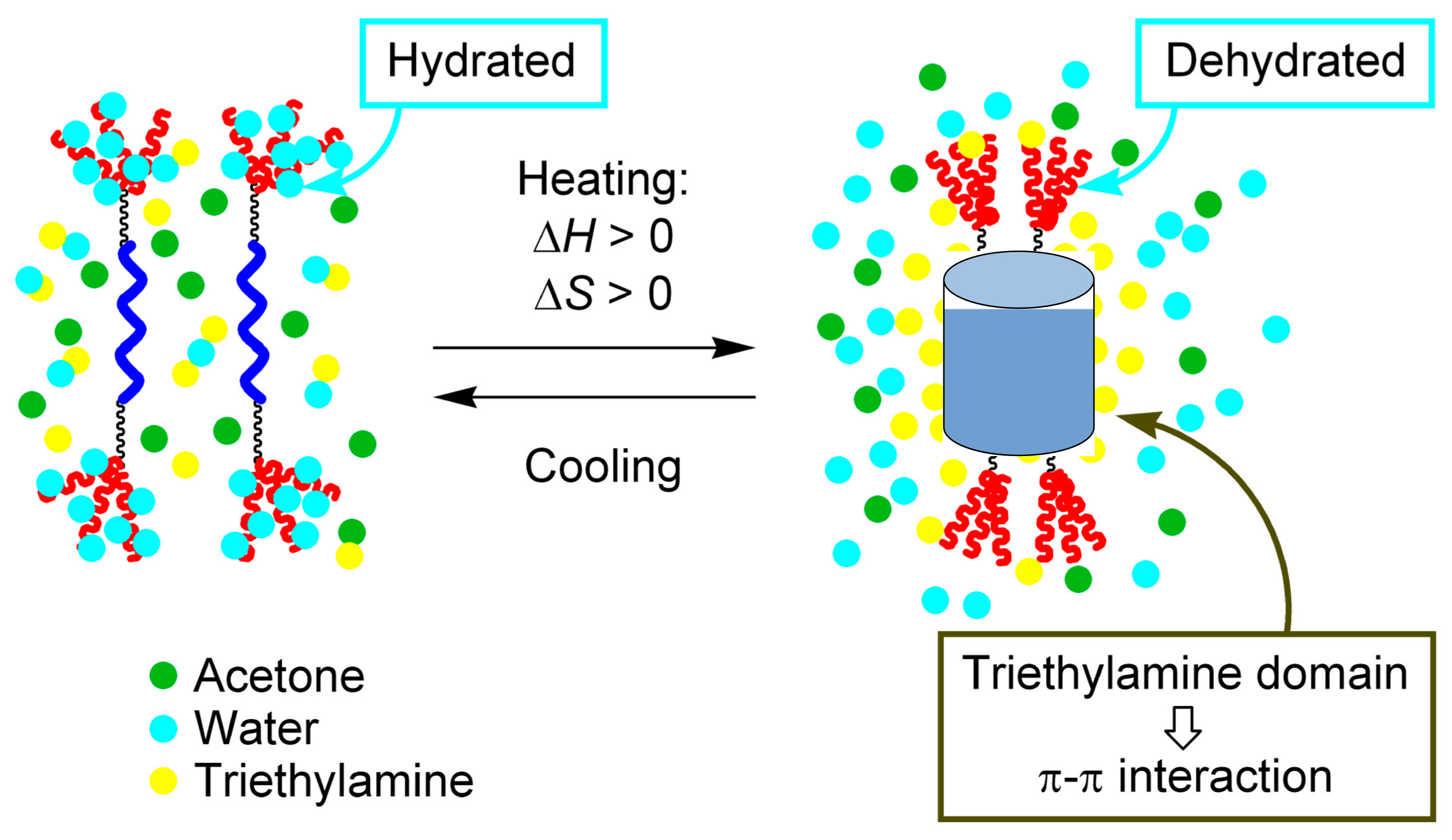
3.4. Inverse Thermo Response of Ethynylhelicene Oligomer with Terminal PEG Groups
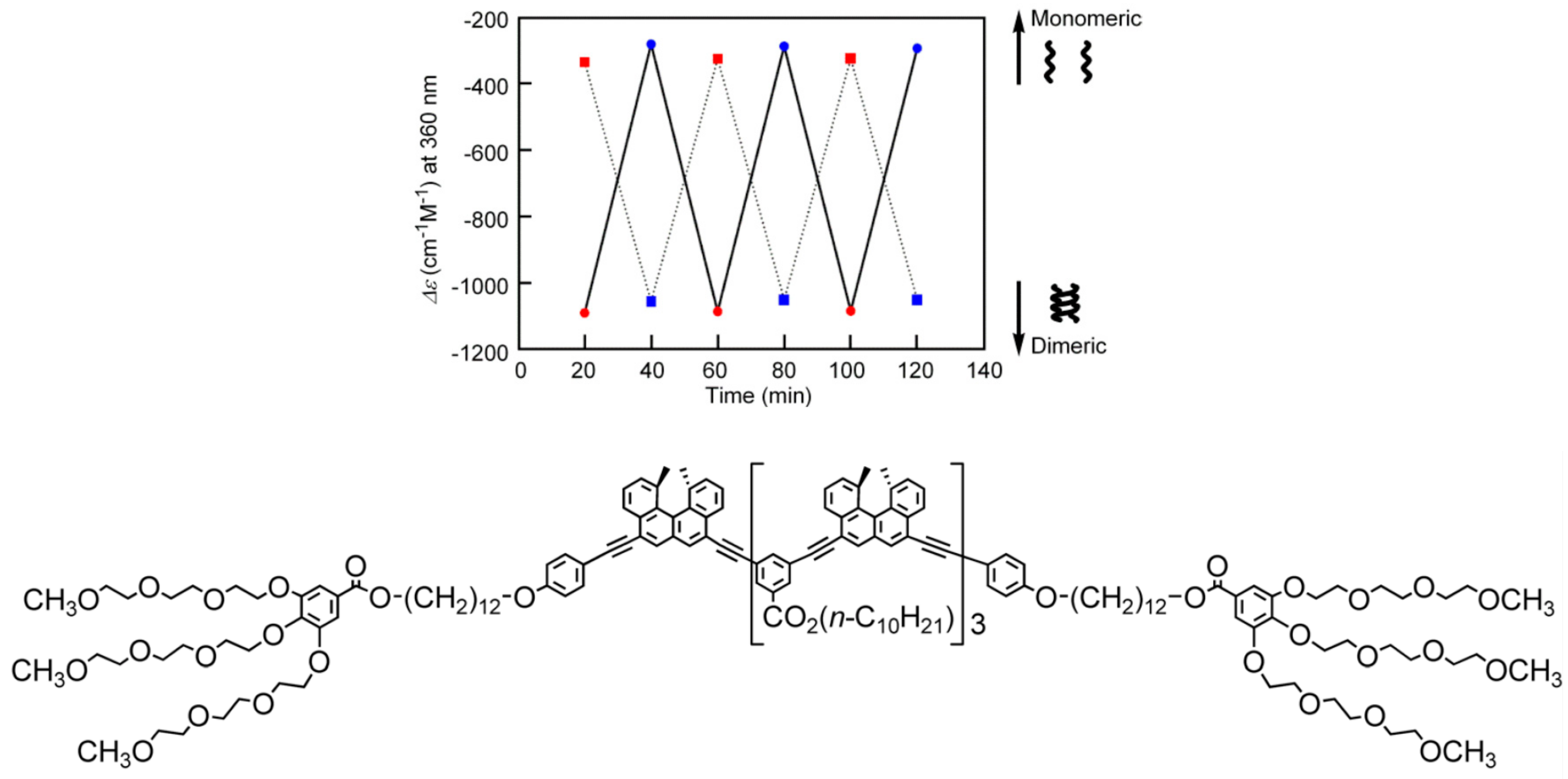
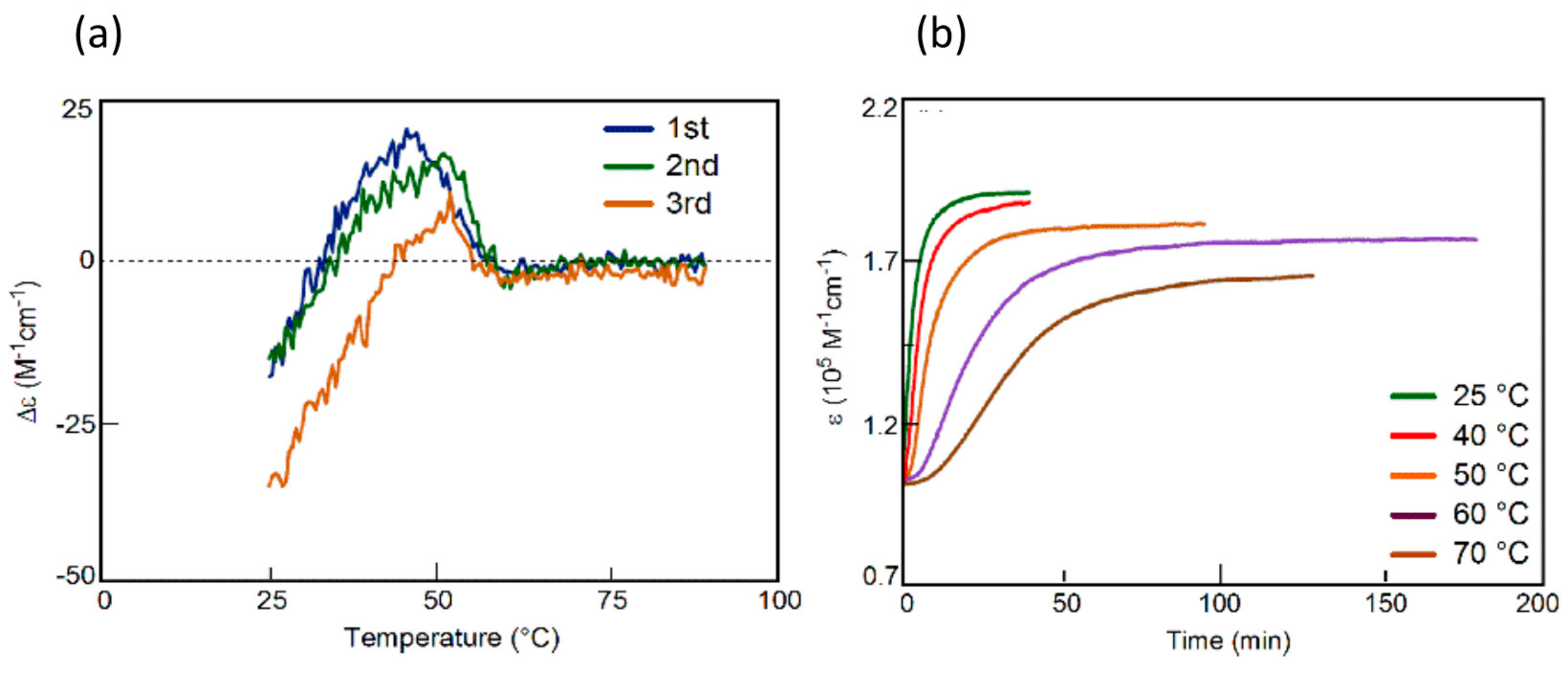
3.5. Thermo Response of Linked and Multi-Domain Ethynylhelicene Oligomers
3.6. Double-Helix Formation by Aminomethylenehelicene and Oxymethylenehelicene Oligomers
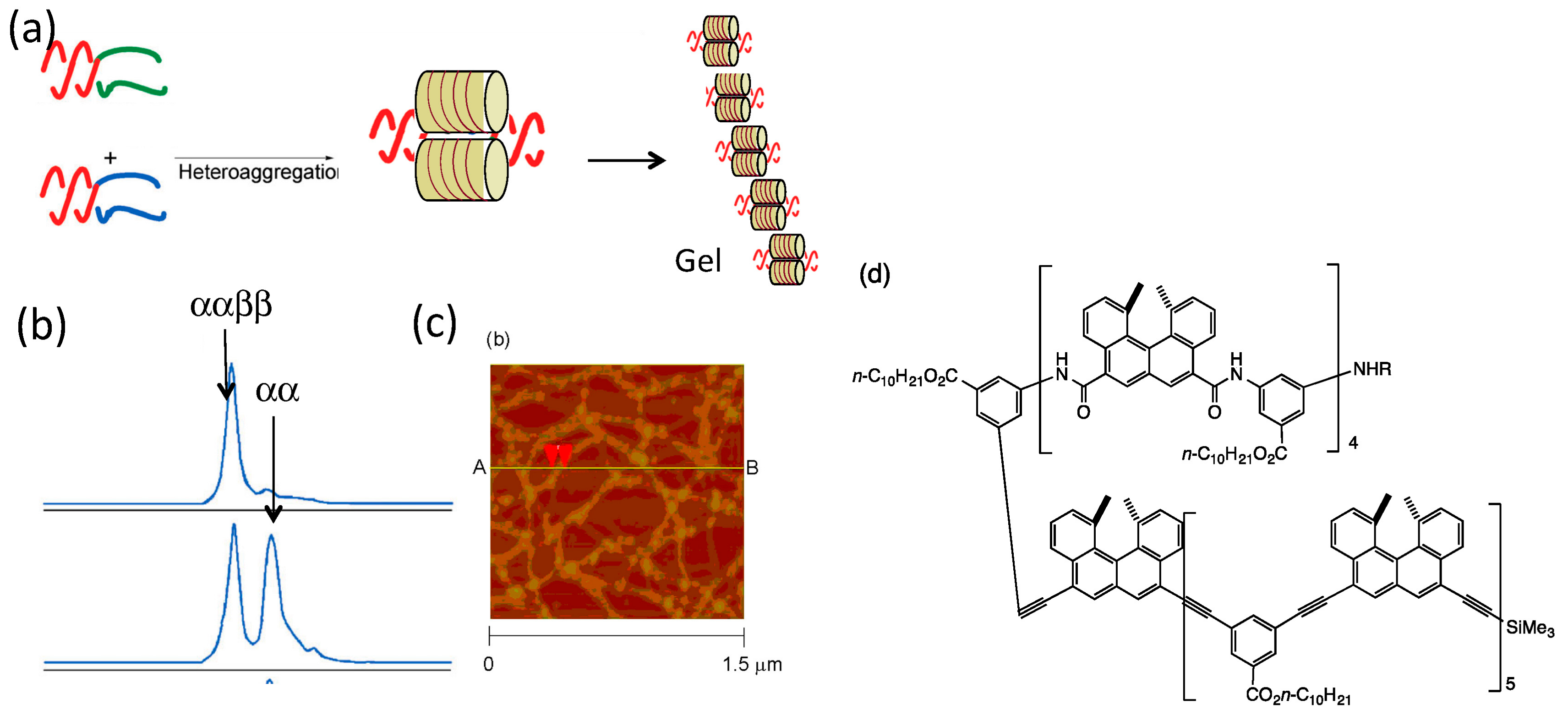
4. Hetero-Double-Helix Cylindrical Molecular Complexes
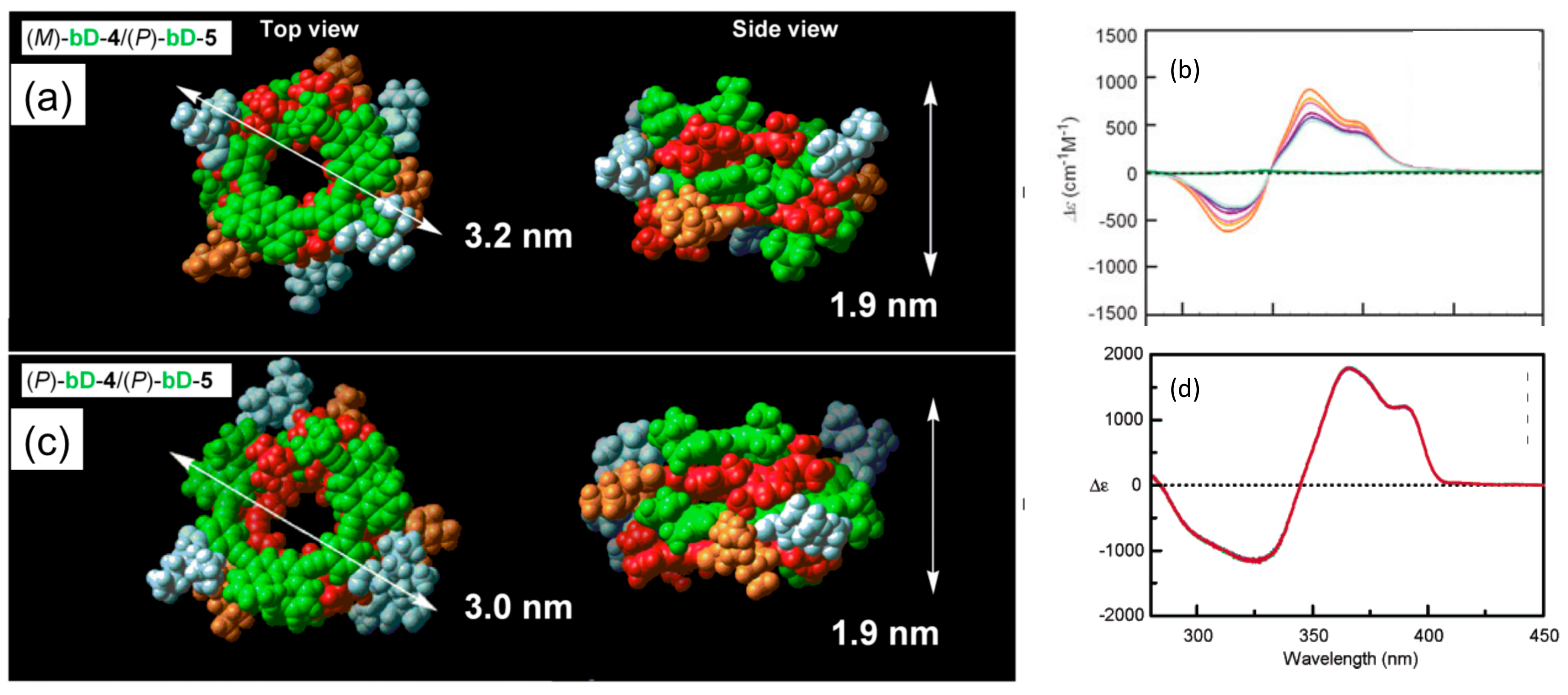
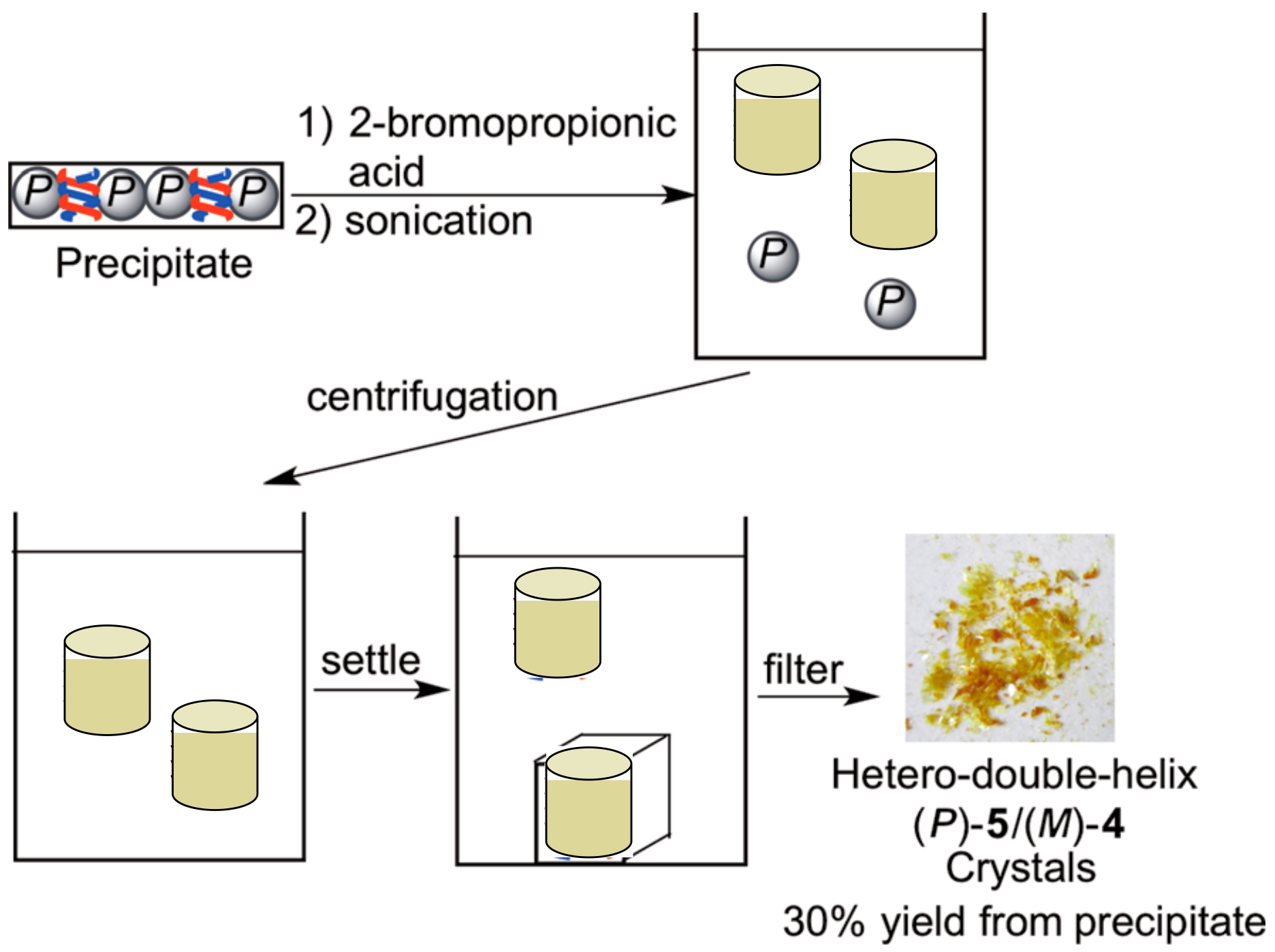
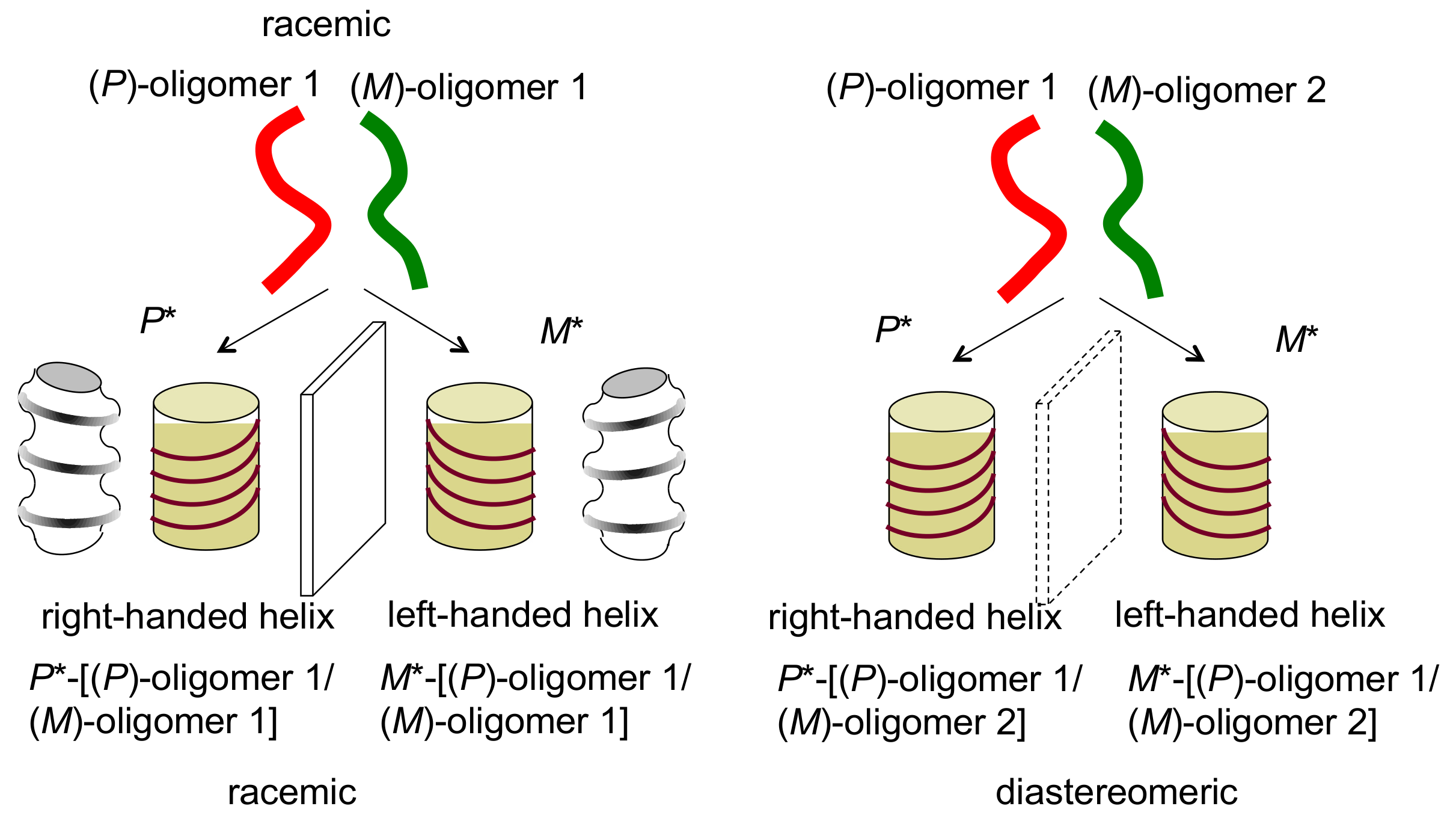
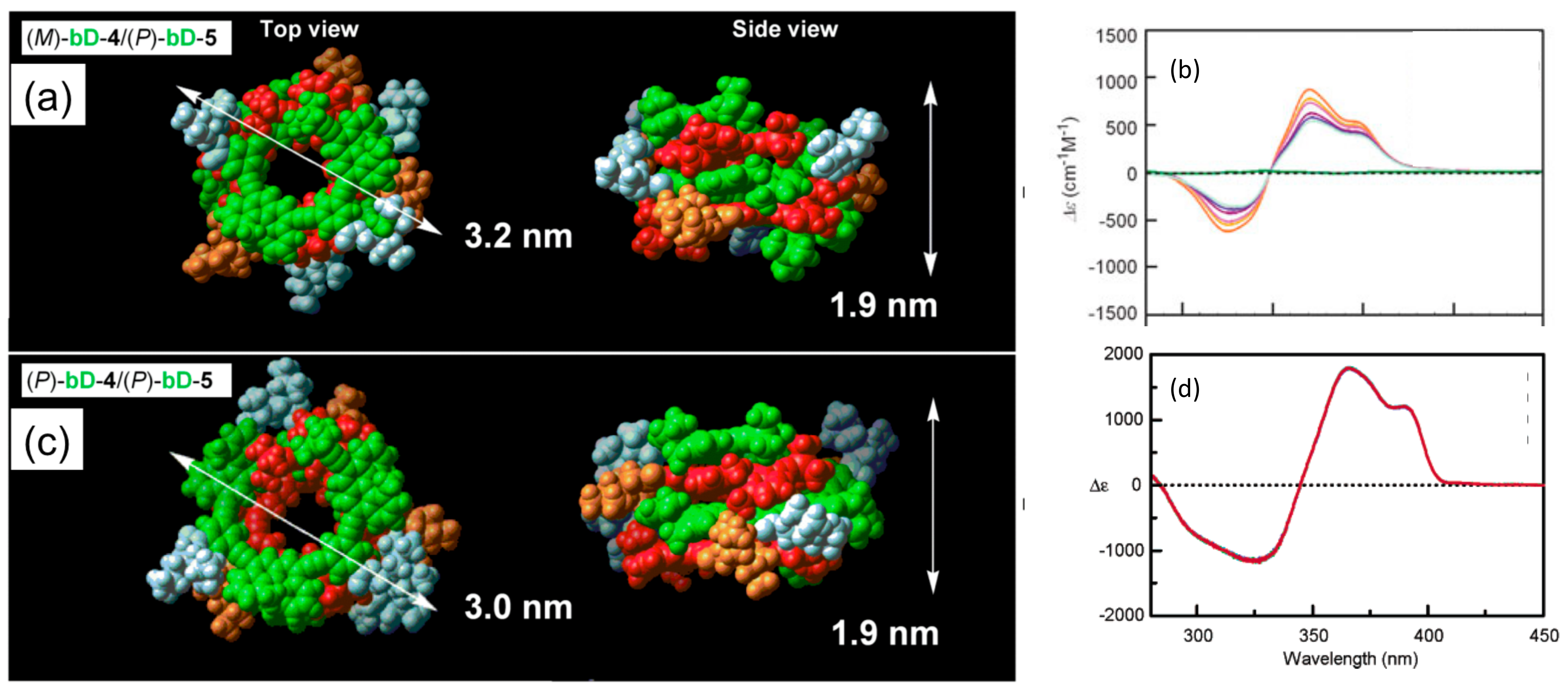
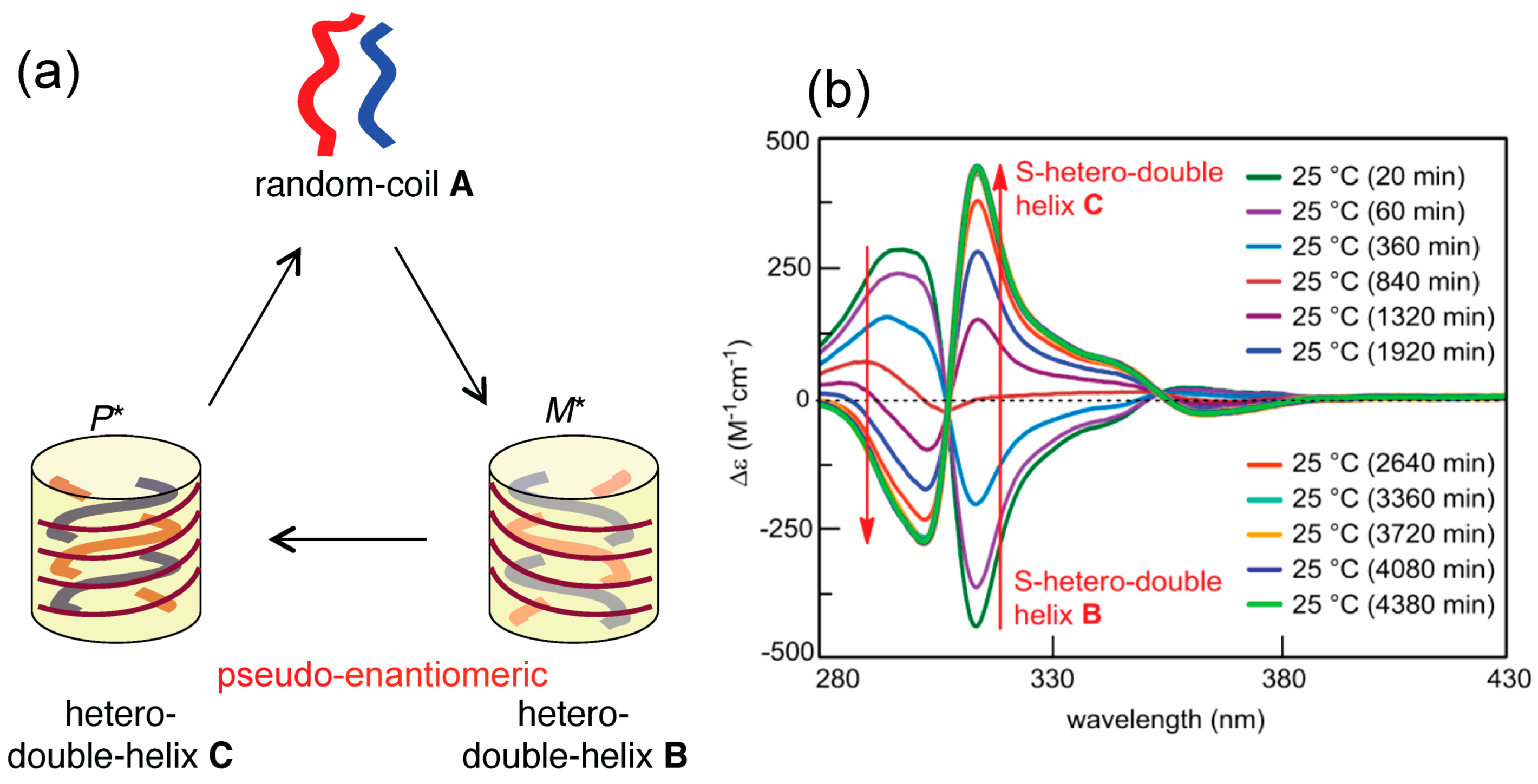
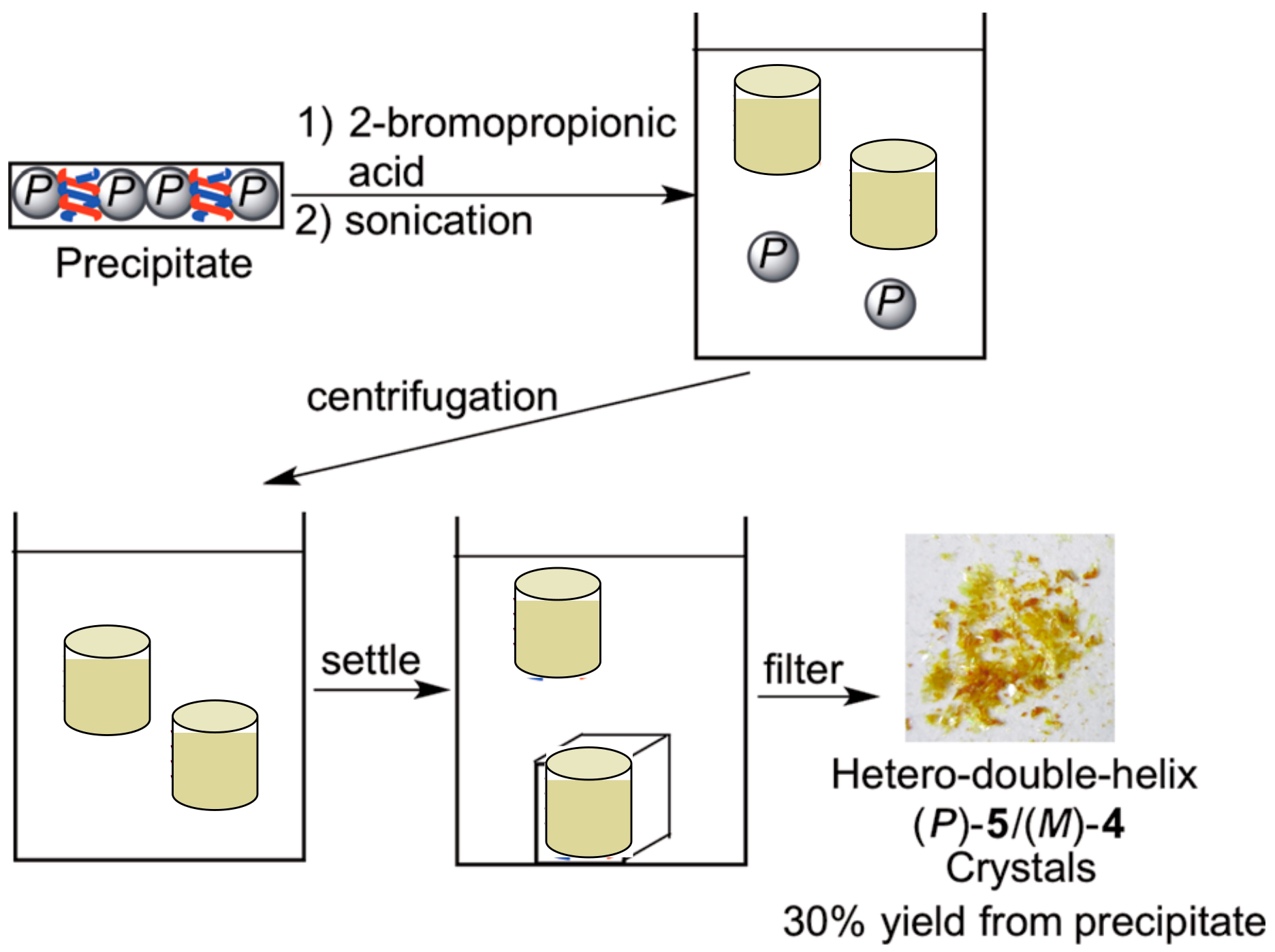
4.1. Hetero-Double-Helix Formation by Pseudo-Enantiomeric Ethynylhelicene Oligomers
4.2. Hetero-Double-Helix Formation from Pseudo-Enantiomeric Aminohelicene Oligomers
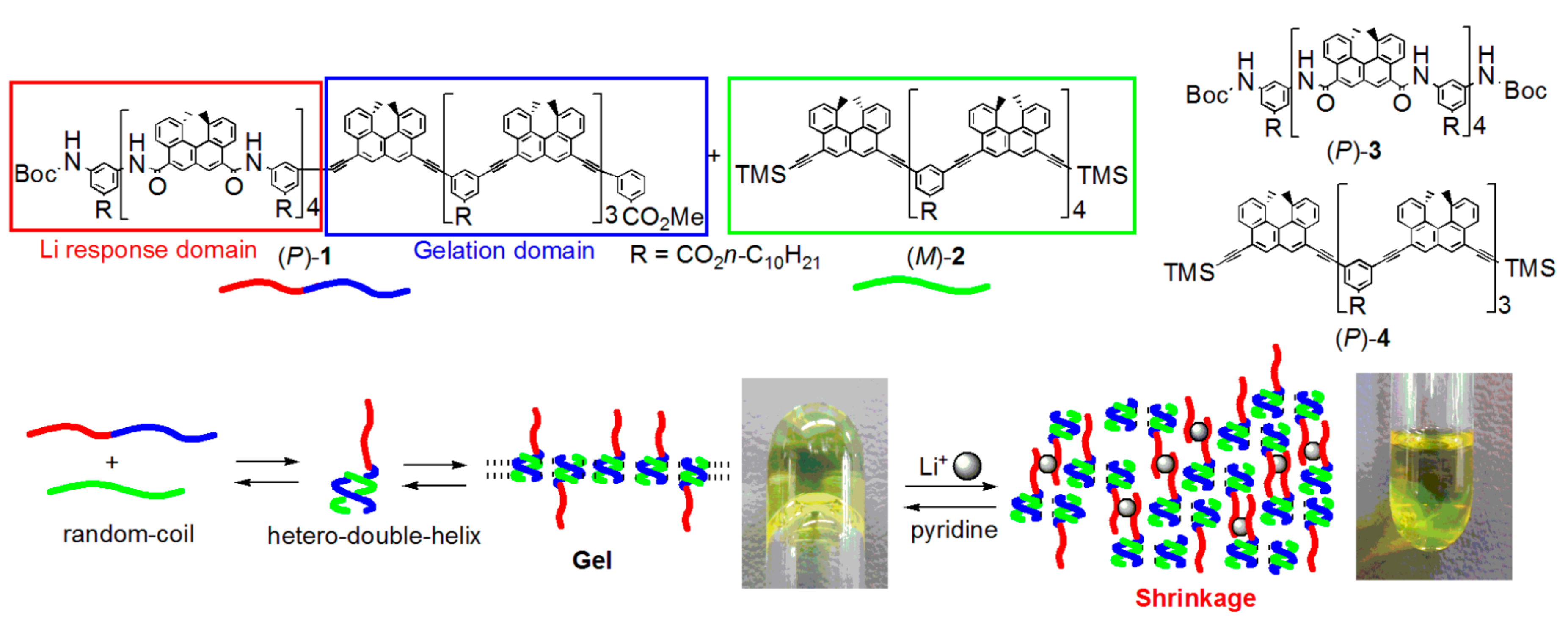
5. Self-Assembly Gel Formation by Hetero-Double-Helix Cylindrical Molecular Complexes
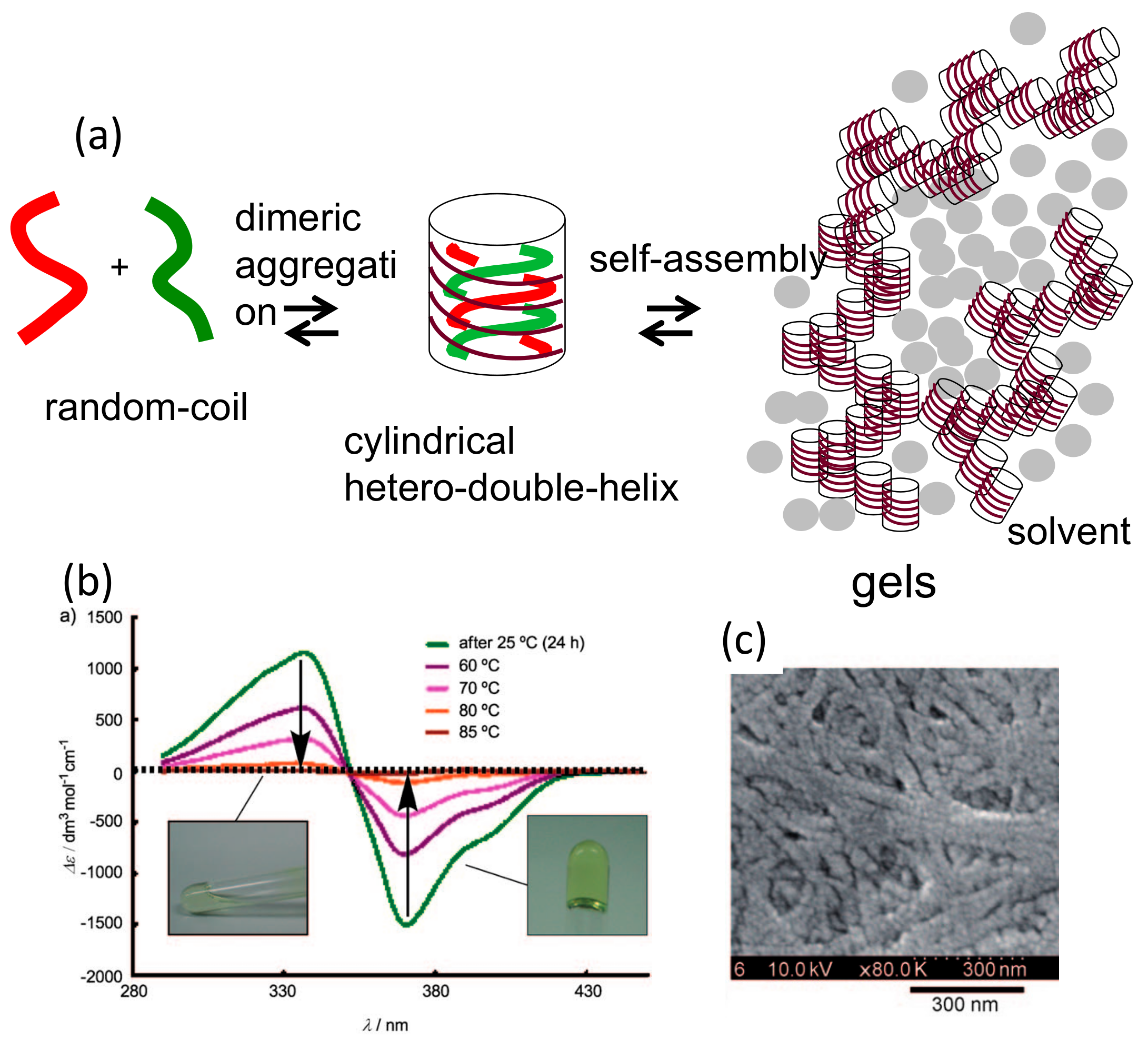
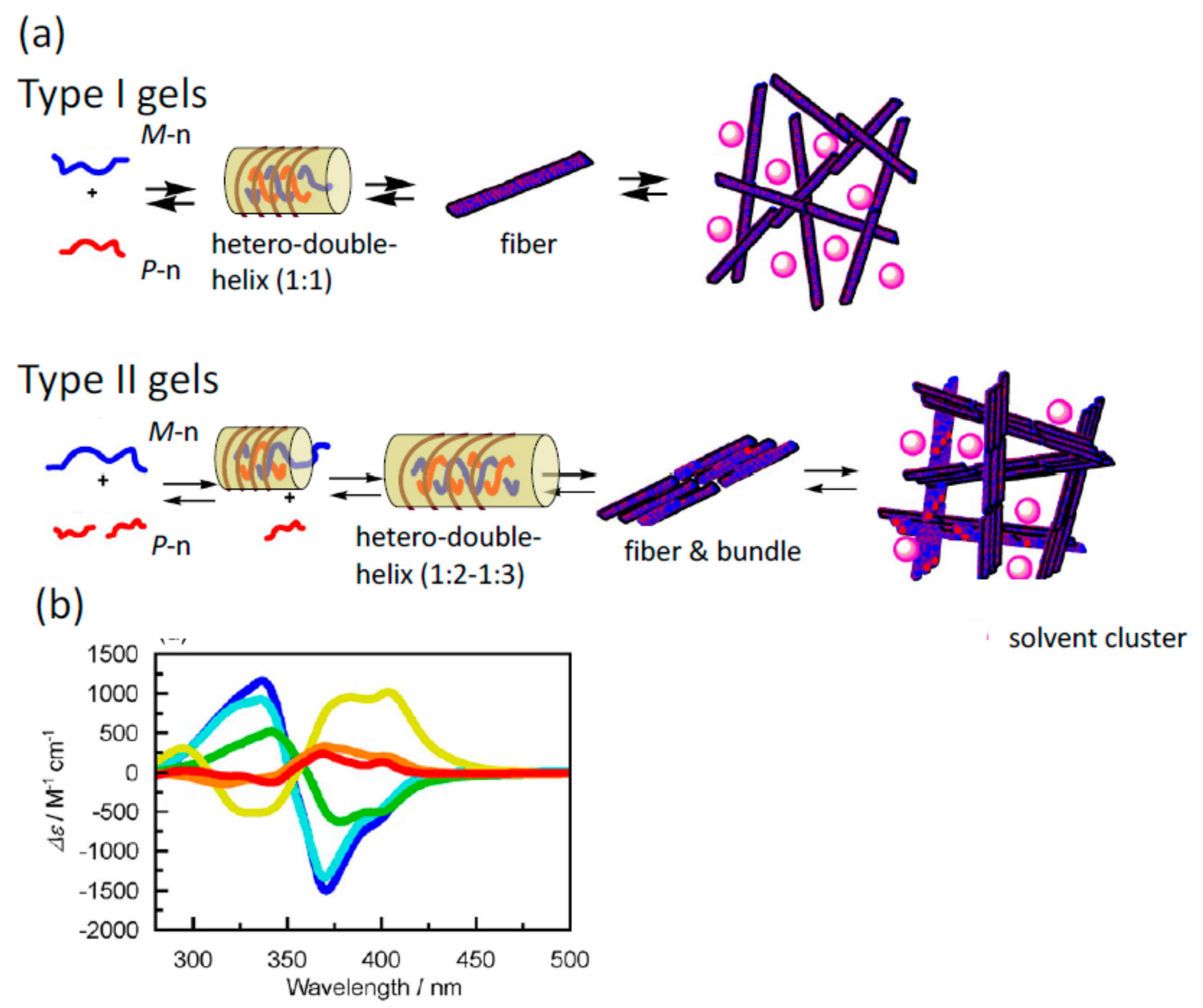
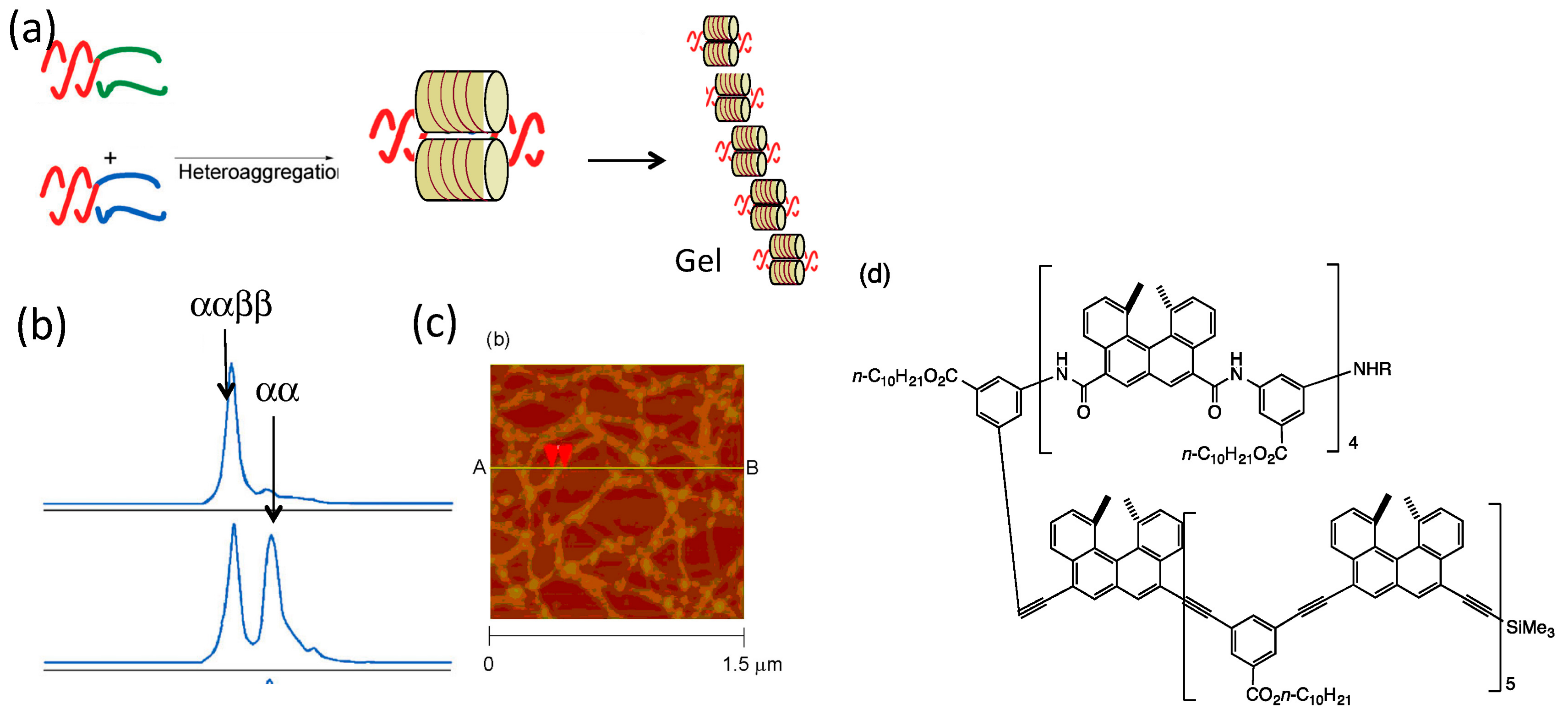
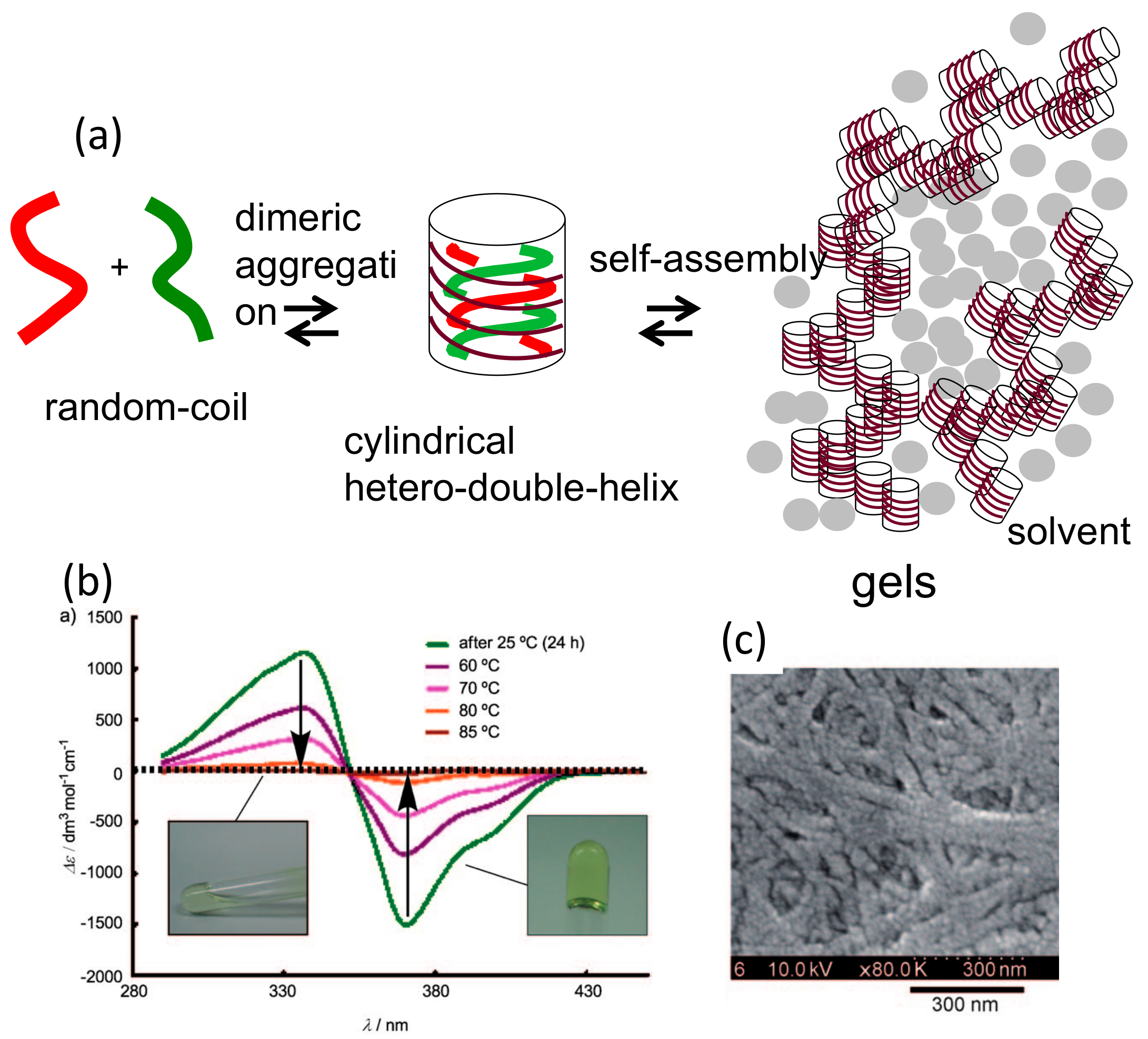
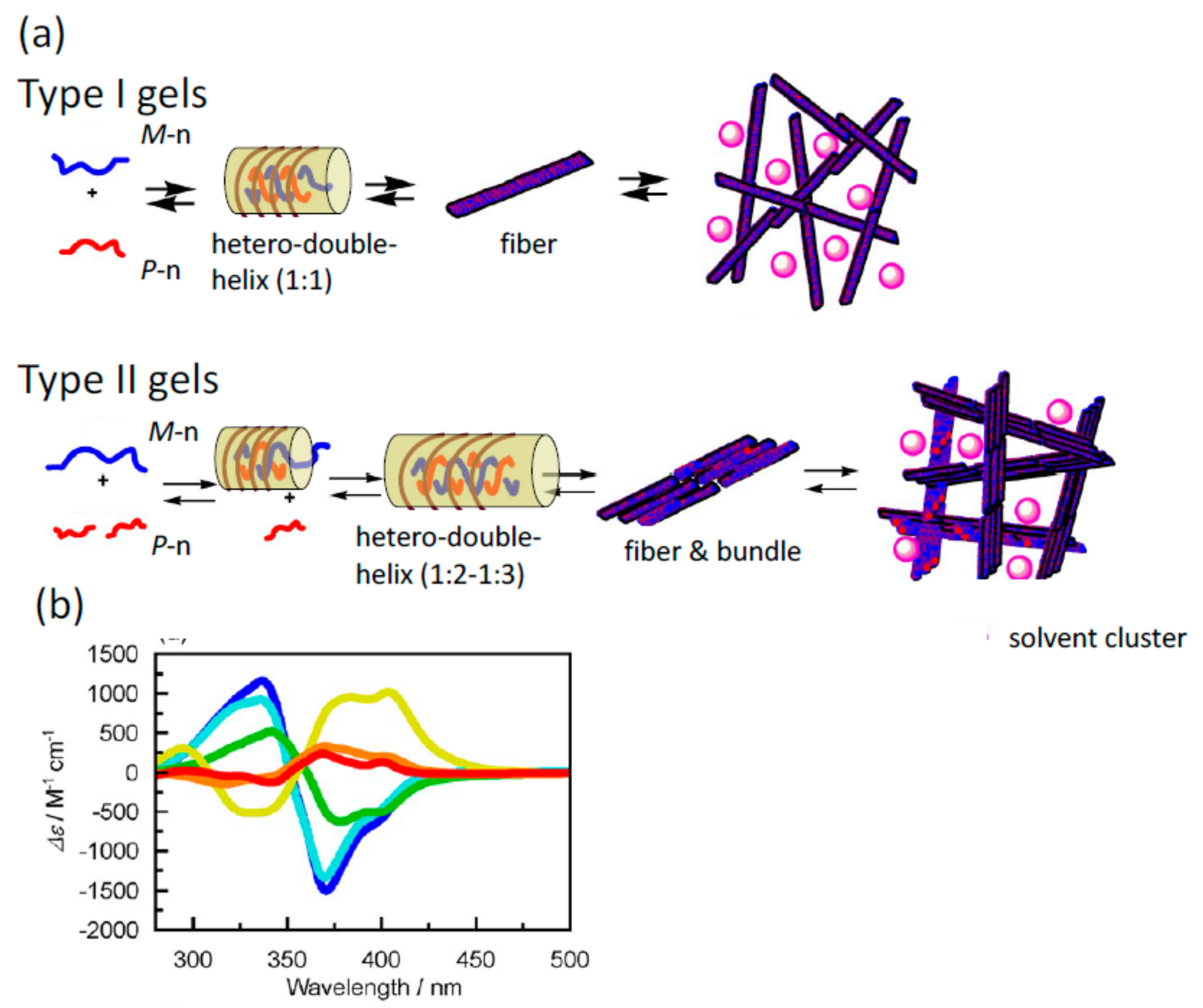
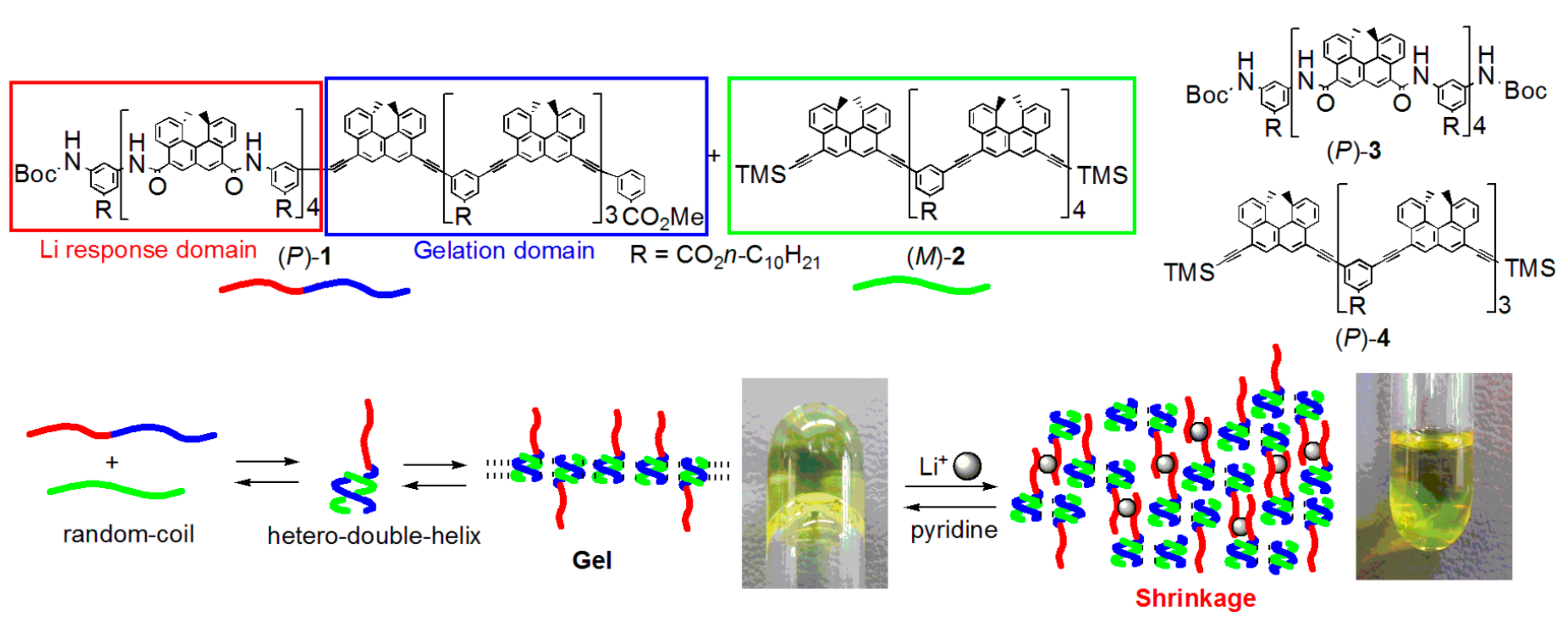
5.1. Self-Assembly Two-Component Gels of Heterogeneous Liquid-Solid Materials Derived from Pseudo-Enantiomeric Ethynylhelicene Oligomers
5.2. Gels Formed by Bi-Domain Oligomers
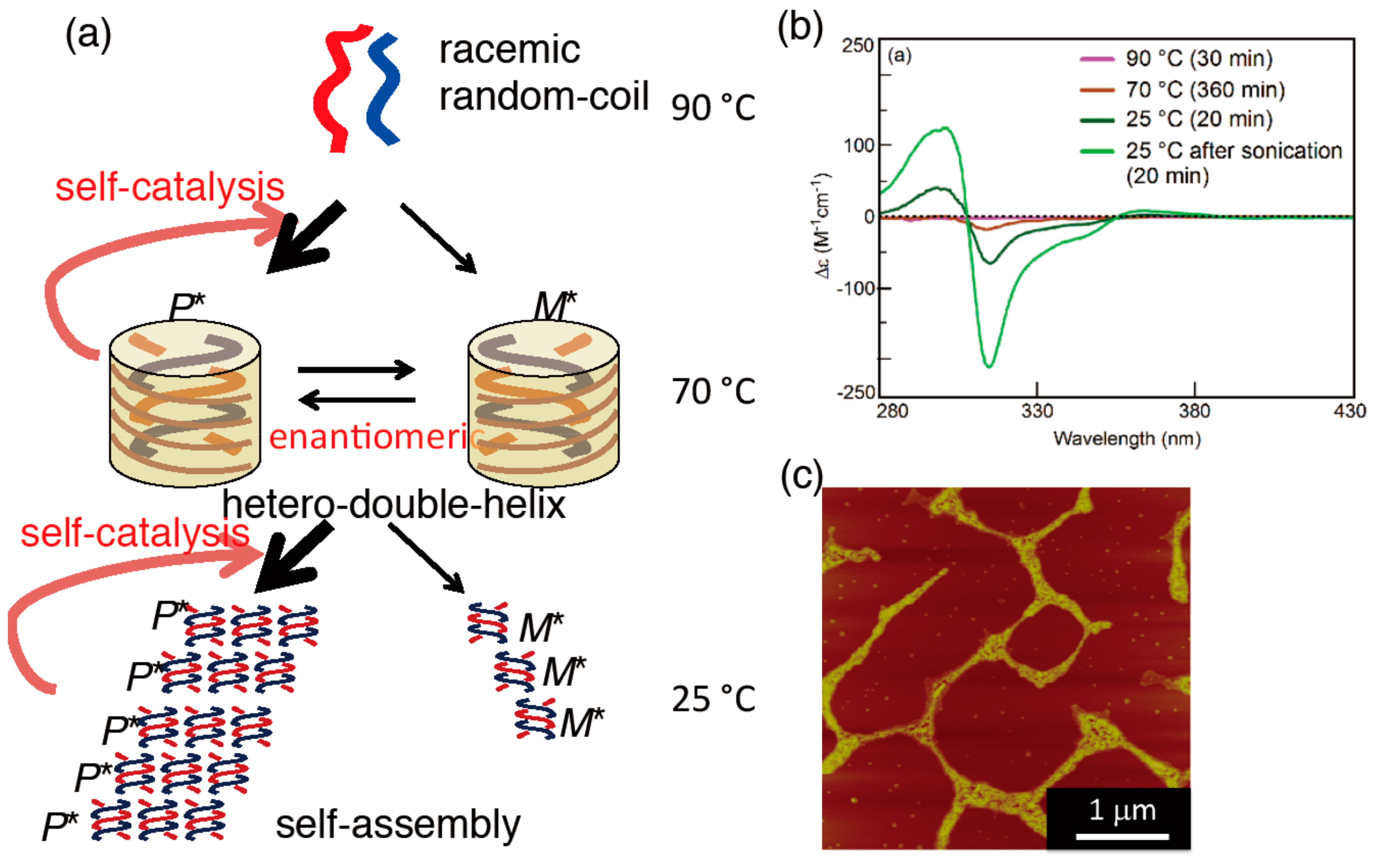
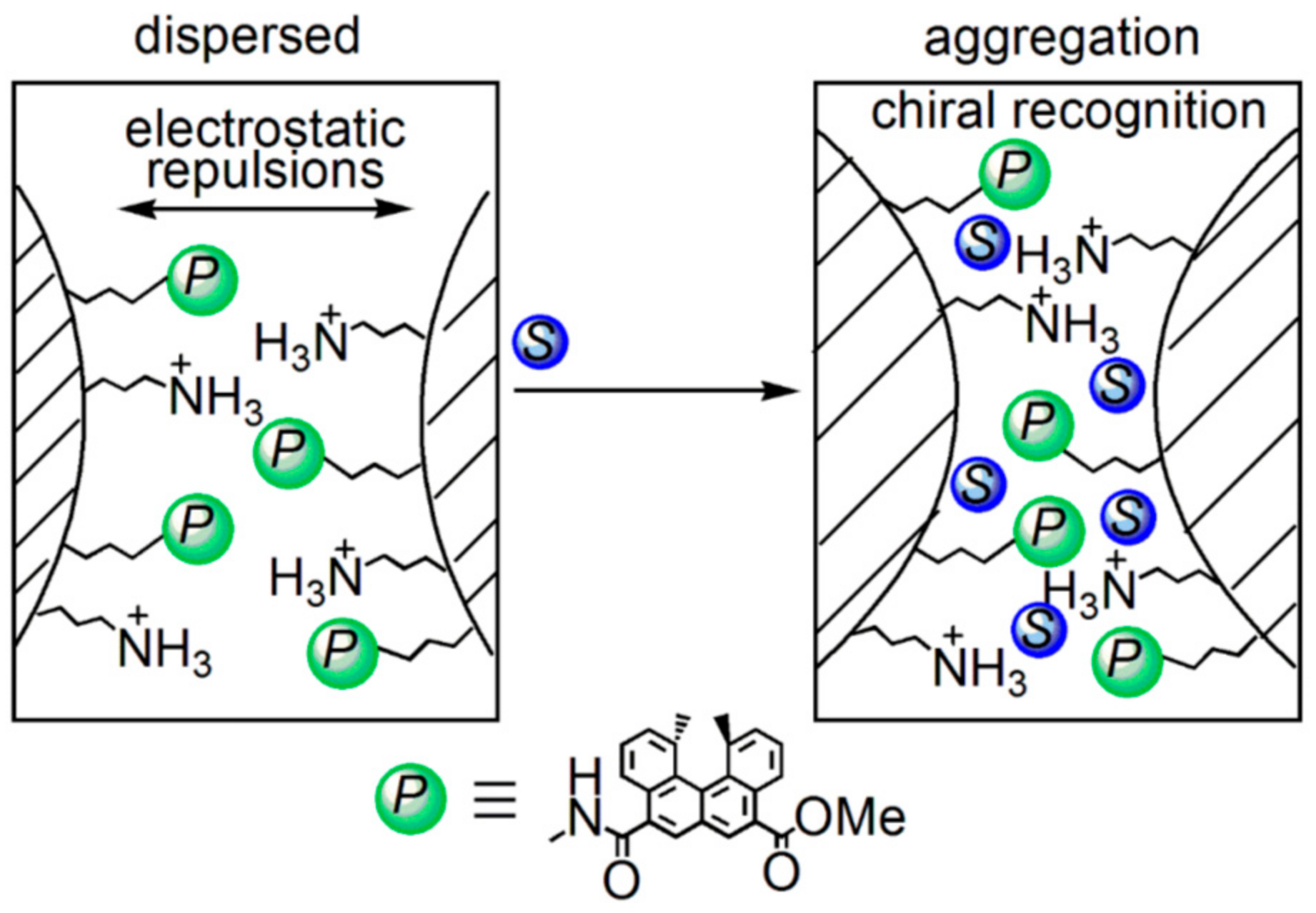
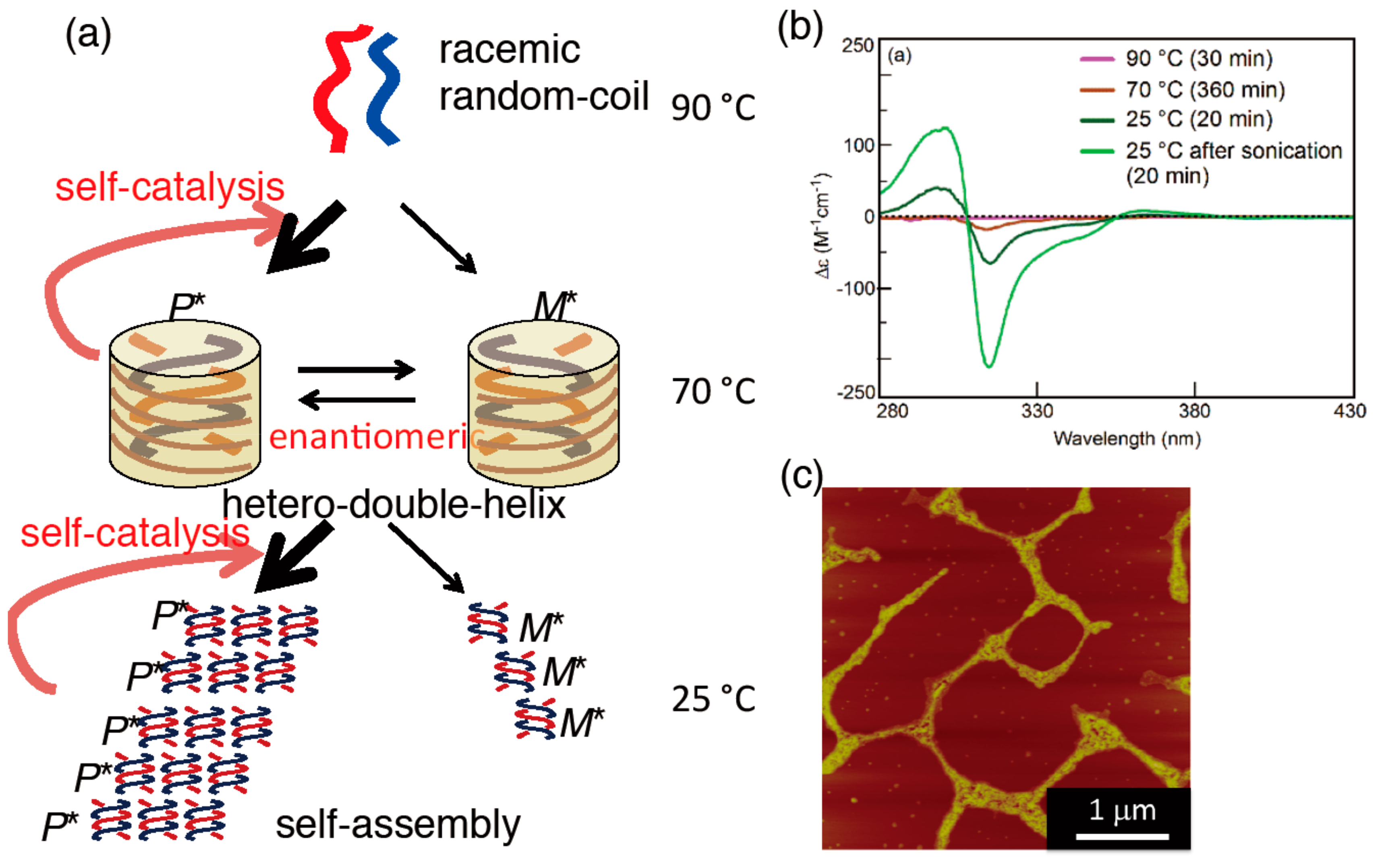
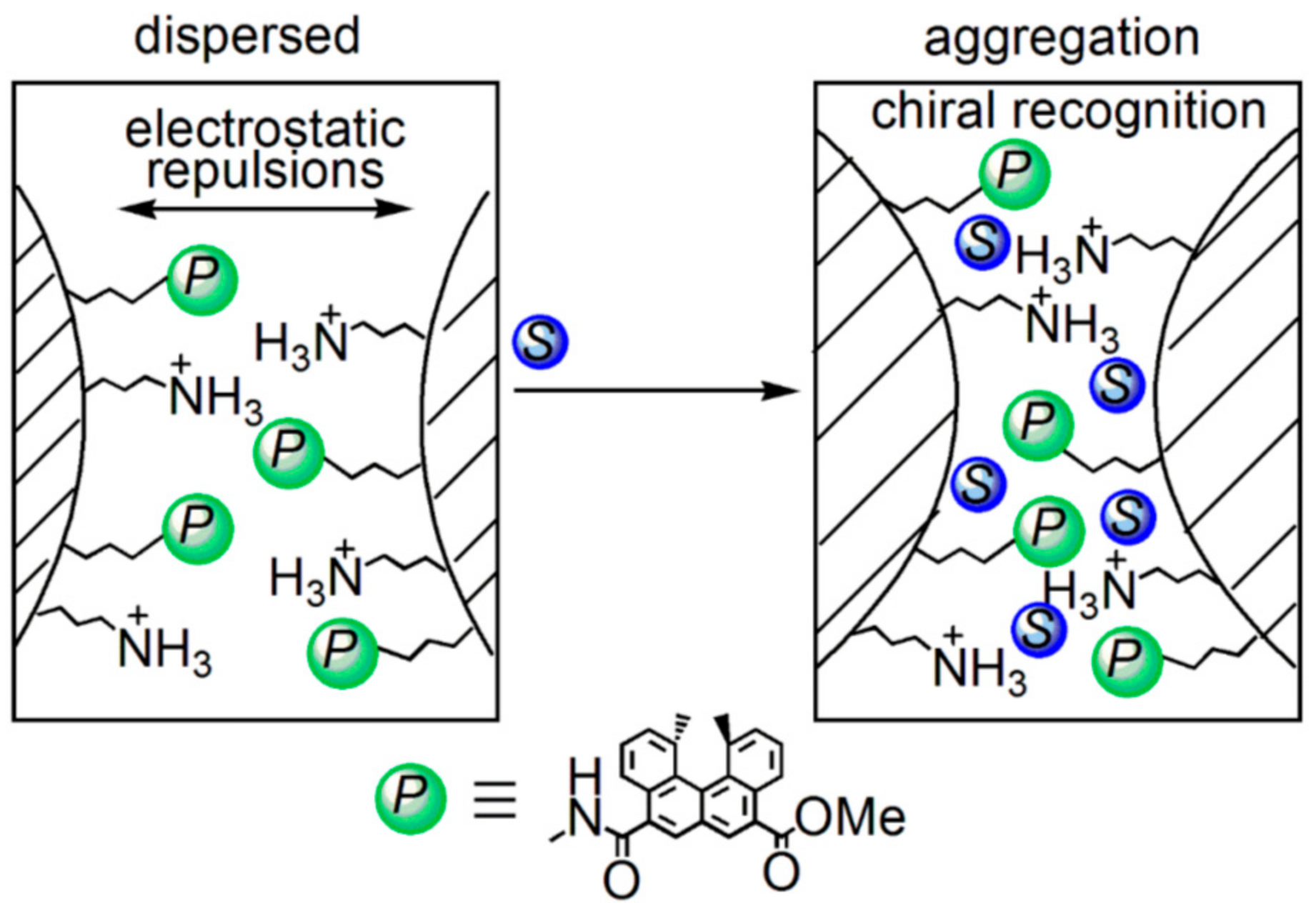
5.3. Chiral Symmetry Breaking Accompanied by Self-Assembly Gel Formation
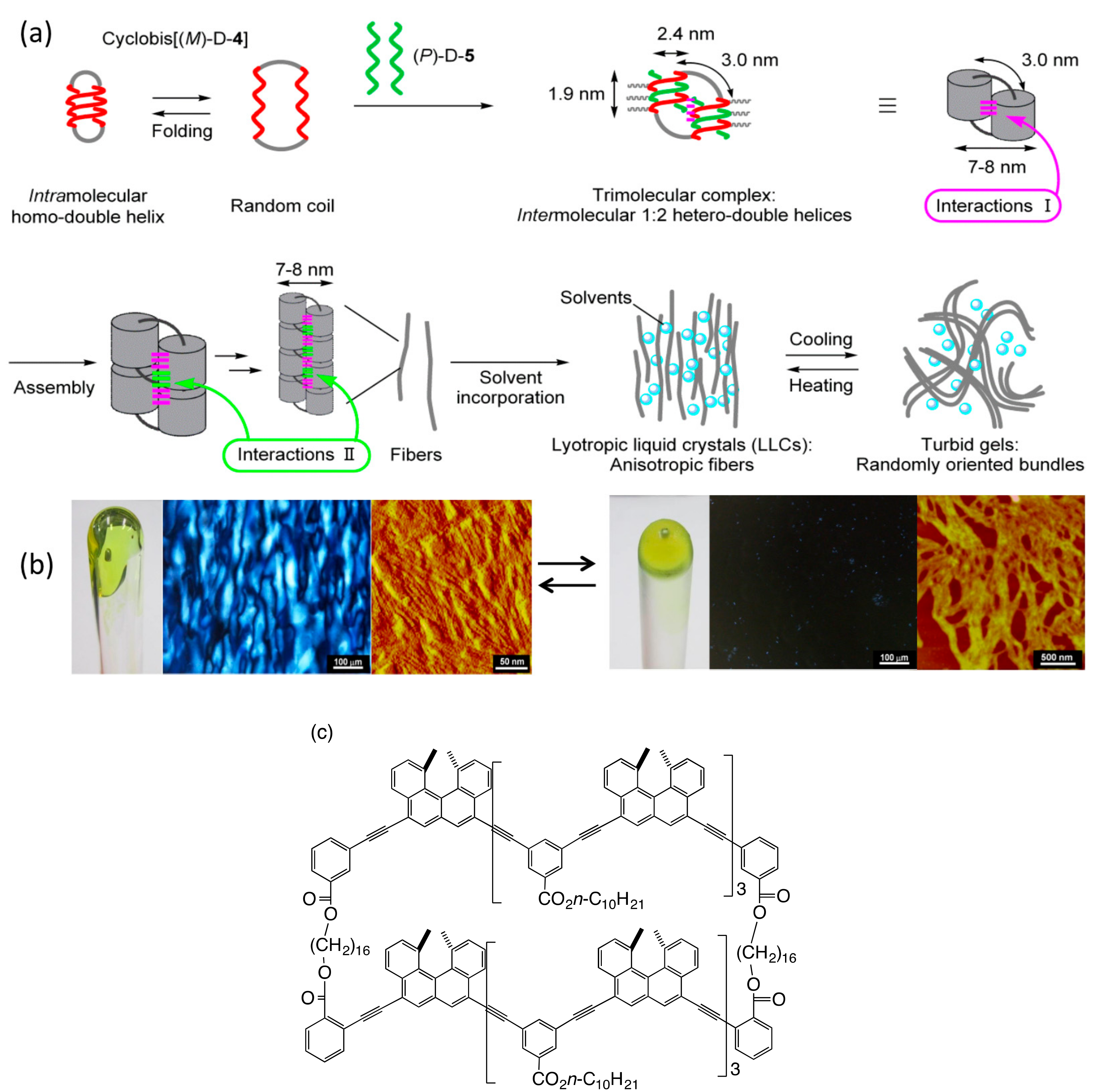
6. Self-Assembly Lipotropic Liquid Crystal Formation by Chiral Cylindrical Molecular Complexes
7. Solid Surface Self-Assembly by Chiral Cylindrical Molecular Complexes
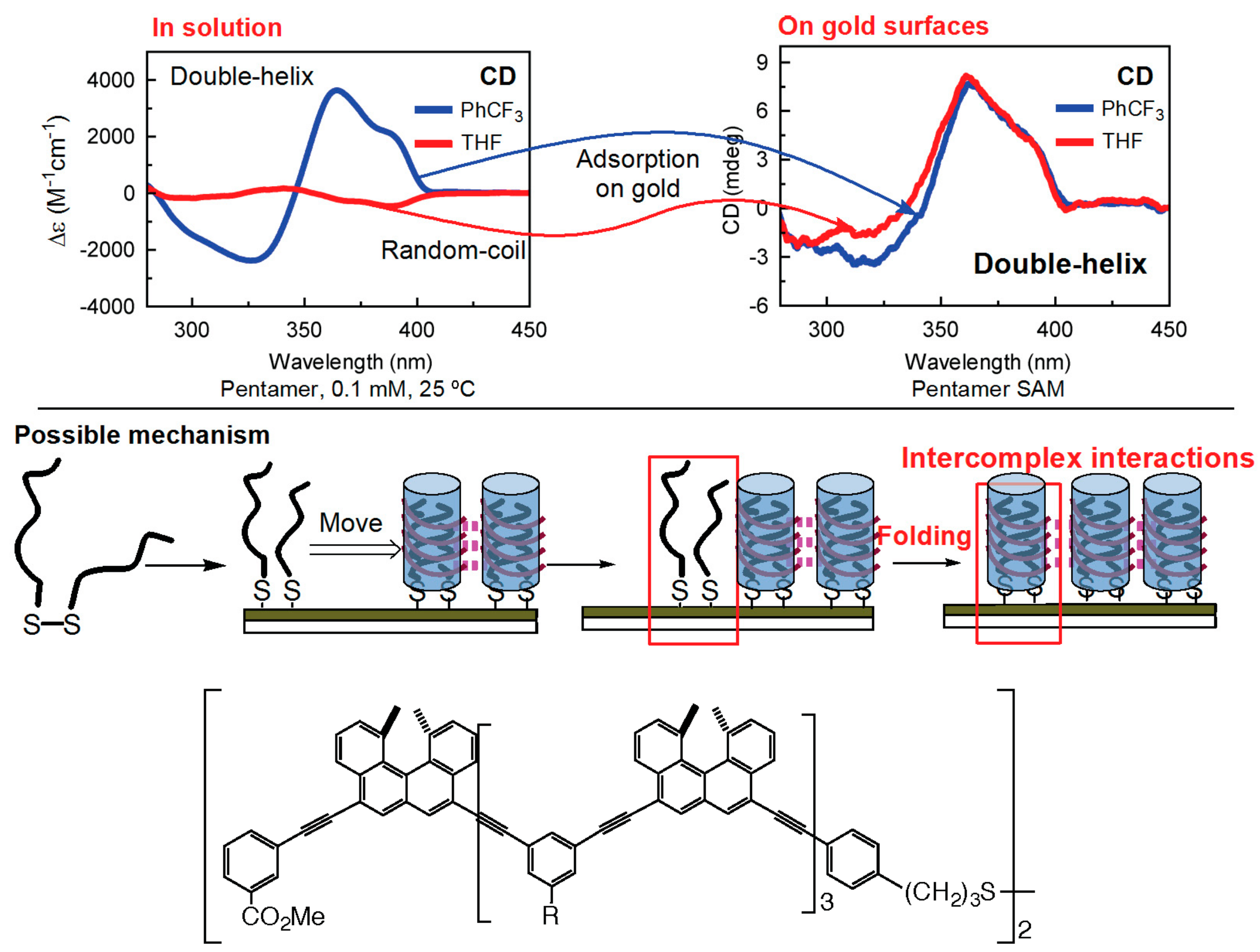
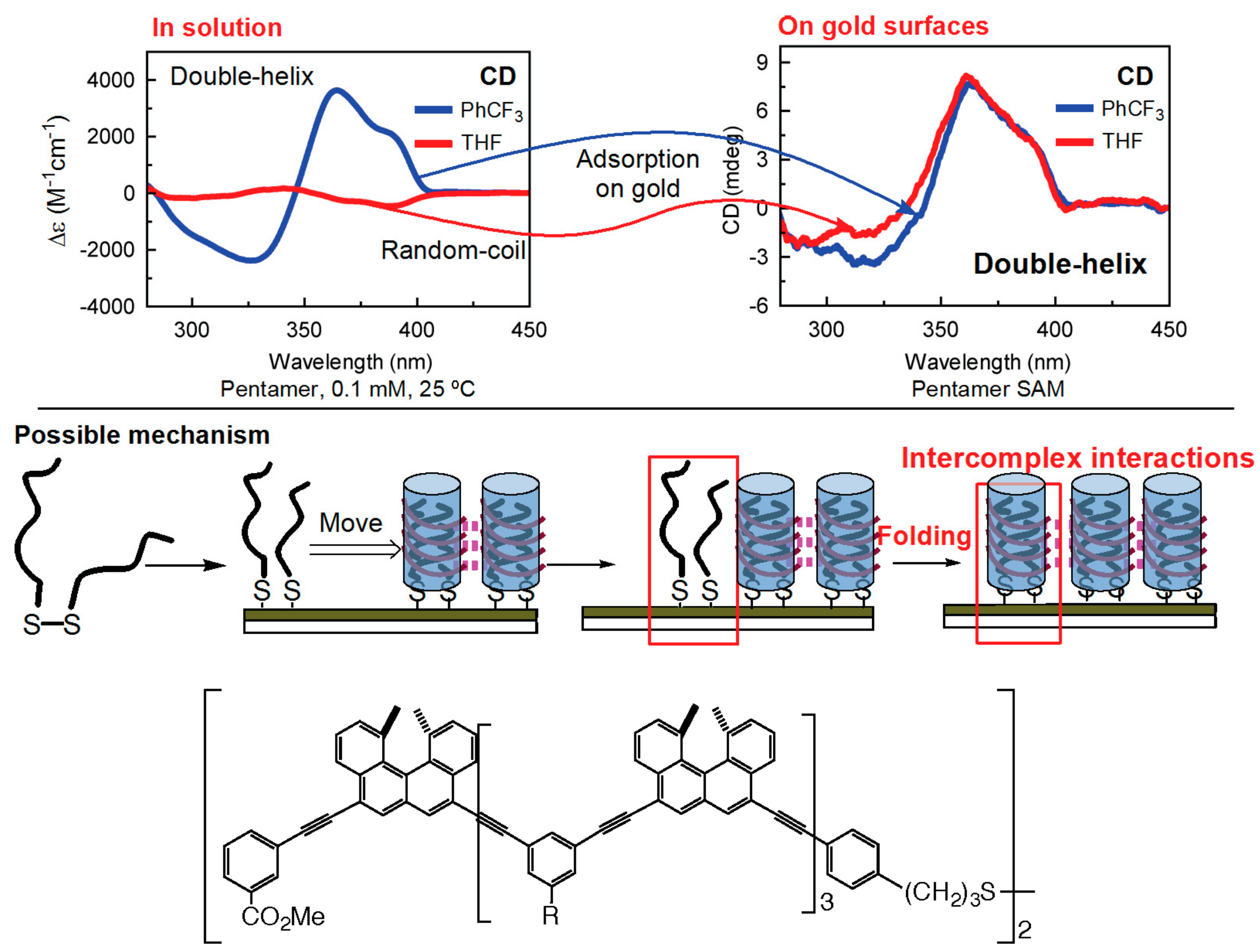
7.1. Homo-Double-Helix Formation on Gold Surface
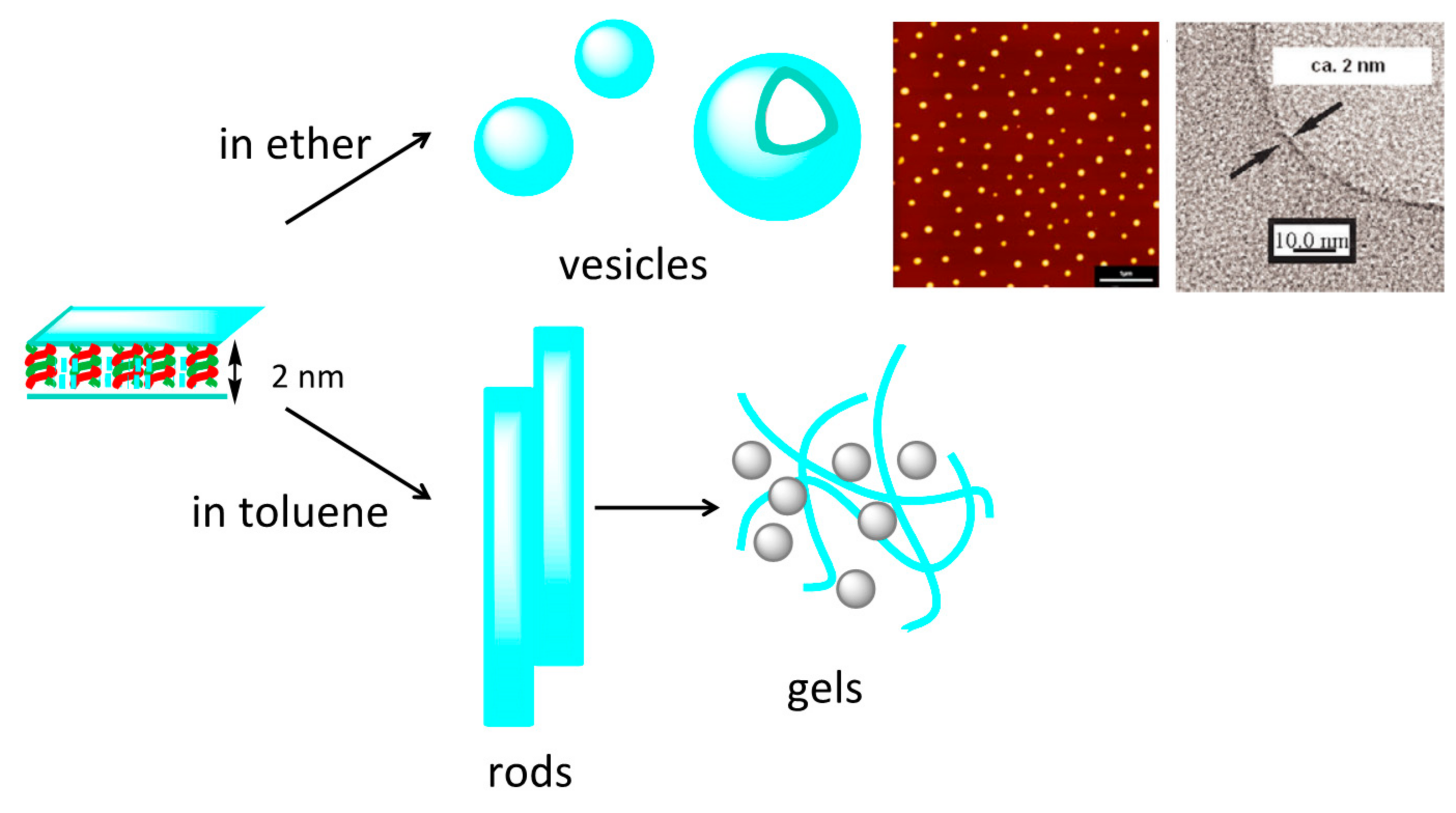
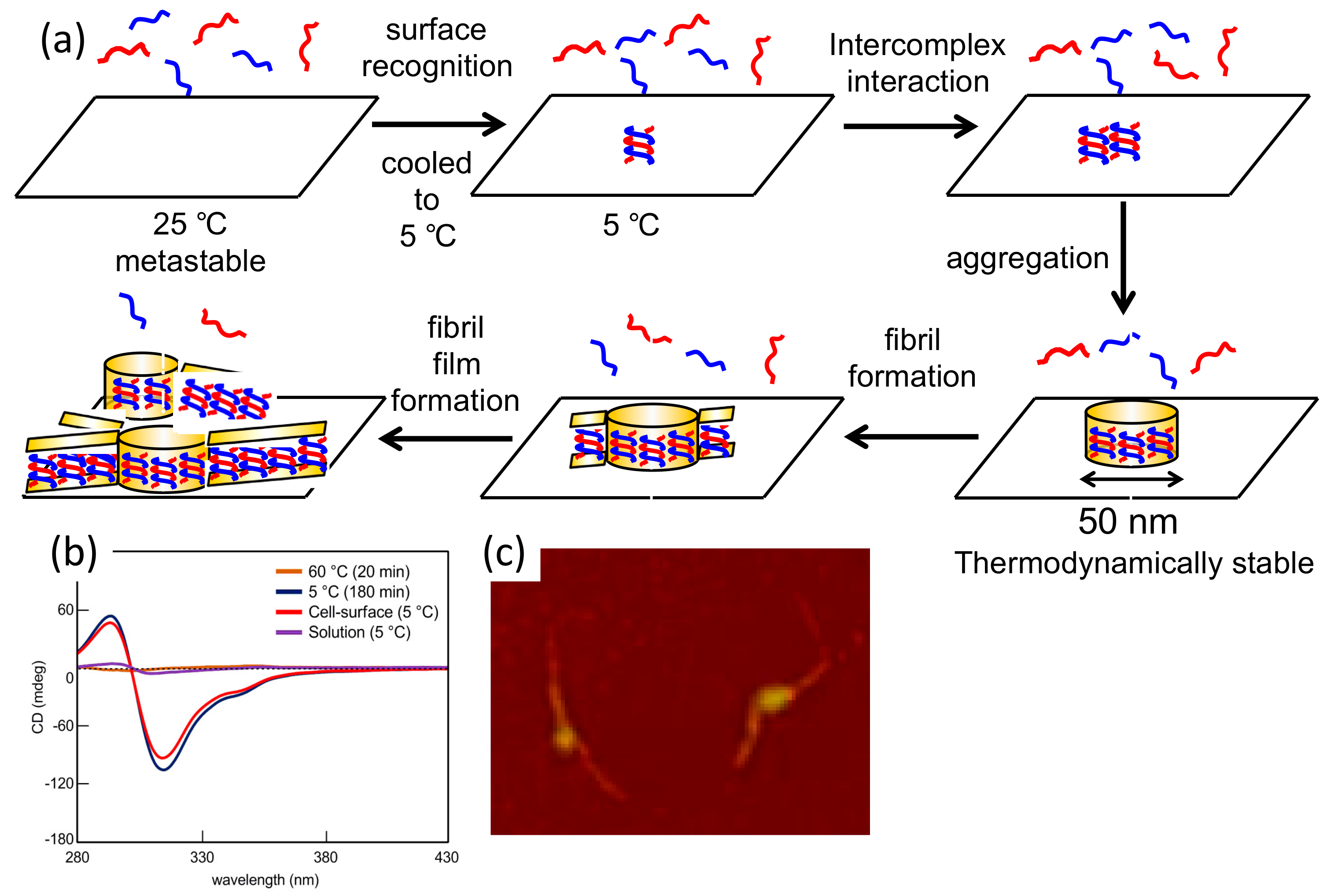
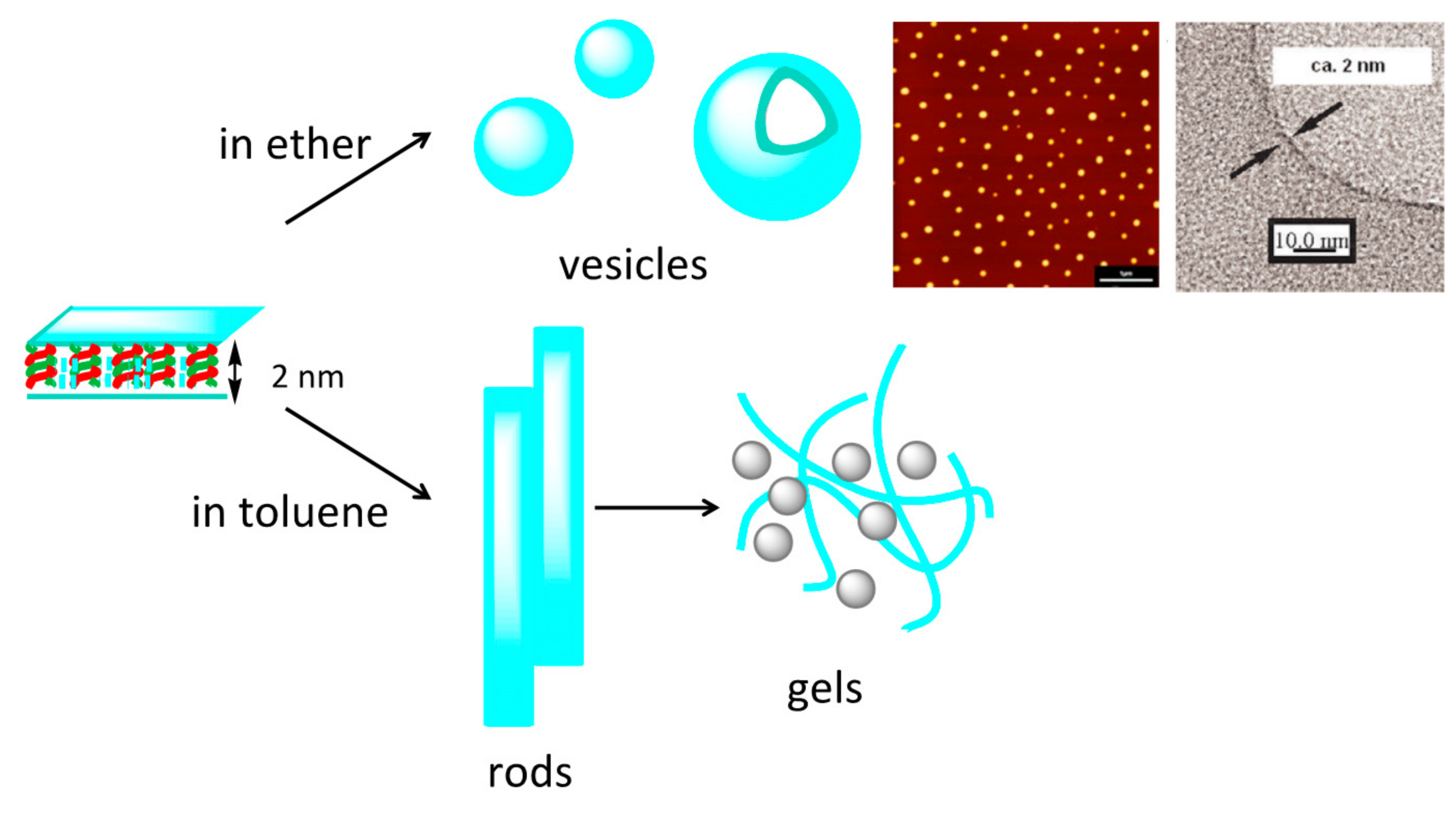
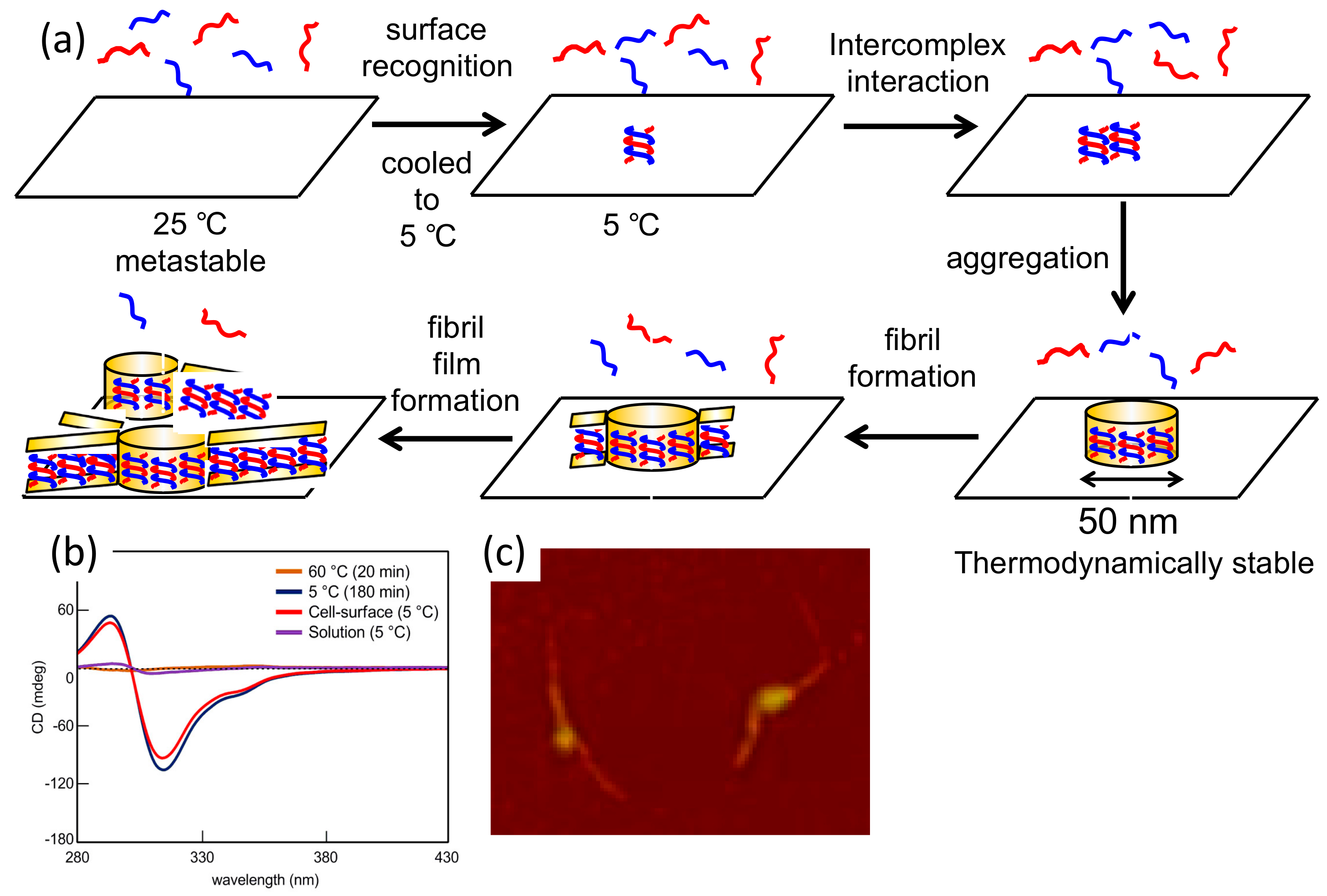
7.2. Formation of Fibril Films on Vesicle Surfaces
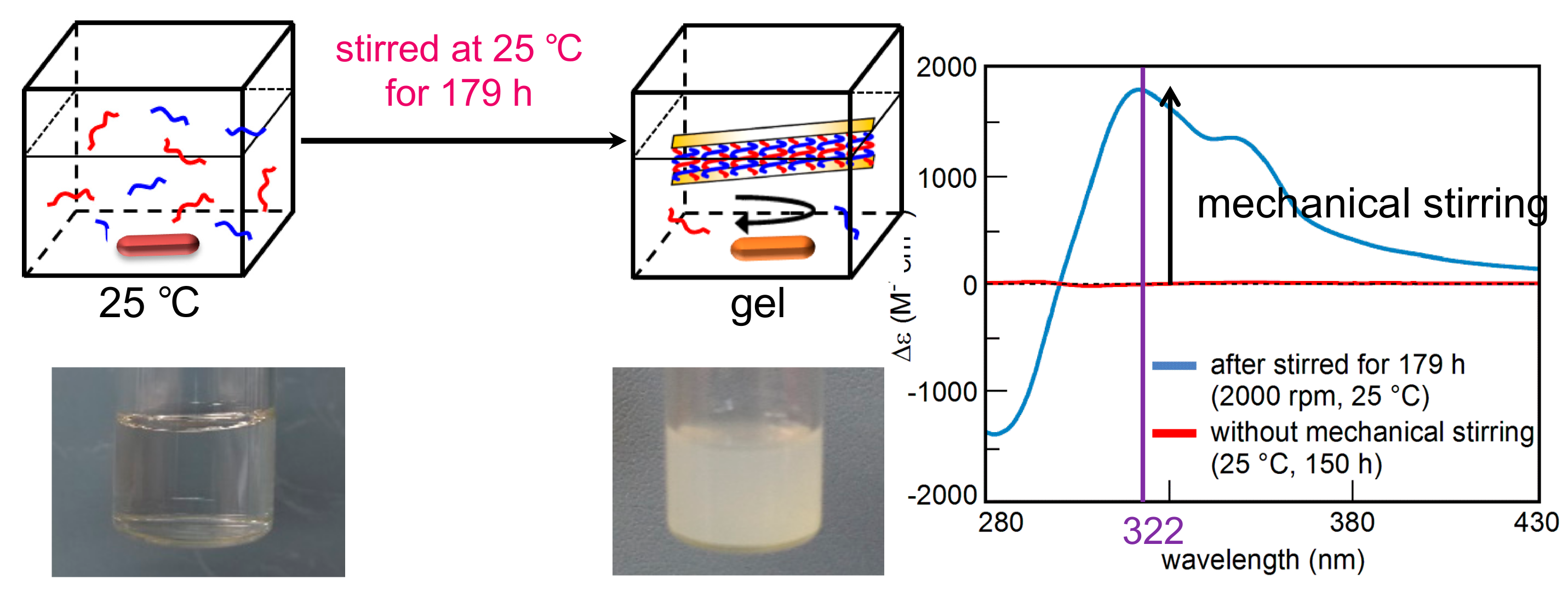
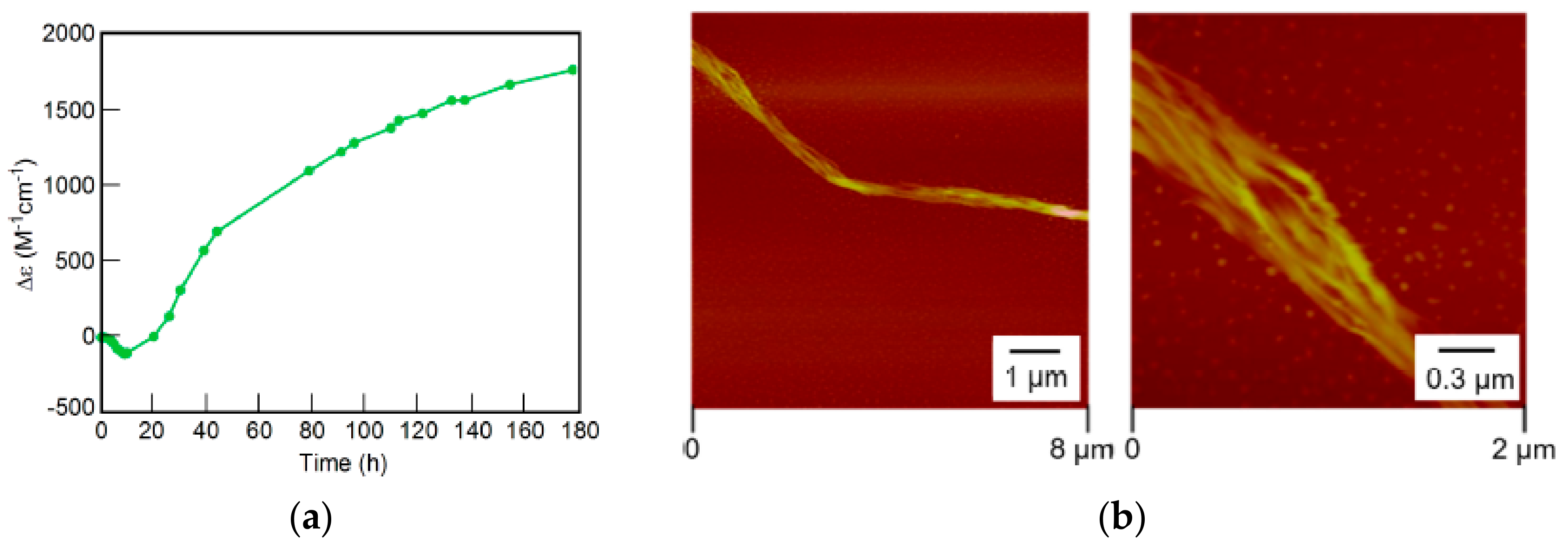
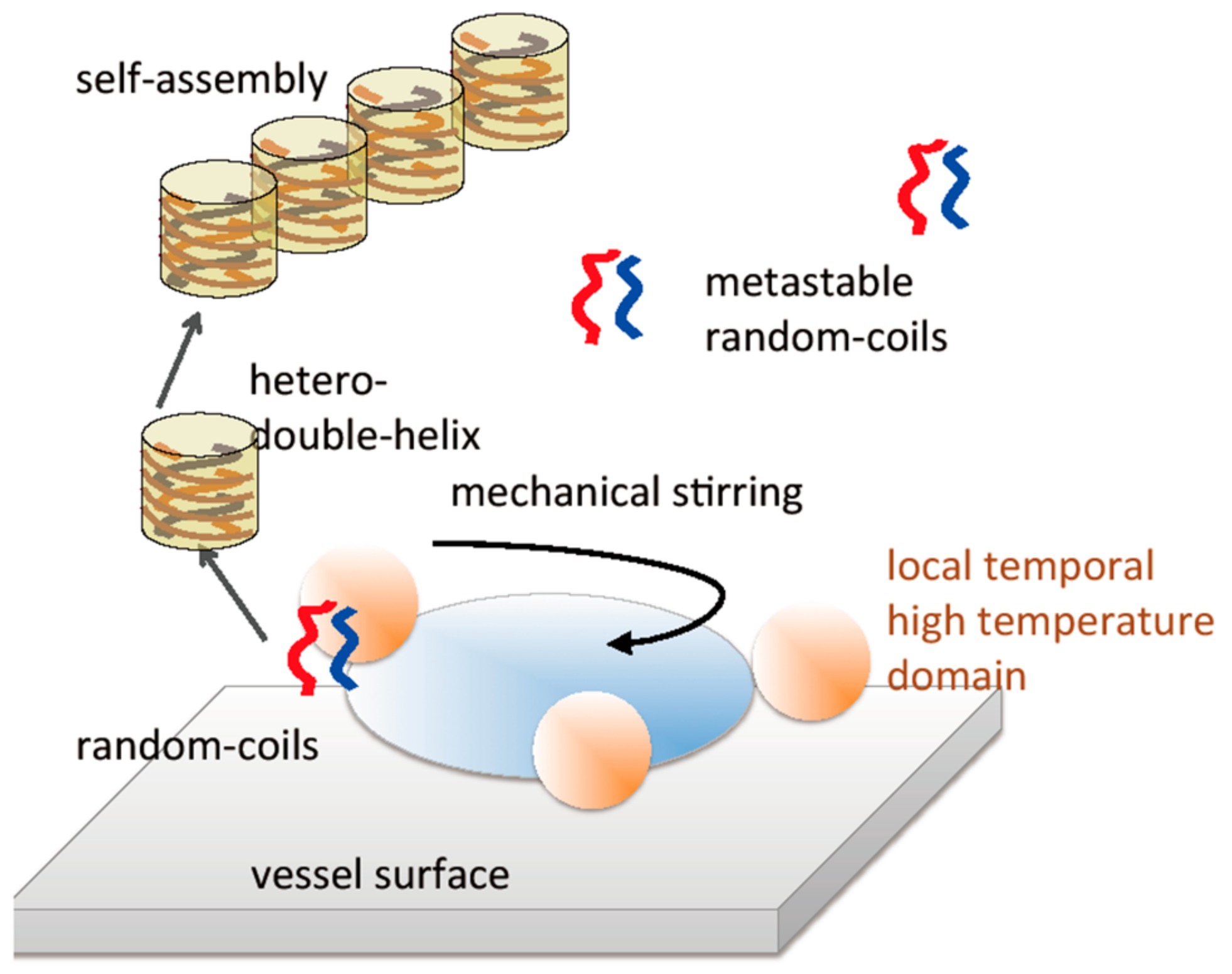
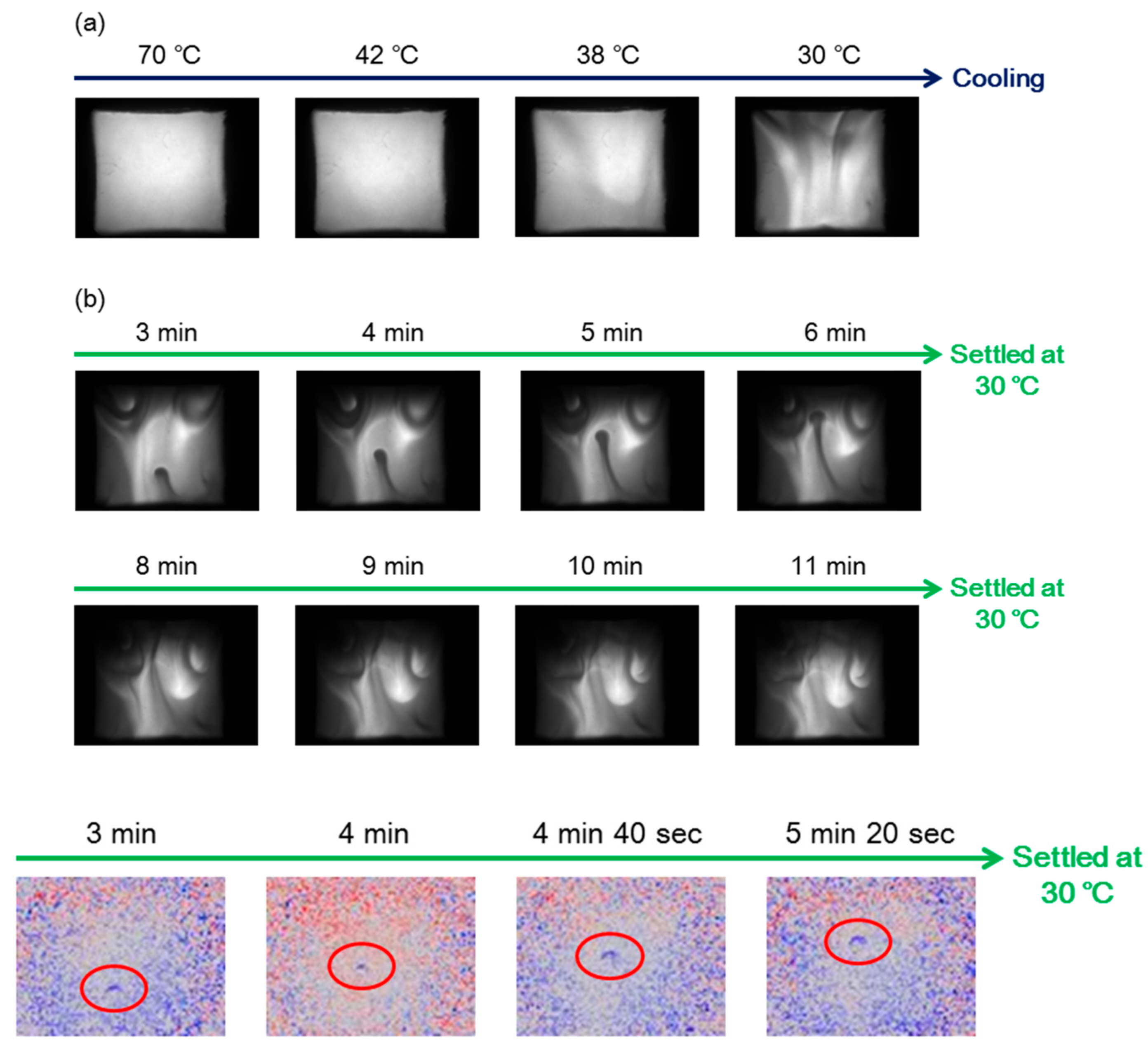
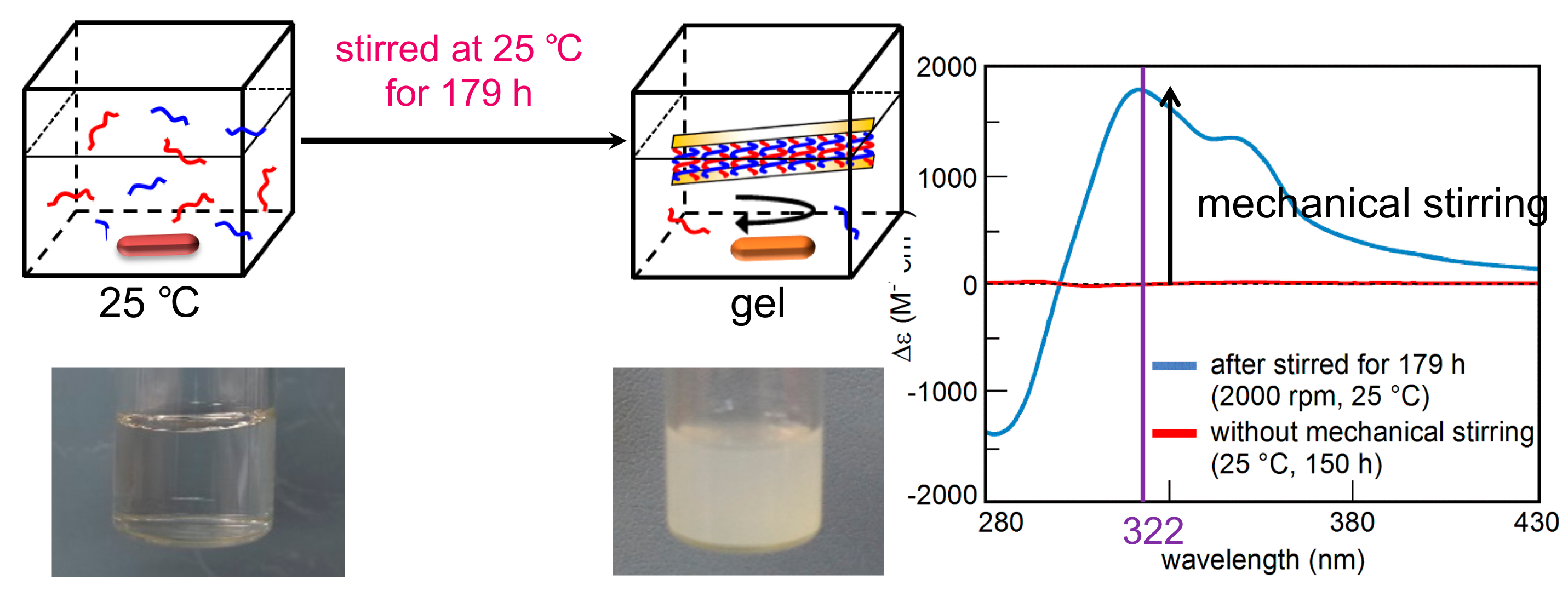
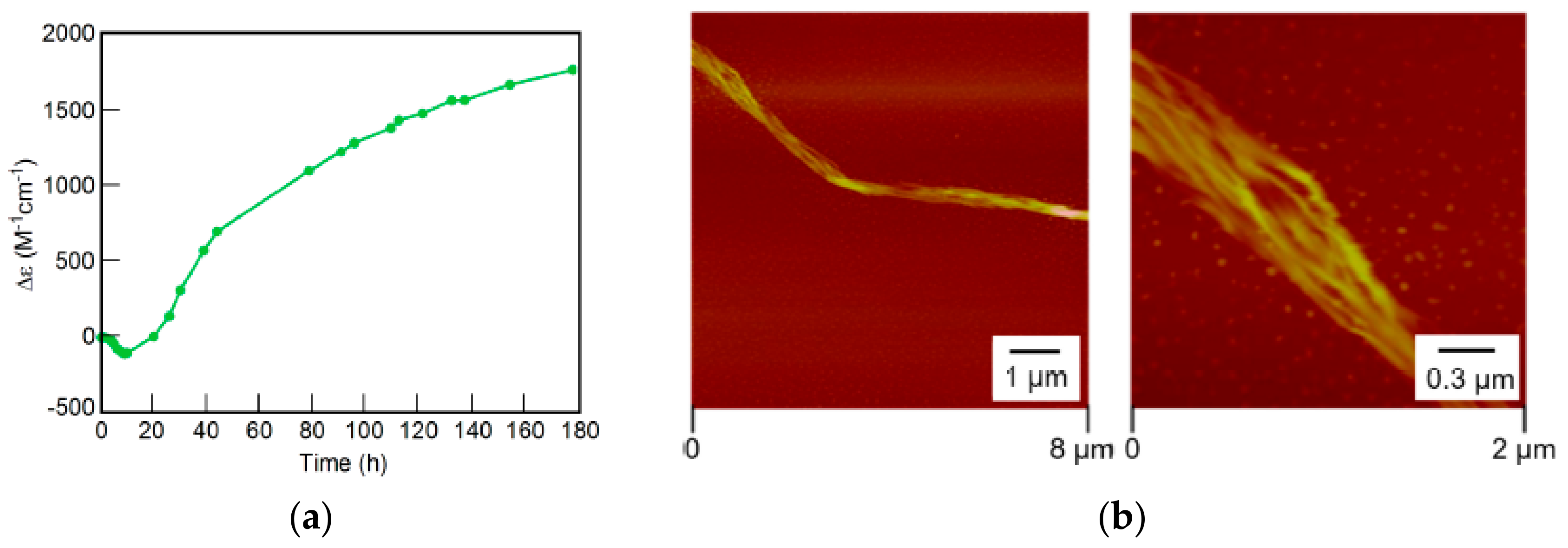
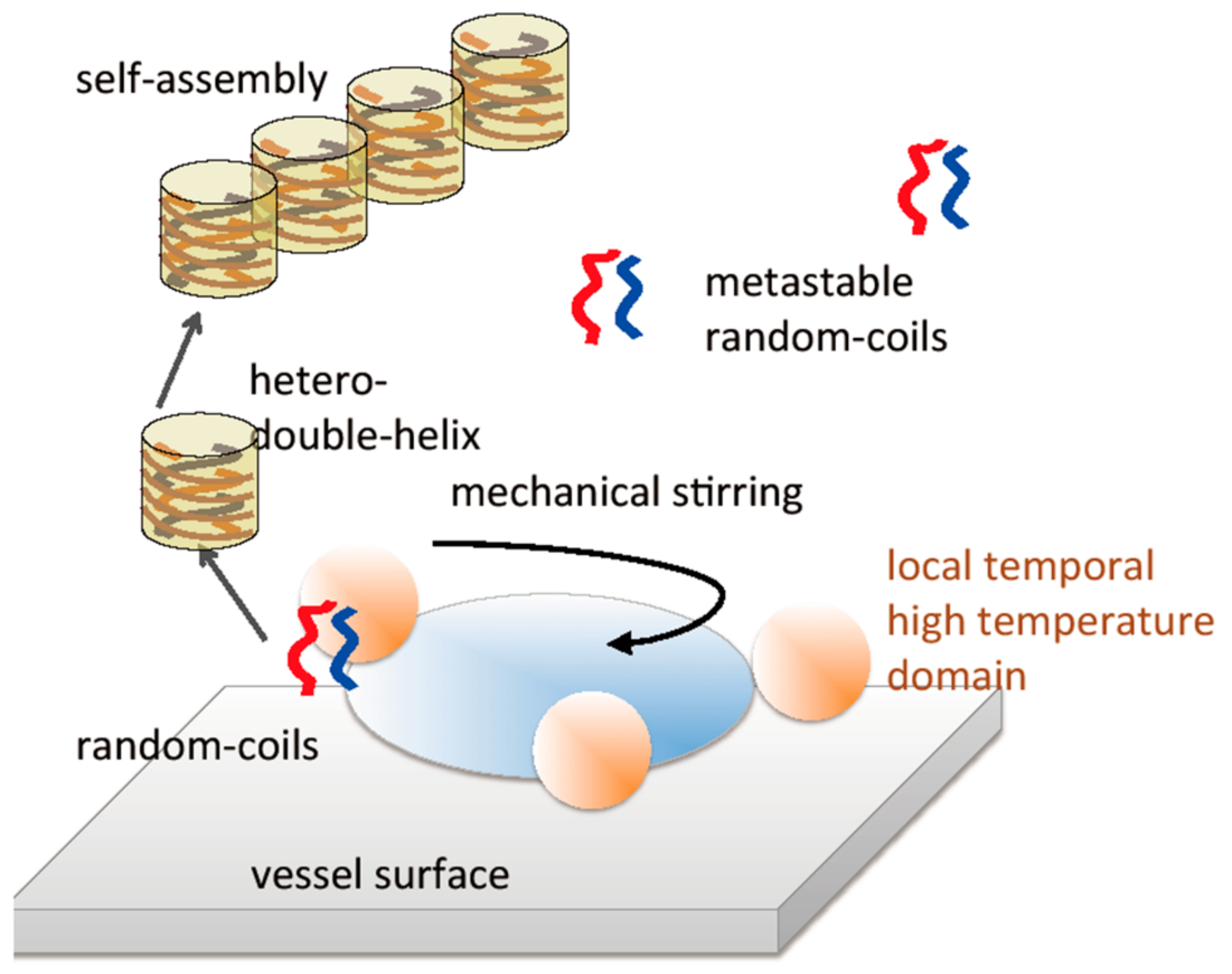
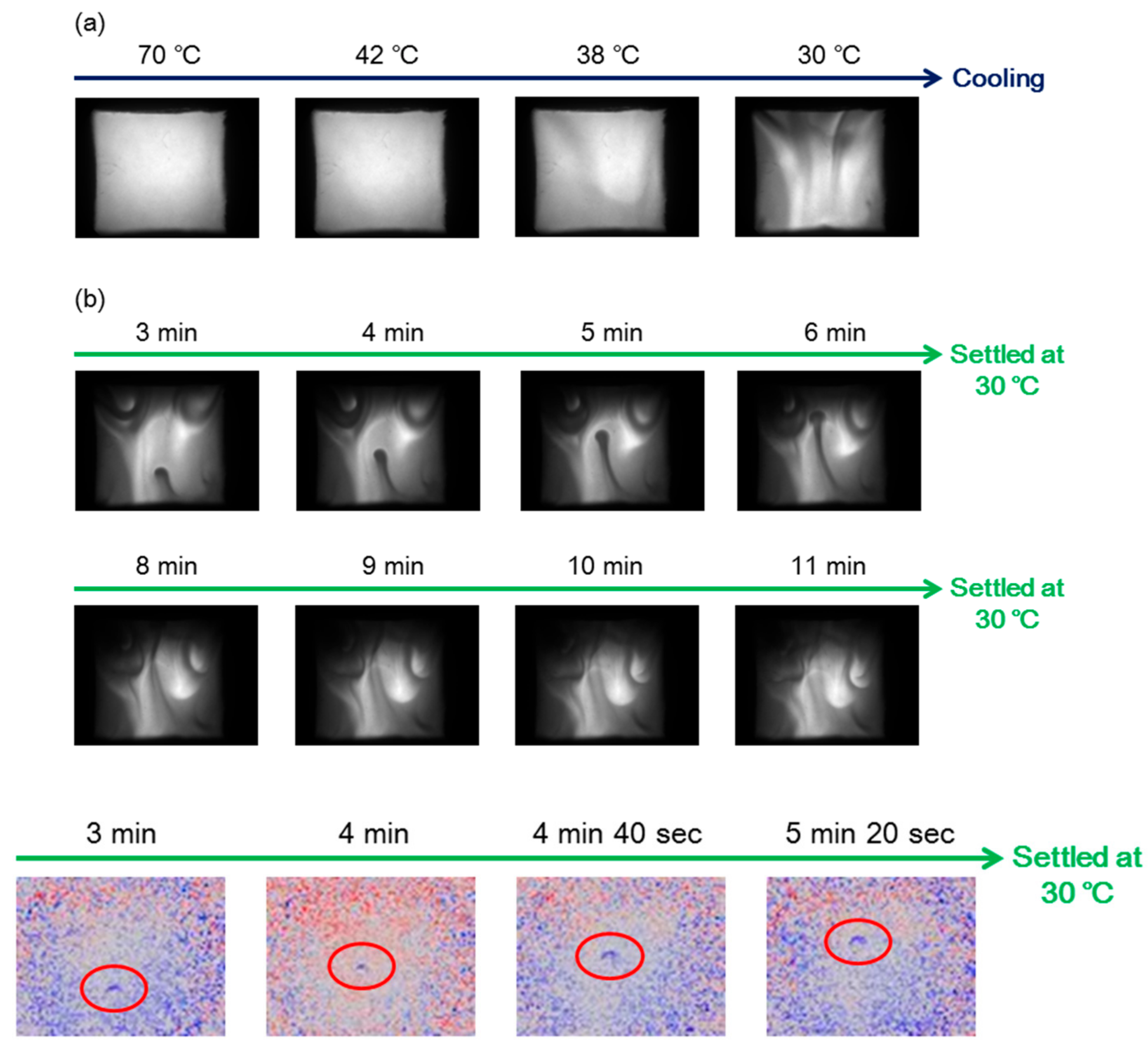
7.3. Self-Assembly Gels Formed by Mechanical Stimulation
8. Self-Assembly of Nanoparticles with Double-Helix Chiral Cylindrical Molecular Complexes
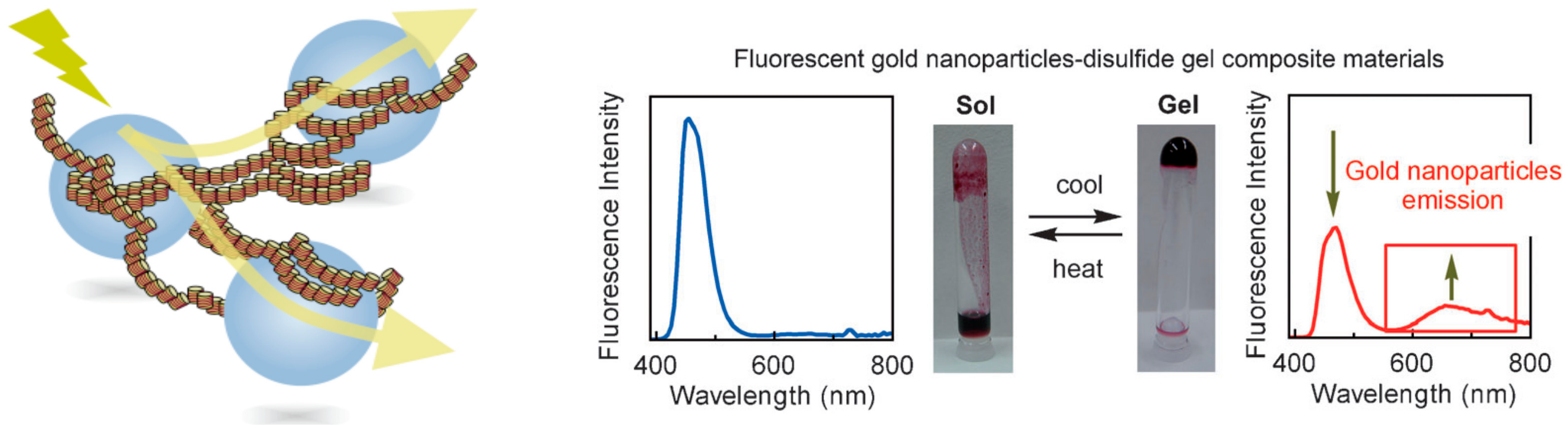
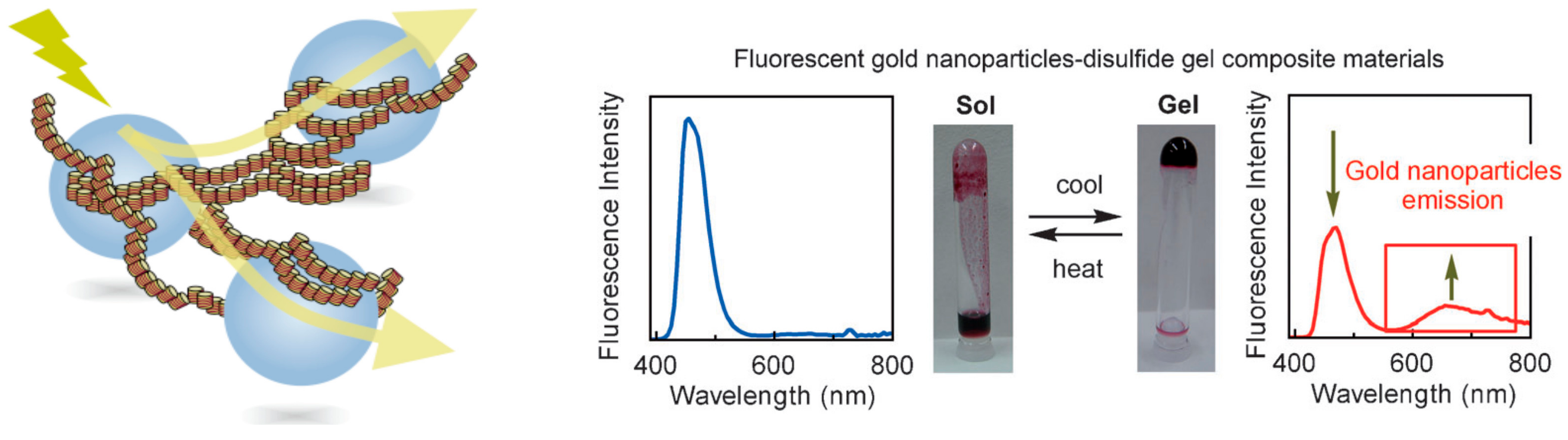
8.1. Light Emission by Composite Materials of Self-Assembly Gels and Gold Nanoparticles
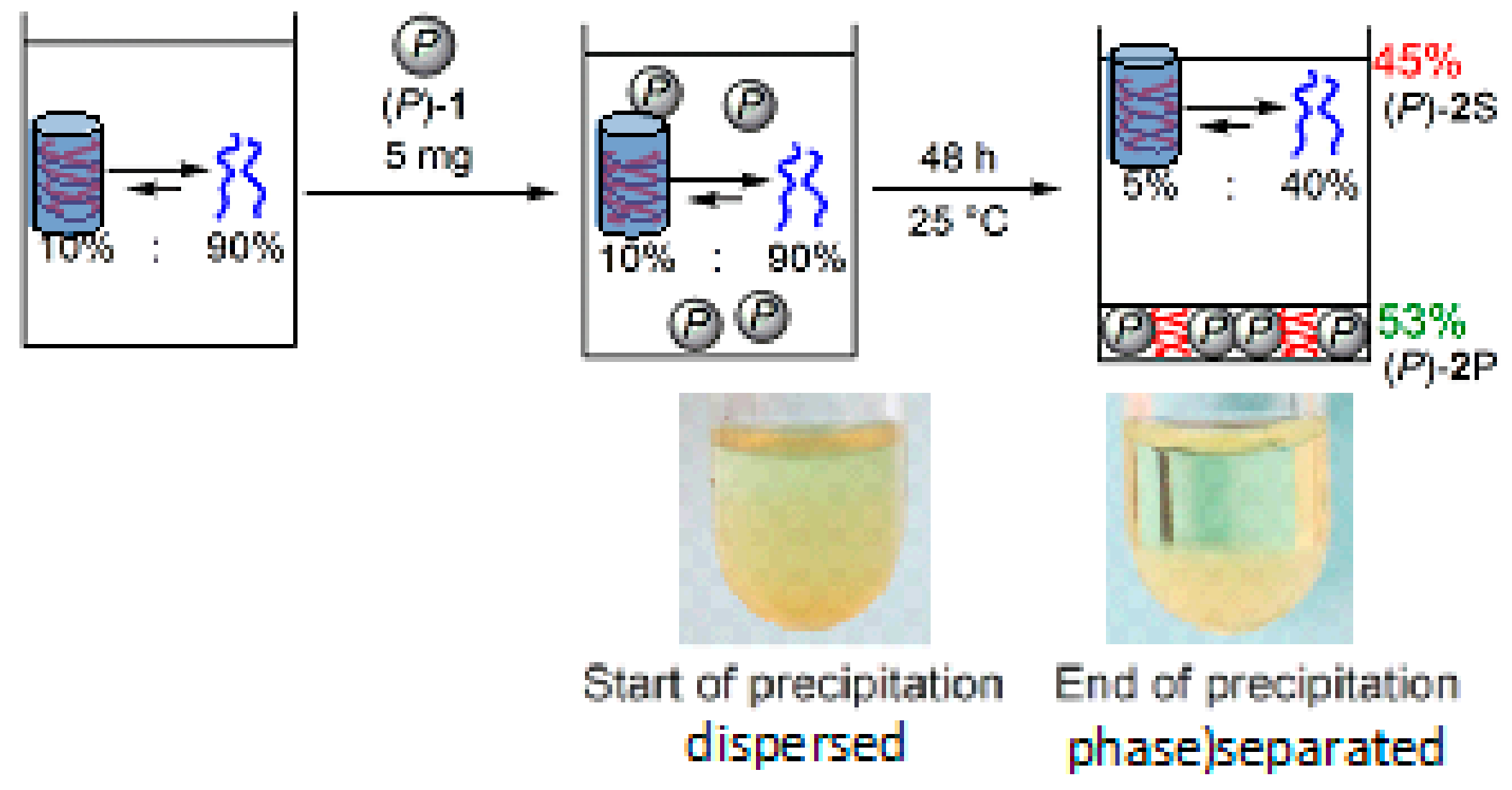
8.2. Molecular Recognition by Helicene-Grafted Silica Nanoparticles
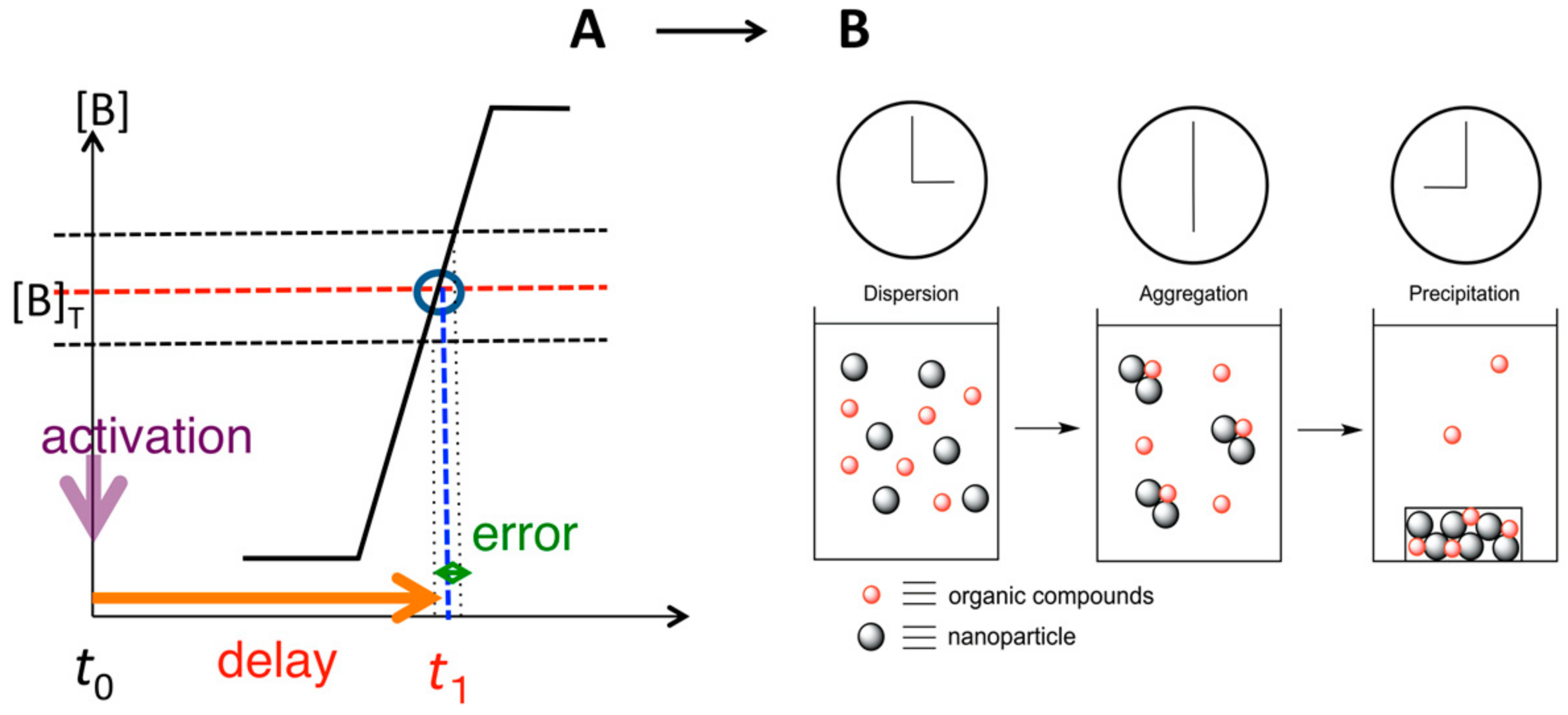
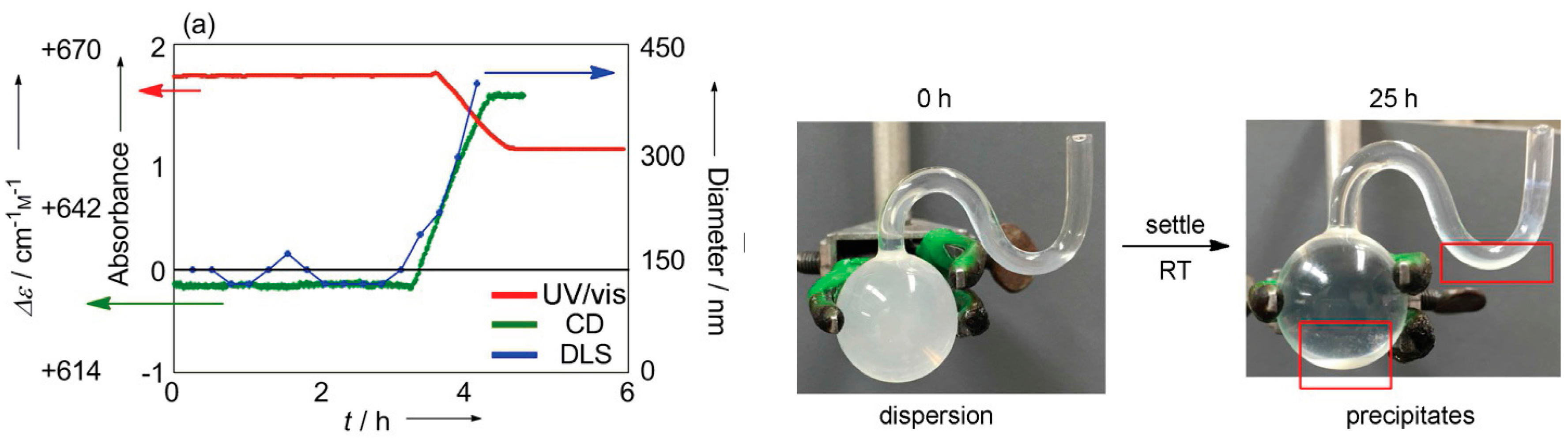
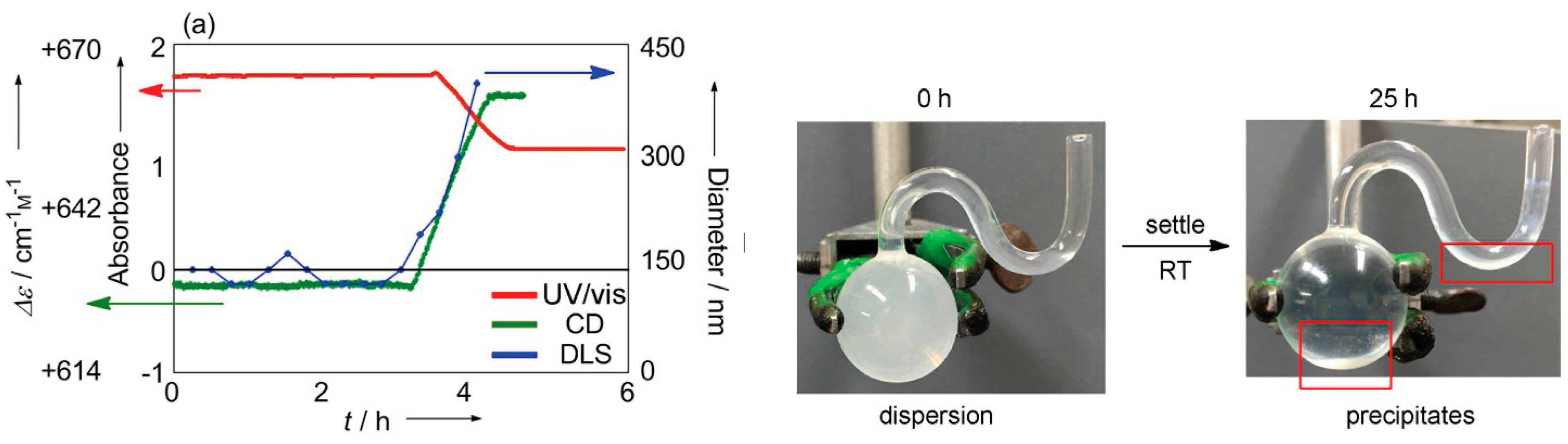
8.3. Materials Clocking by Helicene-Grafted Silica Nanoparticles
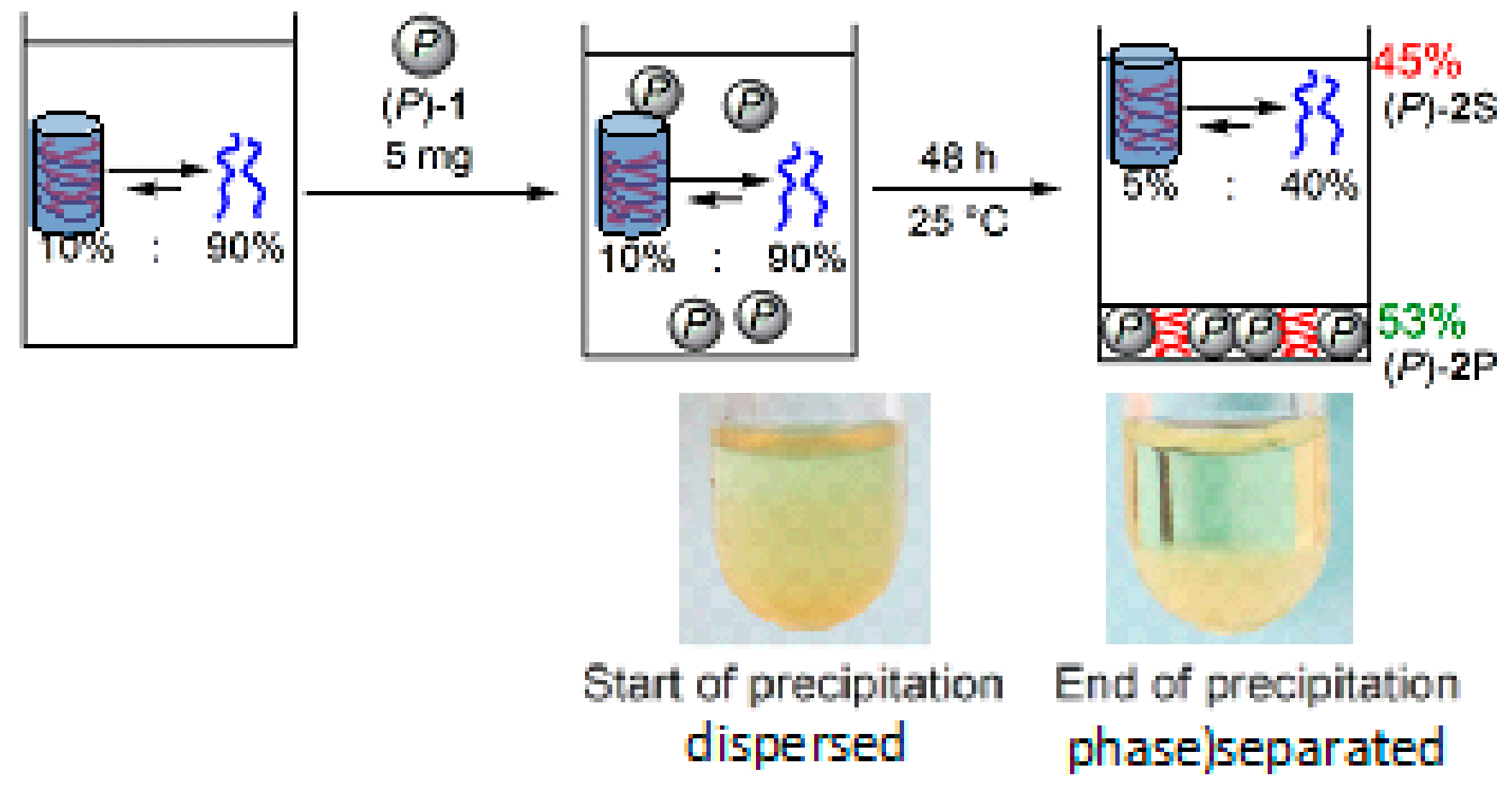
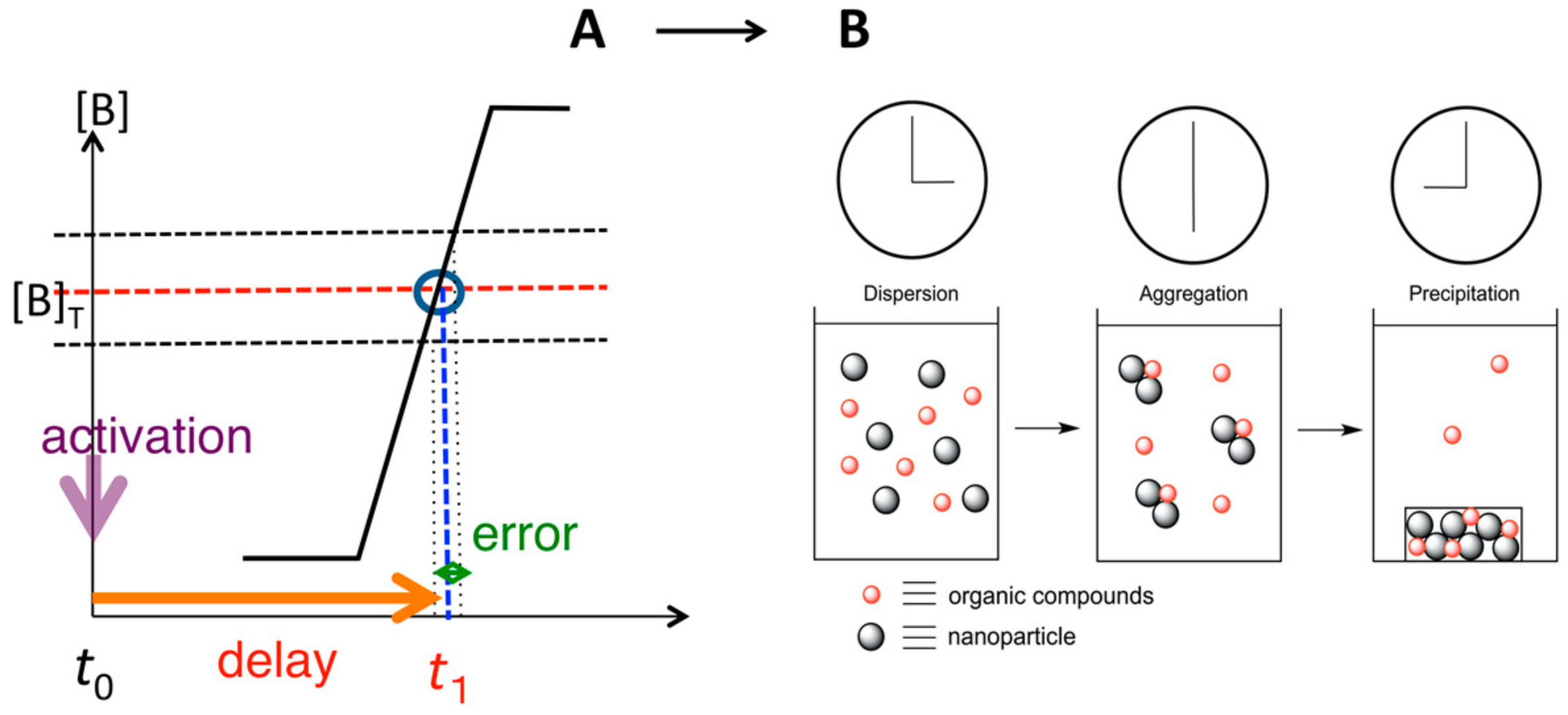
9. Homogeneous-Heterogeneous Transitions in Molecular Dispersed Solutions by Self-Catalysis
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deuter, M.; Bradbery, J.; Turnbull, J. Oxford Advanced Learner’s Dictionary, 9th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Lodish, H.; Berk, A.; Kaiser, C.A.; Krieger, M.; Scott, M.P.; Bretscher, A.; Ploegh, H.; Matsudaira, P. Molecular Cell Biology, 6th ed.; W. H. Freeman and Company: New York, NY, USA, 2008. [Google Scholar]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Robert, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Semrau, S.; Schmidt, T. Membrane heterogeneity—From lipid domains to curvature effects. Soft Matter 2009, 5, 3174–3186. [Google Scholar] [CrossRef]
- Gupta, P.; Sarkar, S.; Das, B.; Bhattecharjee, S.; Tribedi, P. Biofilm, pathogenesis and prevention—A journey to break the wall: A review. Arch. Microbiol. 2016, 198, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bechtloff, B.; Jüsten, P.; Ulrich, J. The kinetics of heterogeneous solid-liquid crystallizations—An overview and examples. Chem. Ing. Tech. 2001, 51, 453–460. [Google Scholar] [CrossRef]
- Orlik, M. Self-organization in nonlinear dynamical systems and its relation to the materials science. J. Solid State Electrochem. 2009, 13, 245–261. [Google Scholar] [CrossRef]
- Busseron, E.; Ruff, Y.; Moulin, E.; Giuseppone, N. Supramolecular self-assemblies as functional nanomaterials. Nanoscale 2013, 5, 7098–7140. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Carter, J.D.; LaBean, T.L. Nanofabrication by DNA self-assembly. Materialstoday 2009, 12, 24–32. [Google Scholar] [CrossRef]
- Min, Y.; Kwak, J.; Soon, A.; Jeong, U. Nonstoichiometric nucleation and growth of multicomponent nanocrystals in solution. Acc. Chem. Res. 2014, 47, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- De Santis, E.; Ryadnov, M.G. Peptide self-assembly for nanomaterials: The old new kid on the block. Chem. Soc. Rev. 2015, 44, 8288–8300. [Google Scholar] [CrossRef] [PubMed]
- Thorkelsson, K.; Bai, P.; Xu, T. Self-assembly and applications of anisotropic nanomaterials: A review. Nano Today 2015, 10, 48–66. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Bhattacharya, S. Multifarious facets of sugar-derived molecular gels: Molecular features, mechanisms of self-assembly and emerging applications. Chem. Soc. Rev. 2015, 44, 5596–5637. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Levin, A.; Adler-Abramovich, L.; Gazit, E. Fmoc-modified amino acids and short peptides: Simple bio-inspired building blocks for the fabrication of functional materials. Chem. Soc. Rev. 2016, 45, 3935–3953. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, J.I.; Francisco, J.S.; Hase, W.L. Chemical Kinetics and Dynamics; Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, 1989. [Google Scholar]
- Van Santen, R.A.; Niemantsverdriet, J.W. Chemical Kinetics and Catalysis; Plenum Press: New York, NY, USA, 1995. [Google Scholar]
- Wright, M.R. Fundamental Chemical Kinetics; Horwood Publishing Limited: Chichester, UK, 1999. [Google Scholar]
- Levine, R.D.; Bernstein, R.B. Molecular Reaction Dynamics; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- De Yoreo, J.J.; Vekilov, P.G. Principles of crystal nucleation and growth. Rev. Mineral. Geochem. 2003, 54, 57–93. [Google Scholar] [CrossRef]
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of nucleation and growth of nanoparticles in solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.; de Paula, J. Physical Chemistry, 10th ed.; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- De Groot, S.R.; Mazur, P. Non-Equilibrium Thermodynamics; Dover Publication: New York, NY, USA, 1984. [Google Scholar]
- Demirel, Y. Non-Equilibrium Thermodynamics: Transport and Rate Processes in Physical, Chemical and Biological Systems, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Epstein, I.R.; Pojman, J.A. An Introduction to Nonlinear Chemical Dynamics; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Scott, S.K. Chemical Chaos; Oxford University Press: Oxford, UK, 1991. [Google Scholar]
- Onsager, L. The effects of shape on the interaction of colloidal particles. Ann. N. Y. Acad. Sci. 1949, 51, 627–659. [Google Scholar] [CrossRef]
- Xiao, X.; Sheng, P. Generalized Onsager theory of liquid crystals. Phys. Rev. E 2013, 88, 062501. [Google Scholar] [CrossRef] [PubMed]
- Goulhen, F.; Hafezi, A.; Uitto, V.-J.; Hinode, D.; Nakamura, R.; Grenier, D.; Mayrand, D. Subcellular Localization and Cytotoxic Activity of the GroEL-Like Protein Isolated from Actinobacillus actinomycetemcomitans. Infect. Immun. 1998, 66, 5307–5313. [Google Scholar] [PubMed]
- Torensma, R.; van der Laan, J.M.; Zantinge, A.; van Bruggen, E.F.J. Reassembly of wall domains of Roman-snail (Helixpomatia) fl-haemocyanin. Biochem. J. 1981, 195, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Self-organization of disc-like molecules: Chemical aspects. Chem. Soc. Rev. 2006, 35, 83–109. [Google Scholar] [CrossRef] [PubMed]
- Wöhrle, T.; Wurzbach, I.; Kirres, J.; Kostidou, A.; Kapernaum, N.; Litterscheidt, J.; Haenle, J.C.; Staffeld, P.; Baro, A.; Giesselmann, F.; et al. Discotic liquid crystals. Chem. Rev. 2016, 116, 1139–1241. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Shigeno, M.; Saito, N.; Yamamoto, K. Synthesis, double-helix formation and higher-assembly formation of chiral polycyclic aromatic compounds: Conceptual development of polyketide aldol synthesis. Chem. Rec. 2014, 14, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Shigeno, M.; Kushida, Y.; Yamaguchi, M. Energy aspects of thermal molecular switching: Molecular thermal hysteresis of helicene oligomers. ChemPhysChem 2015, 16, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Shigeno, M.; Kushida, Y.; Yamaguchi, M. Molecular switching involving metastable states: Molecular thermal hysteresis and sensing of environmental changes by chiral helicene oligomeric foldamers. Chem. Commun. 2016, 52, 4955–4970. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Shigeno, M.; Yamaguchi, M. Structure and property diversity of chiral helicene oligomer. Encycl. Polym. Sci. Technol. 2015. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Arisawa, M.; Shigeno, M.; Saito, N. Equilibrum and nonequilibrium chemical reactions of helicene oligomers in the noncovalent bond formation. Bull. Chem. Soc. Jpn. 2016, 89, 1145–1169. [Google Scholar] [CrossRef]
- Wynberg, H. Some observations on the chemical, photochemical and spectral properties of thiophenes. Acc. Chem. Res. 1971, 4, 65–73. [Google Scholar] [CrossRef]
- Martin, R.H. The helicenes. Angew. Chem. Int. Ed. 1974, 13, 649–660. [Google Scholar] [CrossRef]
- Iwasaki, T.; Nishide, H. Electro- and magneto-responsible chiral polymers. Curr. Org. Chem. 2005, 9, 1665–1684. [Google Scholar] [CrossRef]
- Collins, S.K.; Vachon, M.P. Unlocking the potential of thiaheterohelicenes: Chemical synthesis as the key. Org. Biomol. Chem. 2006, 4, 2518–2524. [Google Scholar] [CrossRef] [PubMed]
- Rajca, A.; Rajca, S.; Pink, M.; Miyasaka, M. Annelated, chiral π-conjugated systems: Tetraphenylenes and helical β-oligothiophenes. Synlett 2007, 12, 1799–1822. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, C.-F. Helicenes: Synthesis and applications. Chem. Rev. 2012, 112, 1463–1535. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M. One hundred years of helicene chemistry. Part 1: Non-stereoselective syntheses of carbohelicenes. Chem. Soc. Rev. 2013, 42, 968–1006. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.; Félix, G.; Peresutti, R. One hundred years of helicene chemistry. Part 2: Stereoselective syntheses and chiral separations of carbohelicenes. Chem. Soc. Rev. 2013, 42, 1007–1050. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M. One hundred years of helicene chemistry. Part 3: Applications and properties of carbohelicenes. Chem. Soc. Rev. 2013, 42, 1051–1095. [Google Scholar] [CrossRef] [PubMed]
- Urbano, A.; Carreno, M.C. Enantioselective synthesis of helicenequinones and -bisquinones. Org. Biomol. Chem. 2013, 11, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Bosson, J.; Gouin, J.; Lacour, J. Cationic triangulenes and helicenes: Synthesis, chemical stability, optical properties and extended applications of these unusual dyes. Chem. Soc. Rev. 2014, 43, 2824–2840. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.; Shen, C.; Crassous, J. Helicene-based transition metal complexes: Synthesis, properties and applications. Chem. Sci. 2014, 5, 3680–3694. [Google Scholar] [CrossRef]
- Isla, H.; Crassous, J. Helicene-based chiroptical switches. Comptes Rendus Chim. 2016, 19, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Okubo, H.; Yamaguchi, M. Synthesis and self-aggregation of cyclic alkynes containing helicene. Org. Lett. 2001, 3, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Terakawa, R.; Yamaguchi, M. Synthesis, π-face-selective aggregation and π-face chiral recognition of configurationally stable C3-symmetric propeller-chiral molecules with a π-core. Chem. Eur. J. 2014, 20, 5601–5607. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, R.; Yamaguchi, M. Chiral recognition in noncovalent bonding interactions between helicenes: Right-handed helix favors right-handed helix over left-handed helix. Org. Biomol. Chem. 2008, 6, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, R.; Yamaguchi, M. Synthesis and structure of built-up organic macromolecules containing helicene. Chem. Rec. 2008, 8, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Nigorikawa, Y.; Saiki, Y.; Nakamura, K.; Yamaguchi, M. Marked effect of aromatic solvent on unfolding rate of helical ethynylhelicene oligomer. J. Am. Chem. Soc. 2004, 126, 14858–14864. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Yamaguchi, M. Helix-dimer–random-coil thermal switching process of ethynylhelicene heptamer highly sensitive to its environment. Chem. Lett. 2007, 36, 58–59. [Google Scholar] [CrossRef]
- Sugiura, H.; Amemiya, R.; Yamaguchi, M. Reversible double-helix-random-coil transition process of bis{hexa(ethynylhelicene)}s. Chem. Asian J. 2008, 3, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Terakawa, R.; Shigeno, M.; Amemiya, R.; Yamaguchi, M. Side chain effect on the double helix formation of ethynylhelicene oligomers. J. Org. Chem. 2011, 76, 4841–4858. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Kobayashi, H.; Yamaguchi, M. “Inverse” thermoresponse: Heat-induced double-helix formation of an ethynylhelicene oligomer with tri(ethylene glycol) termini. Chem. Sci. 2016, 7, 3574–3580. [Google Scholar] [CrossRef]
- Ichinose, W.; Shigeno, M.; Yamaguchi, M. Multiple states of dimeric aggregates formed by (amido–ethynyl)helicene bi-domain compound and (amido–ethynyl–amido)helicene tri-domain compound. Chem. Eur. J. 2012, 18, 12644–12654. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, W.; Ito, J.; Yamaguchi, M. Tetrameric ααββ aggregate formation by stereoisomeric bi-domain helicene oligomers. Angew. Chem. Int. Ed. 2013, 52, 5290–5294. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, W.; Ito, J.; Yamaguchi, M. Heteroaggregation between isomeric amido-ethynyl-amidohelicene tri-domain oligomers. J. Org. Chem. 2012, 77, 10655–10667. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, R.; Saito, N.; Yamaguchi, M. Hetero-double-helix formation by an ethynylhelicene oligomer possessing perfluorooctyl side chains. J. Org. Chem. 2008, 73, 7137–7144. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Shigeno, M.; Yamaguchi, M. Two-component fibers/gels and vesicles formed from hetero-double-helices of pseudoenantiomeric ethynylhelicene oligomers with branched side chains. Chem. Eur. J. 2012, 18, 8994–9004. [Google Scholar] [CrossRef] [PubMed]
- Shigeno, M.; Sato, M.; Kushida, Y.; Yamaguchi, M. Aminomethylenehelicene oligomers possessing flexible two-atom linker form a stimuli-responsive double-helix in solution. Asian J. Org. Chem. 2014, 3, 797–804. [Google Scholar] [CrossRef]
- Shigeno, M.; Kushida, Y.; Yamaguchi, M. Heating/cooling stimulus induces three-state molecular switching of pseudoenantiomeric aminomethylenehelicene oligomers: Reversible nonequilibrium thermodynamic processes. J. Am. Chem. Soc. 2014, 136, 7972–7980. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.K. Lost in translation? Chirality effects in the self-assembly of nanostructured gel-phase materials. Chem. Soc. Rev. 2009, 38, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Fichman, G.; Gazit, E. Self-assembly of short peptides to form hydrogels: Design of building blocks, physical properties and technological applications. Acta Biomater. 2014, 10, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Rasale, D.B.; Das, A.K. Chemical reactions directed peptide self-assembly. Int. J. Mol. Sci. 2015, 16, 10797–10820. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, R.; Mizutani, M.; Yamaguchi, M. Two-component gel formation by pseudoenantiomeric ethynylhelicene oligomers. Angew. Chem. Int. Ed. 2010, 49, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Oyamada, N.; Mizutani, M.; An, Z.; Saito, N.; Yamaguchi, M.; Kasuya, M.; Kurihara, K. Two types of two-component gels formed from pseudoenantiomeric ethynylhelicene oligomers. Langmuir 2012, 28, 11939–11947. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, W.; Miyagawa, M.; Yamaguhi, M. Reversible shrinkage of self-assembled two-component organogels by lithium salts: Synthesis of gelation property and lithium salt response using bi-domain helicene oligomer. Chem. Mater. 2013, 25, 4036–4043. [Google Scholar] [CrossRef]
- Tranter, G.E. The parity-violating energy differences between the enantiomers of α-amino acids. Chem. Phys. Lett. 1985, 120, 93–96. [Google Scholar] [CrossRef]
- Mason, S.F.; Tranter, G.E. The electroweak origin of biomolecular handedness. Proc. R. Soc. Lond. 1985, 397, 45–65. [Google Scholar] [CrossRef]
- Kondepudi, D.K.; Nelson, G.W. Weak neutral currents and the origin of biomolecular chirality. Nature 1985, 314, 438–441. [Google Scholar] [CrossRef]
- Kondepudi, D.K. Selection of molecular chirality by extremely weak chiral interactions under far-from-equilibrium conditions. BioSystems 1987, 20, 75–83. [Google Scholar] [CrossRef]
- Kondepudi, D.K.; Asakura, K. Chiral autocatalysis, spontaneous symmetry breaking and stochastic behavior. Acc. Chem. Res. 2001, 34, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Kushida, Y.; Sawato, T.; Shigeno, M.; Saito, N.; Yamaguchi, M. Deterministic and stochastic chiral symmetry breaking exhibited by racemic aminomethylenehelicene oligomers. Chem. Eur. J. 2017, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Mann, S. Self-assembly and transformation of hybrid nano-objects and nanostructures under equilibrium and non-equilibrium conditions. Nat. Mater. 2009, 8, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Yan, X.; Han, C.; Huang, F. Characterization of supramolecular gels. Chem. Soc. Rev. 2013, 42, 6697–6722. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Steed, J.W. Supramolecular gel phase crystallization: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Kanie, K.; Matsubara, M.; Muramatsu, A.; Yamaguchi, M. Dynamic and reversible polymorphism of self-assembled lyotropic liquid crystalline systems derived from cyclic bis(ethynylhelicene) oligomers. J. Am. Chem. Soc. 2015, 137, 6594–6601. [Google Scholar] [CrossRef] [PubMed]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1169. [Google Scholar] [CrossRef] [PubMed]
- Vericat, C.; Vela, M.E.; Corthey, G.; Pensa, E.; Cortés, E.; Fonticelli, M.H.; Ibanez, F.; Benitez, G.E.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiolates on metals: A review article on sulfur-metal chemistry and surface structures. RSC Adv. 2014, 4, 27730–27754. [Google Scholar] [CrossRef]
- Casalini, S.; Bortolotti, C.A.; Leonardi, F.; Biscarini, F. Self-assembled monolayers in organic electronics. Chem. Soc. Rev. 2017, 46, 40–71. [Google Scholar] [CrossRef] [PubMed]
- Pieters, G.; Prins, L.J. Catalytic self-assembled monolayers on gold. New J. Chem. 2012, 36, 1931–1939. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sugiura, H.; Amemiya, R.; Aikawa, H.; An, Z.; Yamaguchi, M.; Mizukami, M.; Kurihara, K. Formation of double helix self-assembled monolayers of ethynylhelicene oligomer disulfides on gold surfaces. Tetrahedron 2011, 67, 5972–5978. [Google Scholar] [CrossRef]
- Moores, B.; Drolle, E.; Attwood, S.J.; Simons, J.; Leonenko, Z. Effect of Surfaces on Amyloid Fibril Formation. PLoS ONE 2011, 6, e25954. [Google Scholar] [CrossRef] [PubMed]
- Brodoceanu, D.; Bauer, C.T.; Kroner, E.; Arzt, E.; Kraus, T. Hierarchical bioinspired adhesive surfaces-a review. Bioinspir. Biomim. 2016, 11, 051001. [Google Scholar] [CrossRef] [PubMed]
- Shigeno, M.; Sawato, T.; Yamaguchi, M. Fibril film formation of pseudoenantiomeric oxymethylenehelicene oligomers at the liquid–solid interface: Structural changes, aggregation and discontinuous heterogeneous nucleation. Chem. Eur. J. 2015, 21, 17676–17682. [Google Scholar] [CrossRef] [PubMed]
- Helen, W.; de Leonardis, P.; Ulijn, R.V.; Gough, J.; Tirelli, N. Mechanosensitive peptide gelation: Mode of agitation controls mechanical properties and nano-scale morphology. Soft Matter 2011, 7, 1732–1740. [Google Scholar] [CrossRef]
- Maity, S.; Kumar, P.; Haldar, D. Sonication-induced instant amyloid-like fibril formation and organogelation by a tripeptide. Soft Matter 2011, 7, 5239–5245. [Google Scholar] [CrossRef]
- Okano, K.; Arteaga, O.; Ribo, J.M.; Yamashita, T. Emergence of chiral environments by effect of flows: The case of an ionic oligomer and Congo red dye. Chem. Eur. J. 2011, 17, 9288–9292. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-W.; Ma, J.-T.; Chen, C.-F. Structure–property relationship of a class of efficient organogelators and their multistimuli responsiveness. Tetrahedron 2011, 67, 85–91. [Google Scholar] [CrossRef]
- Okano, K.; Taguchi, M.; Fujiki, M.; Yamashita, T. Circularly polarized luminescence of rhodamine B in a supramolecular chiral medium formed by a vortex flow. Angew. Chem. Int. Ed. 2011, 50, 12474–12477. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Srivastava, A. Mechano-responsive gelation of water by a short alanine-derivative. Soft Matter 2014, 10, 4863–4868. [Google Scholar] [CrossRef] [PubMed]
- Van Herpt, J.T.; Stuart, M.C.A.; Browne, W.R.; Feringa, B.L. Mechanically induced gel formation. Langmuir 2013, 29, 8763–8767. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, A.J.P.; Nieuwenhuizen, M.M.L.; Rodríguez-Llansola, F.; Palmans, A.R.A.; Meijer, E.W. Mechanically induced gelation of a kinetically trapped supramolecular polymer. Macromolecules 2014, 47, 8429–8436. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, T.; Shi, L.; Tang, Z.; Liu, M. Strong circularly polarized luminescence from the supramolecular gels of an achiral gelator: Tunable intensity and handedness. Chem. Sci. 2015, 6, 4267–4272. [Google Scholar] [CrossRef] [PubMed]
- Sawato, T.; Saito, N.; Shigeno, M.; Yamaguchi, M. Mechanical stirring induces heteroaggregate formation and self-assembly of pseudoenantiomeric oxymethylene helicene oligomers in solution. ChemSelect 2017, 2, 2205–2211. [Google Scholar] [CrossRef]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, E.; Comenge, J.; Paramelle, D.; Volk, M.; Chen, Q.; Lévy, R. Characterizing self-assembled monolayers on gold nanoparticles. Bioconjug. Chem. 2017, 28, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; An, Z.; Saito, N.; Yamaguchi, M. Fluorescent gold nanoparticles: Synthesis of composite materials of two-component disulfide gels and gold nanoparticles. Chem. Eur. J. 2013, 19, 10580–10588. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, W.; Miyagawa, M.; An, Z.; Yamaguchi, M. Optical resolution of aromatic alcohols using silica nanoparticles grafted with helicene. Org. Lett. 2012, 14, 3123–3125. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Ichinose, W.; Yamaguchi, M. Equilibrium shift in solution: Molecular shape recognition and precipitation of a synthetic double helix using helicene-grafted silica nanoparticles. Chem. Eur. J. 2014, 20, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Yamaguchi, M. Helicene-grafted silica nanoparticles capture hetero-double-helix Iintermediates during self-assembly gelation. Chem. Eur. J. 2015, 21, 8408–8415. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Arisawa, M.; Yamaguchi, M. Equilibrium shift induced by chiral nanoparticle precipitation in rhodium-catalyzed disulfide exchange reaction. Tetrahedron 2015, 71, 4920–4926. [Google Scholar] [CrossRef]
- Hardin, P.E. Molecular genetic analysis of circadian timekeeping in Drosophila. Adv. Genet. 2011, 74, 141–173. [Google Scholar] [CrossRef] [PubMed]
- Green, C.B.; Takahashi, J.S.; Bass, J. The meter of metabolism. Cell 2008, 134, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Schibler, U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011, 13, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Paschos, G.; Curtis, A.M.; Musiek, E.S.; McLoughlin, S.C.; FitzGerald, G.A. Knitting up the raveled sleave of care. Sci. Transl. Med. 2013, 5, 212rv3. [Google Scholar] [CrossRef] [PubMed]
- Umemura, Y.; Koike, N.; Matsumoto, T.; Yoo, S.-H.; Chen, Z.; Yasuhara, N.; Takahashi, J.S.; Yagita, K. Transcriptional program of Kpna2/Importin-α2 regulates cellular differentiation-coupled circadian clock development in mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5039–E5048. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Yamaguchi, M. Material clocking by silica nanoparticle precipitation in solution phase that is tunable by organic molecules. ChemPlusChem 2015, 80, 1502–1507. [Google Scholar] [CrossRef]
- Kushida, Y.; Sawato, T.; Saito, N.; Shigeno, M.; Satozono, H.; Yamaguchi, M. Spatially heterogeneous nature of self-catalytic reaction in hetero-double helix formation of helicene oligomers. ChemPhysChem 2016, 17, 3283–3288. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (M)-1 | (M)-2 | (M)-3 | (M)-4 | (M)-5 | (M)-6 | (M)-7 | (M)-8 | |
|---|---|---|---|---|---|---|---|---|
| (P)-1 | C | S | S | S | S | S | Type II | |
| (P)-2 | C | S | S | S | S | |||
| (P)-3 | 1 | 2.5 | 2.5-5.0 | 0.25–0.5 | 1 | 1 | ||
| (P)-4 | 1 | 0.05–0.1 | 0.05–0.15 | 1 | ||||
| (P)-5 | 0.25 | 0.1 | Type I | |||||
| (P)-6 | 0.25 | |||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, N.; Yamaguchi, M. Synthesis and Self-Assembly of Chiral Cylindrical Molecular Complexes: Functional Heterogeneous Liquid-Solid Materials Formed by Helicene Oligomers. Molecules 2018, 23, 277. https://doi.org/10.3390/molecules23020277
Saito N, Yamaguchi M. Synthesis and Self-Assembly of Chiral Cylindrical Molecular Complexes: Functional Heterogeneous Liquid-Solid Materials Formed by Helicene Oligomers. Molecules. 2018; 23(2):277. https://doi.org/10.3390/molecules23020277
Chicago/Turabian StyleSaito, Nozomi, and Masahiko Yamaguchi. 2018. "Synthesis and Self-Assembly of Chiral Cylindrical Molecular Complexes: Functional Heterogeneous Liquid-Solid Materials Formed by Helicene Oligomers" Molecules 23, no. 2: 277. https://doi.org/10.3390/molecules23020277