Carbonic Anhydrase Inhibitors as Novel Drugs against Mycobacterial β-Carbonic Anhydrases: An Update on In Vitro and In Vivo Studies

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. In Vitro Inhibition Studies of M. tuberculosis β-CAs
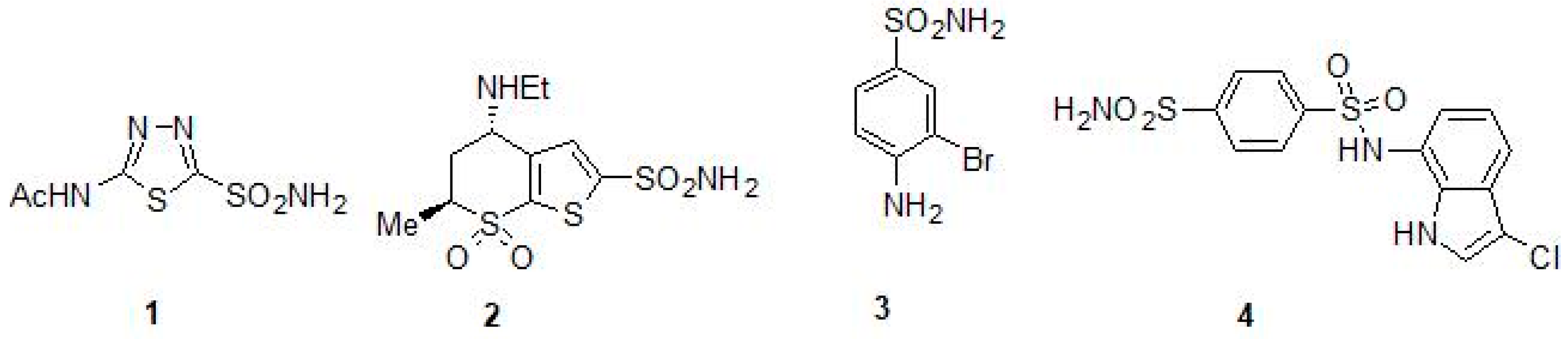
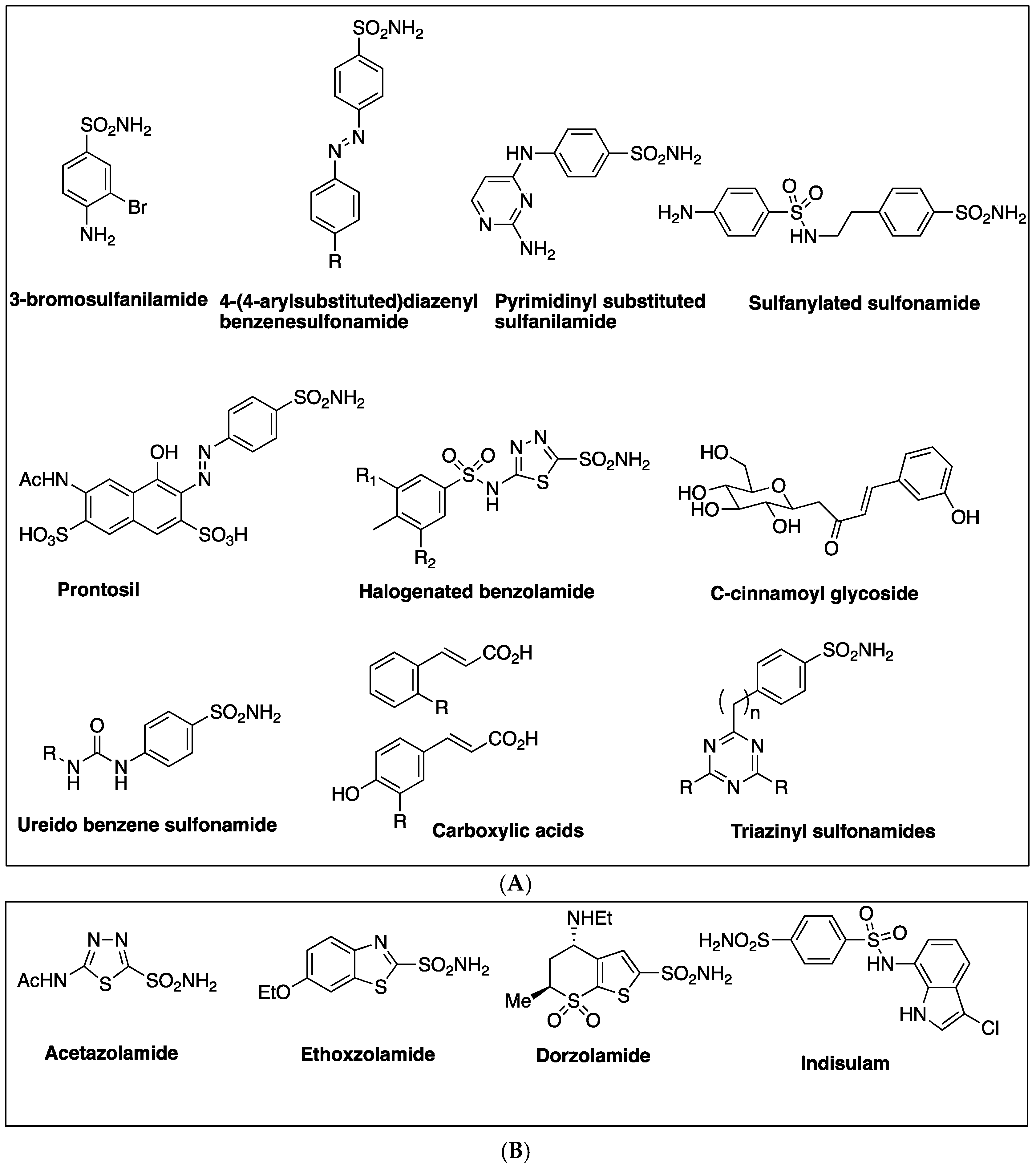
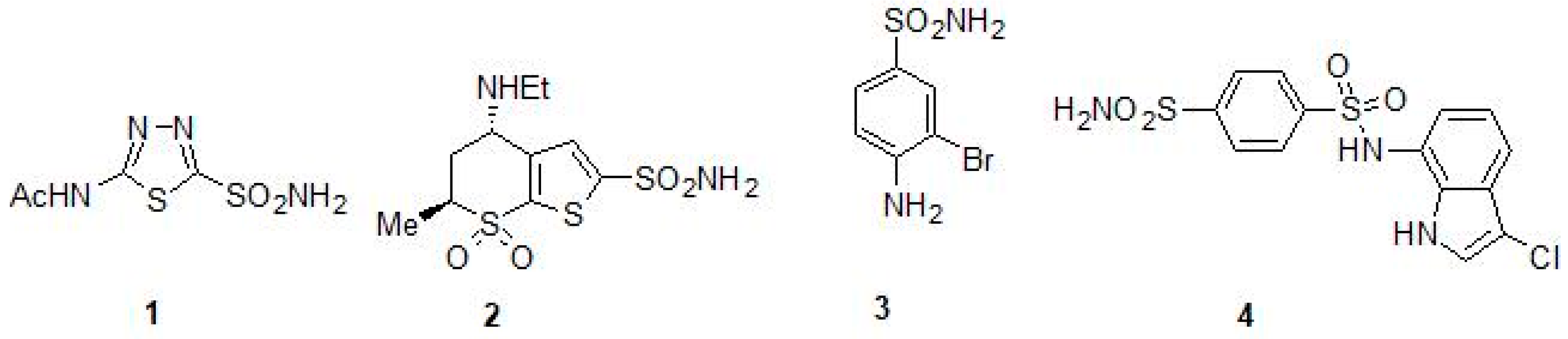
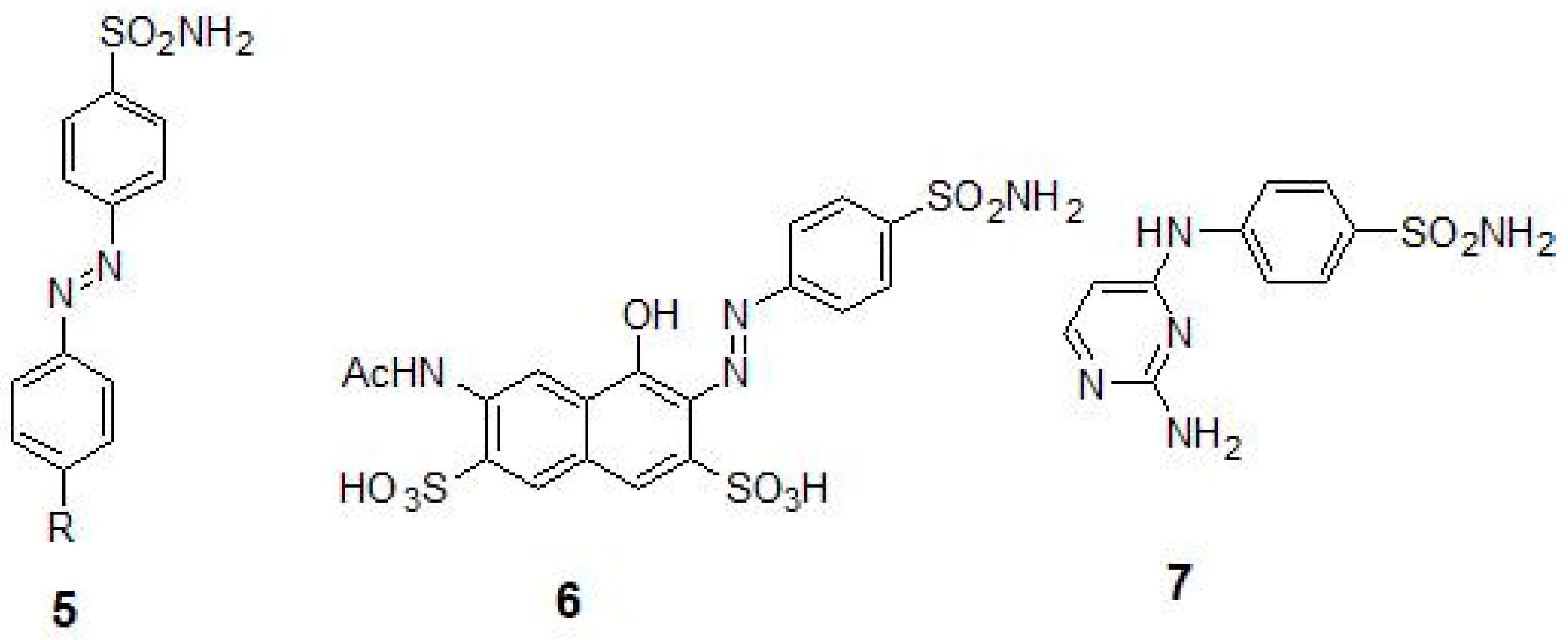
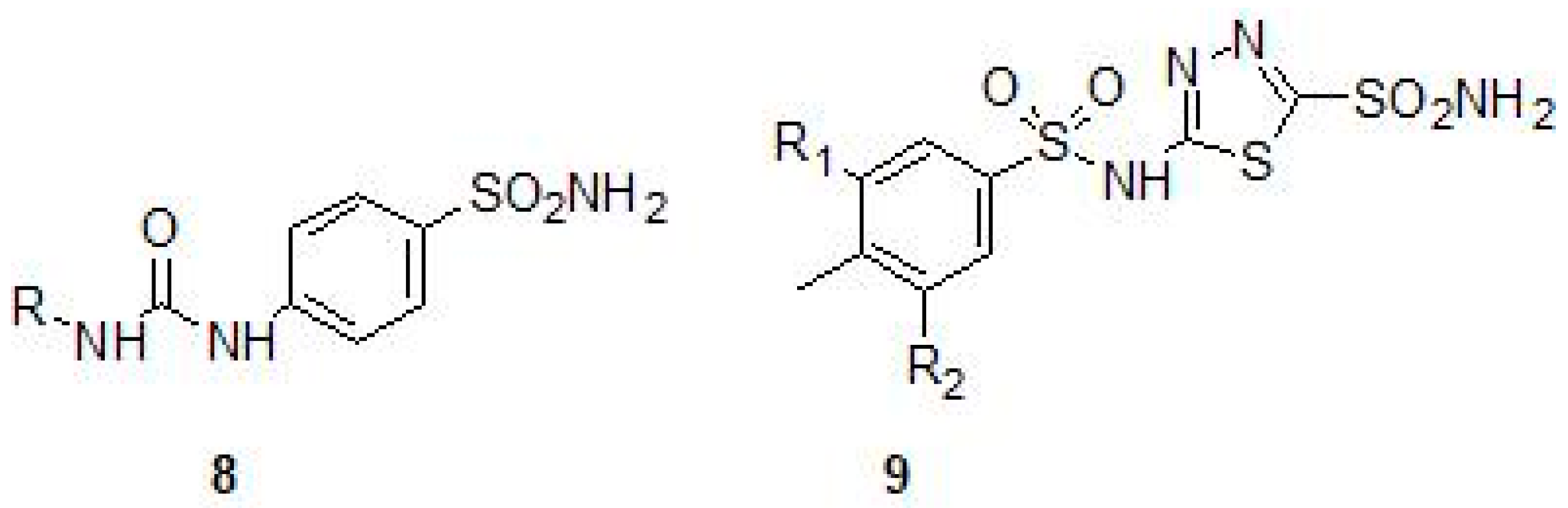
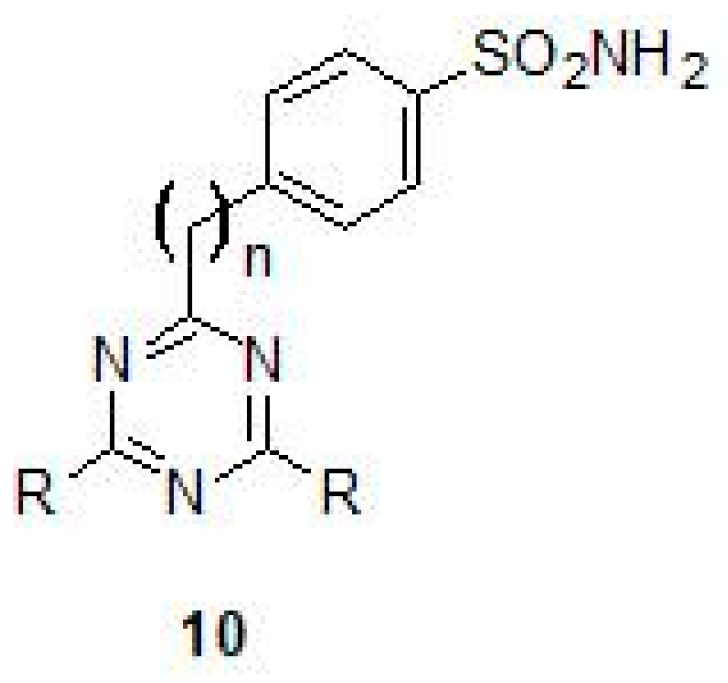
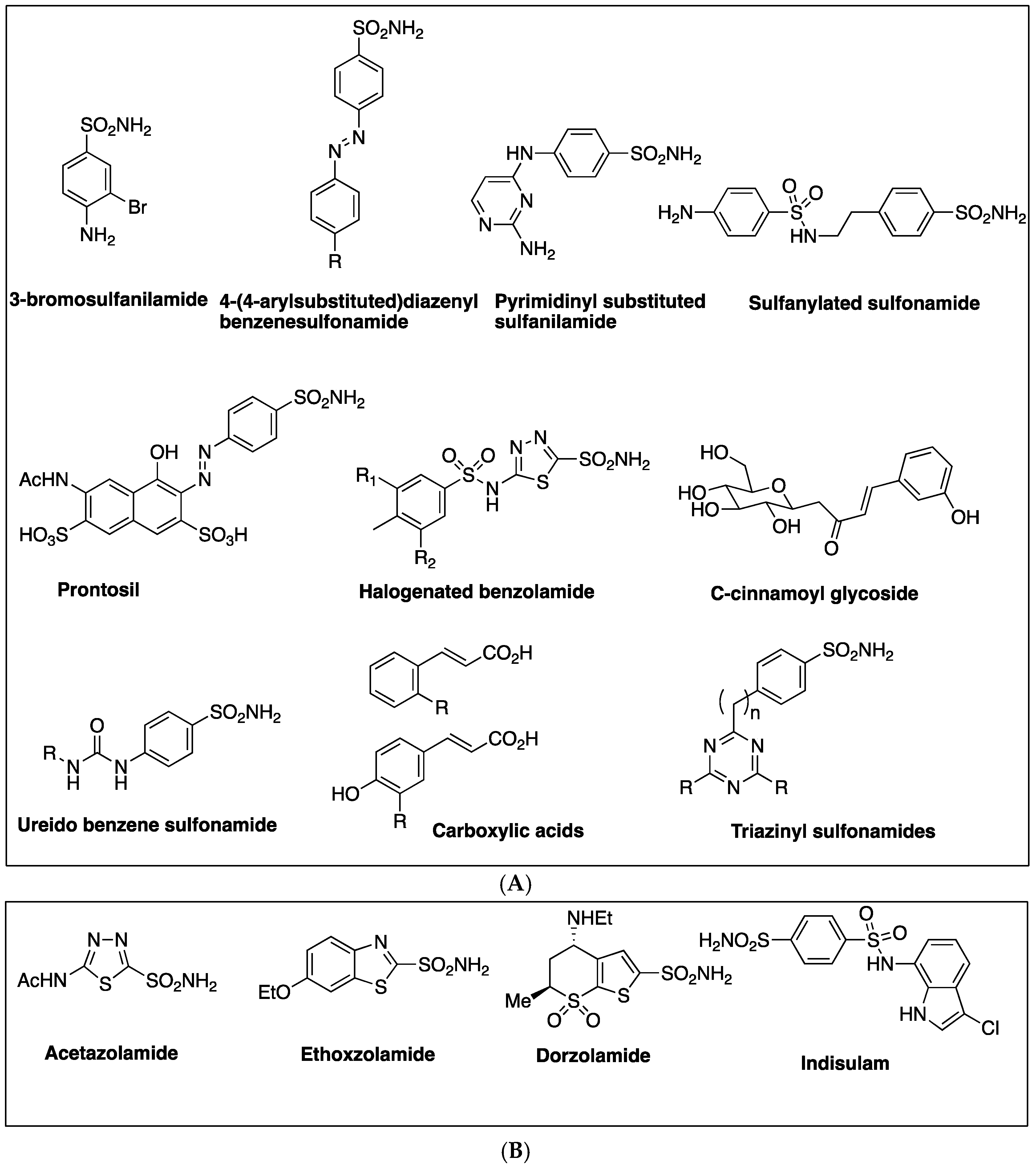
2.1. Sulfonamides as Inhibitors of M. tuberculosis β-CAs
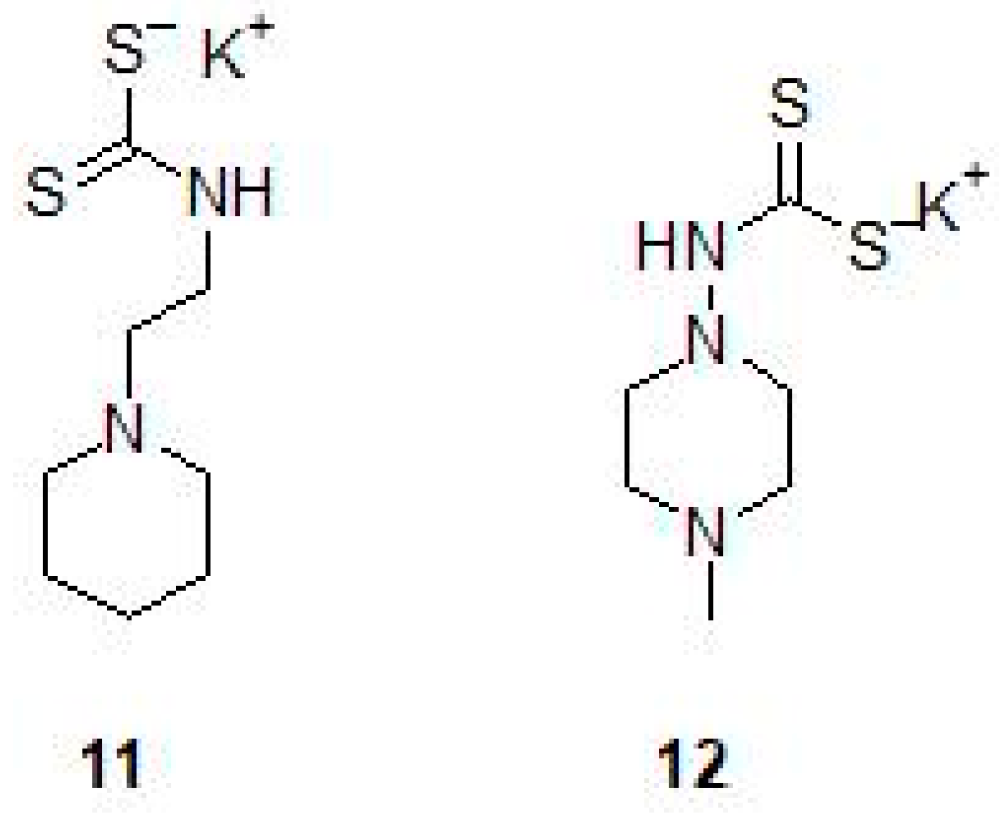
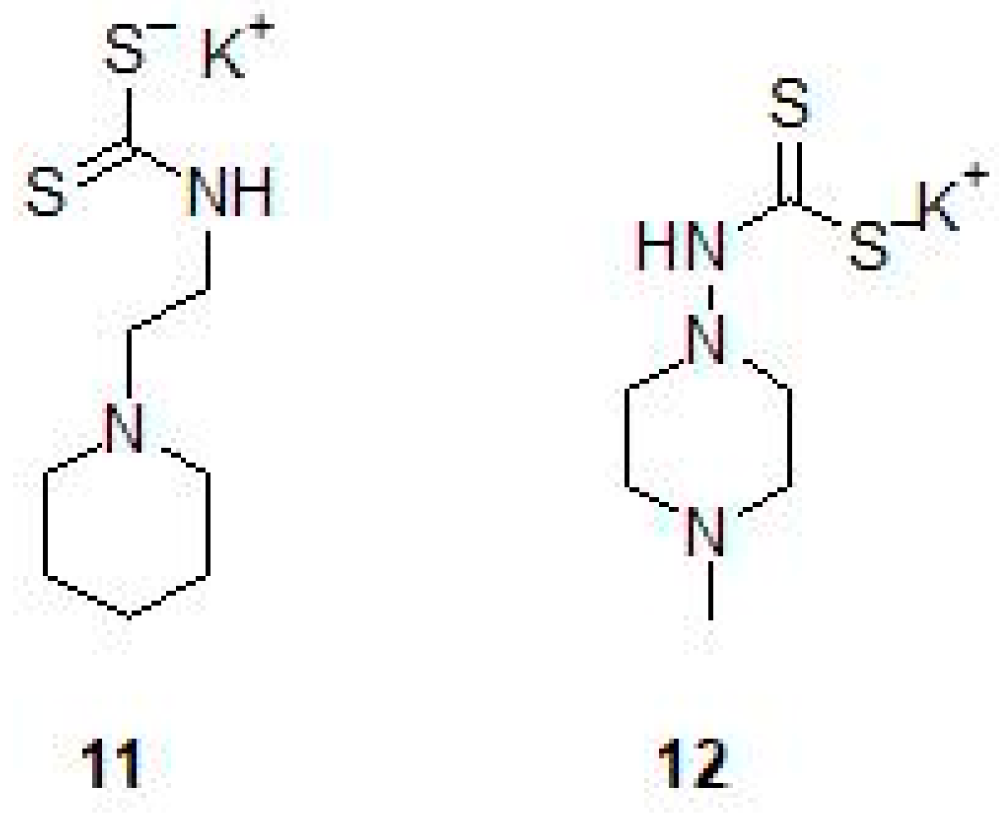
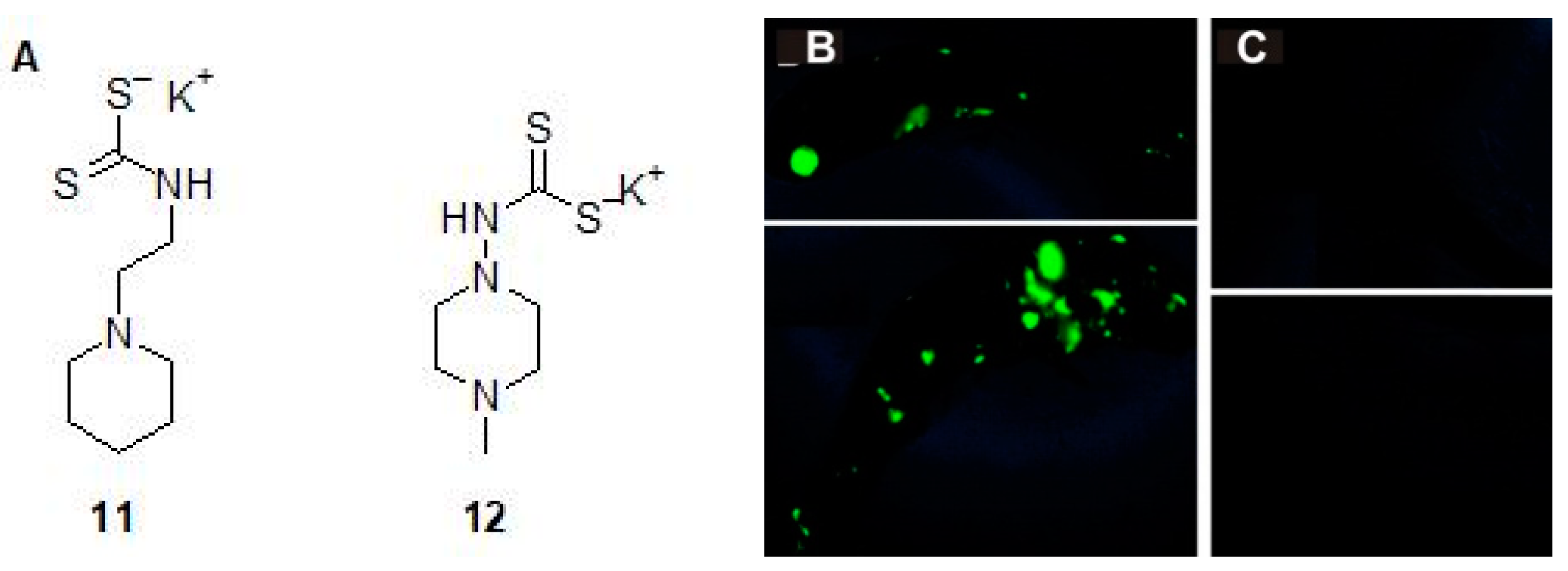
2.2. Mono and Dithiocarbamates
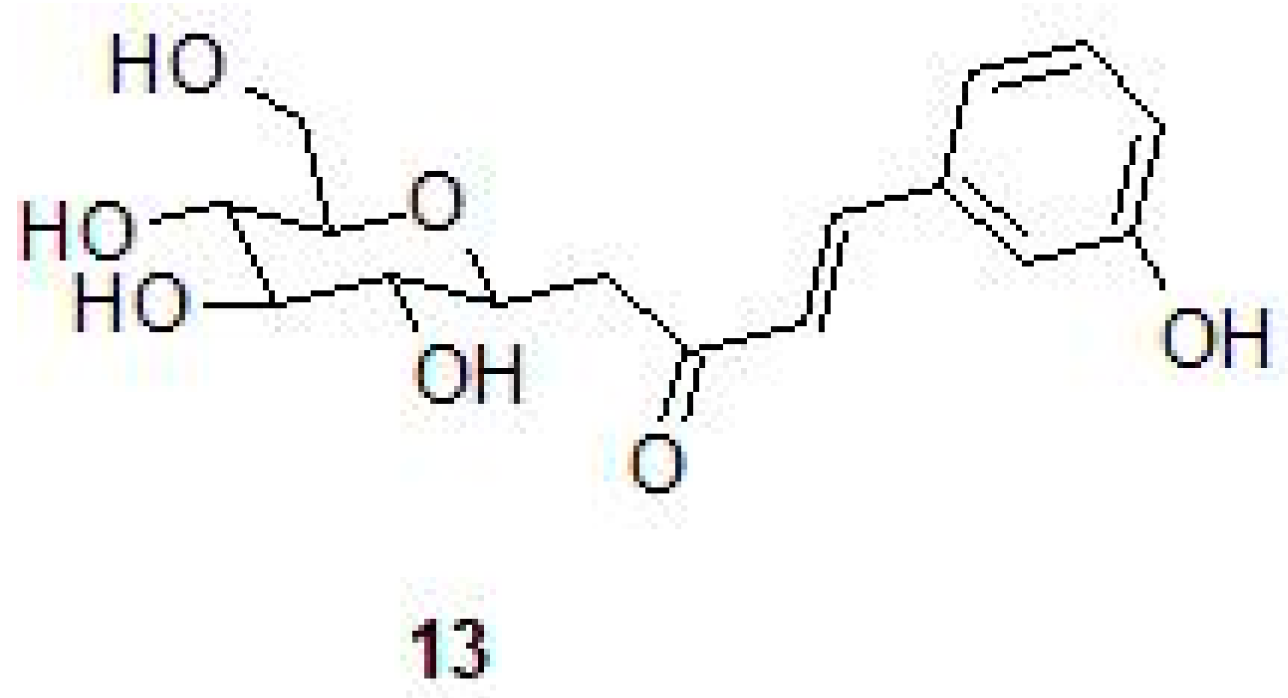
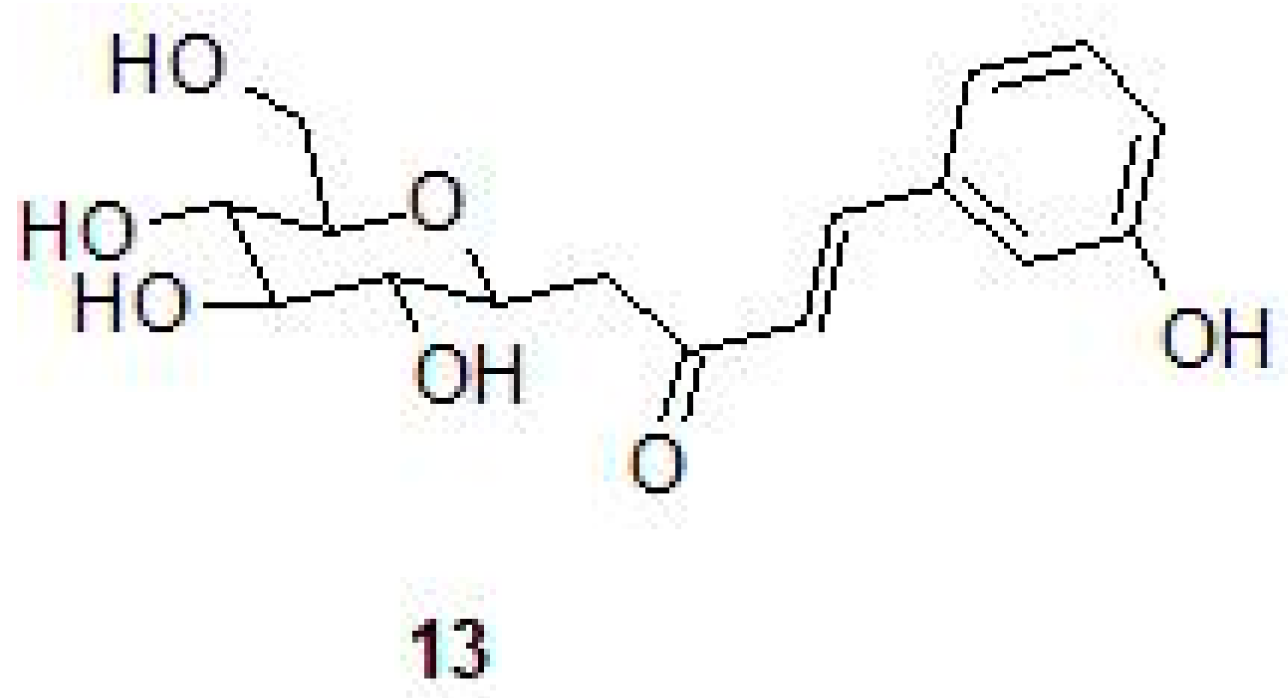
2.3. Phenolic Natural Products and Phenolic Acids
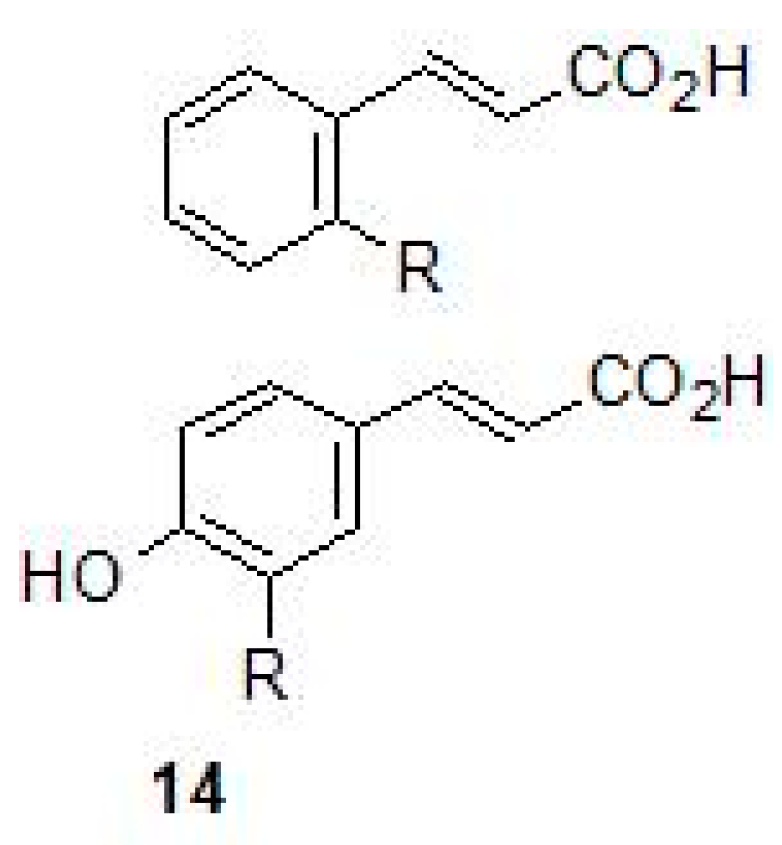
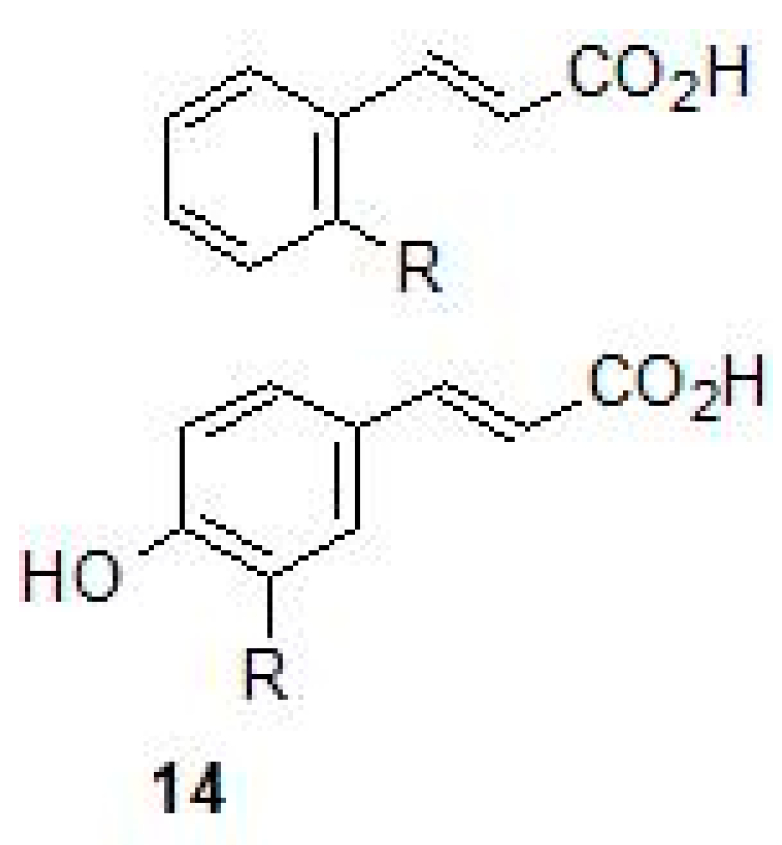
2.4. Carboxylic Acids
3. In Vitro Inhibition of Mycobacterial strains Using CA Inhibitors
4. CA Inhibitors and In Vivo Inhibition of Mycobacteria
5. Future Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CA | carbonic anhydrase |
| Mtb | Mycobacterium tuberculosis |
| MIC | minimal inhibitory concentration |
| CAI | carbonic anhydrase inhibitor |
| TB | tuberculosis |
| MTC | Mycobacterium tuberculosis complex |
| NTM | nontuberculous mycobacteria |
| EZA | ethoxzolamide |
| DTCs | dithiocarbamates |
| ATZ | acetazolamide |
| DZA | dorzolamide |
| eDNA | extracellular DNA |
References
- Stahl, D.A.; Urbance, J.W. The division between fast- and slow-growing species corresponds to natural relationships among the mycobacteria. J. Bacteriol. 1990, 172, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Kim, N.H.; Song, K.H.; Choe, P.G.; Kim, E.S.; Park, S.W.; Kim, H.B.; Kim, N.J.; Kim, E.C.; Park, W.B.; et al. Differentiating rapid- and slow-growing mycobacteria by difference in time to growth detection in liquid media. Diagn. Microbiol. Infect. Dis. 2013, 75, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Gupta, A.; Prakash, P.; Anupurba, S.; Tripathi, R.; Srivastava, G.N. Differentiation of Mycobacterium tuberculosis complex from non-tubercular mycobacteria by nested multiplex PCR targeting IS6110, MTP40 and 32kD alpha antigen encoding gene fragments. BMC Infect. Dis. 2016, 16, 123. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018; pp. 1–277. ISBN 978-92-4-156564-6. [Google Scholar]
- WHO. World Health Organization Lepracy: Fact Sheet on Leprosy; World Health Organization: Geneva, Switzerland, 2017; pp. 1–5. [Google Scholar]
- Daley, C.L.; Griffith, D.E. Pulmonary non-tuberculous mycobacterial infections. Int. J. Tuberc. Lung Dis. 2010, 14, 665–671. [Google Scholar] [PubMed]
- Adrados, B.; Julián, E.; Codony, F.; Torrents, E.; Luquin, M.; Morató, J. Prevalence and concentration of non-tuberculous mycobacteria in cooling towers by means of quantitative PCR: A prospective study. Curr. Microbiol. 2011, 62, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.C.; Khera-Butler, T.; James, P.; Oakley, B.B.; Erenso, G.; Aseffa, A.; Knight, R.; Wellington, E.M.; Courtenay, O. Environmental reservoirs of pathogenic mycobacteria across the Ethiopian biogeographical landscape. PLoS ONE 2017, 12, e0173811. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O. Ecology of nontuberculous mycobacteria--where do human infections come from? Semin. Respir. Crit. Care Med. 2013, 34, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, K.; Singh, S. Non-tuberculous mycobacteria in TB-endemic countries: Are we neglecting the danger? PLoS Negl. Trop. Dis. 2010, 4, e615. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.M.; Odell, J.A. Nontuberculous mycobacterial pulmonary infections. J. Thorac. Dis. 2014, 6, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Faria, S.; Joao, I.; Jordao, L. General Overview on Nontuberculous Mycobacteria, Biofilms, and Human Infection. J. Pathog. 2015, 809014. [Google Scholar] [CrossRef] [PubMed]
- Misch, E.A.; Saddler, C.; Davis, J.M. Skin and Soft Tissue Infections Due to Nontuberculous Mycobacteria. Curr. Infect. Dis. Rep. 2018, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghafli, H.; Al-Hajoj, S. Nontuberculous Mycobacteria in Saudi Arabia and Gulf Countries: A Review. Can. Respir. J. 2017, 5035932. [Google Scholar] [CrossRef] [PubMed]
- Nishiuchi, Y.; Iwamoto, T.; Maruyama, F. Infection Sources of a Common Non-tuberculous Mycobacterial Pathogen, Mycobacterium avium Complex. Front. Med. 2017, 27. [Google Scholar] [CrossRef] [PubMed]
- Fedrizzi, T.; Meehan, C.J.; Grottola, A.; Giacobazzi, E.; Fregni Serpini, G.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; Bertorelli, R.; De Sanctis, V.; et al. Genomic characterization of Nontuberculous Mycobacteria. Sci. Rep. 2017, 7, 45258. [Google Scholar] [CrossRef] [PubMed]
- Stinear, T.P.; Seemann, T.; Harrison, P.F.; Jenkin, G.A.; Davies, J.K.; Johnson, P.D.; Abdellah, Z.; Arrowsmith, C.; Chillingworth, T.; Churcher, C.; et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008, 18, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Vissa, V.D.; Brennan, P.J. The genome of Mycobacterium leprae: A minimal mycobacterial gene set. Genome Biol. 2001, 2, Reviews1023. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Eiglmeier, K.; Parkhill, J.; James, K.D.; Thomson, N.R.; Wheeler, P.R.; Honoré, N.; Garnier, T.; Churcher, C.; Harris, D.; et al. Massive gene decay in the leprosy bacillus. Nature 2001, 409, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Showalter, H.D.; Denny, W.A. A roadmap for drug discovery and its translation to small molecule agents in clinical development for tuberculosis treatment. Tuberculosis 2008, 88, S3–S17. [Google Scholar] [CrossRef]
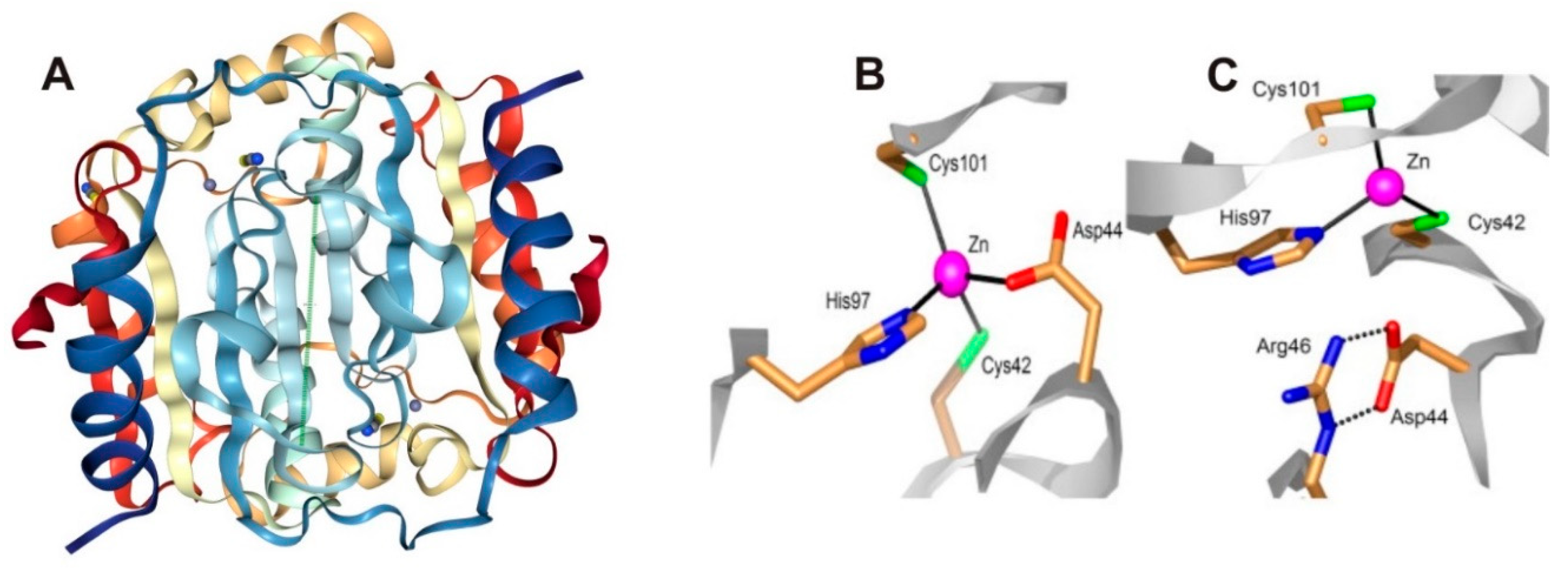
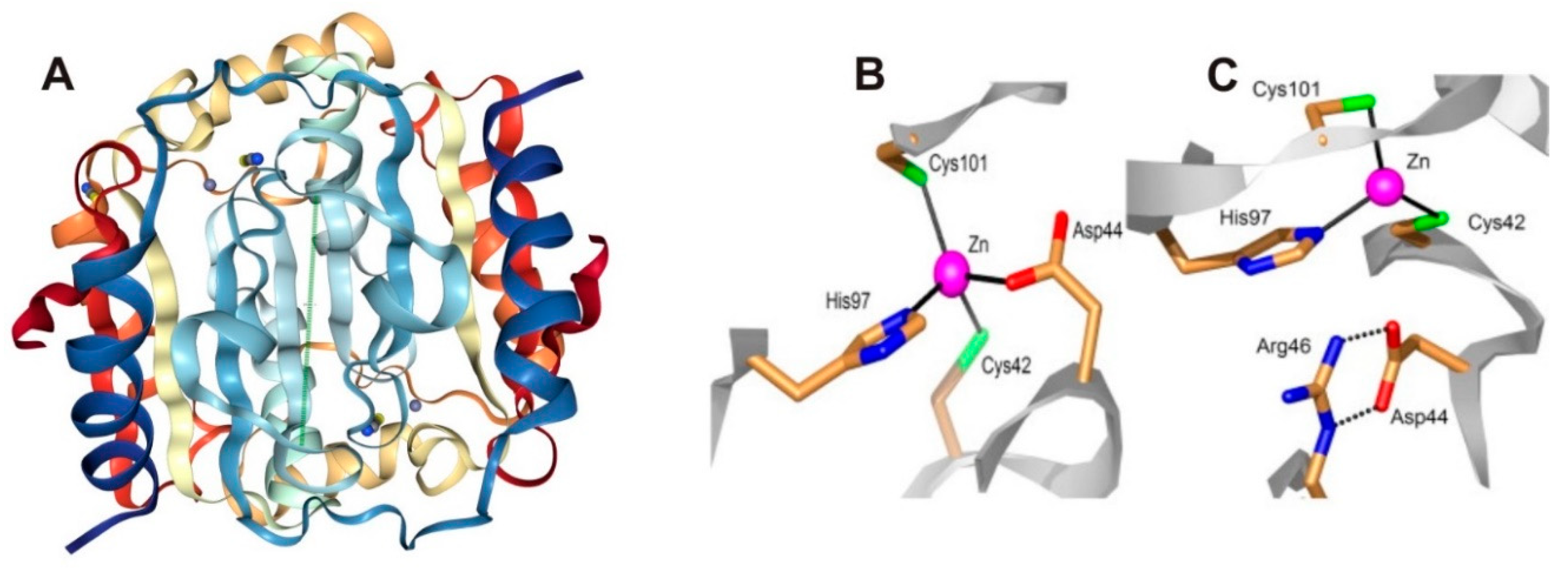
- Covarrubias, A.S.; Larsson, A.M.; Högbom, M.; Lindberg, J.; Bergfors, T.; Björkelid, C.; Mowbray, S.L.; Unge, T.; Jones, T.A. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 18782–18789. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.S.; Bergfors, T.; Jones, T.A.; Högbom, M. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 4993–4999. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.-Y.; Stephan, K.; Scozzafava, A.; Montero, J.-L.; Supuran, C.T. Targeting bacterial metalloenzymes: A new strategy for the development of anti-infective agents. Anti-Infect. Agents Med. Chem. 2008, 7, 169–179. [Google Scholar] [CrossRef]
- Kapopoulou, A.; Lew, J.M.; Cole, S.T. The MycoBrowser portal: A comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011, 91, 8–13. [Google Scholar] [CrossRef] [PubMed]
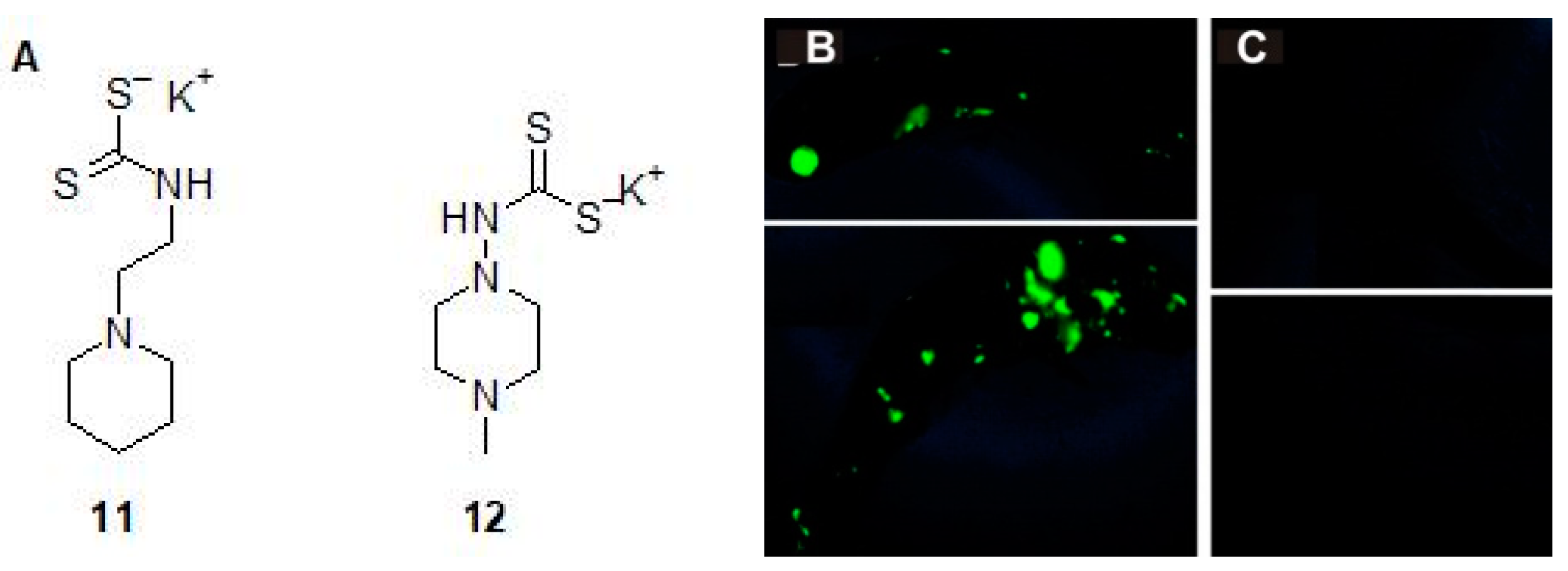
- Aspatwar, A.; Hammarén, M.; Koskinen, S.; Luukinen, B.; Barker, H.; Carta, F.; Supuran, C.T.; Parikka, M.; Parkkila, S. beta-CA-specific inhibitor dithiocarbamate Fc14-584B: A novel antimycobacterial agent with potential to treat drug-resistant tuberculosis. J. Enzyme Inhib. Med. Chem. 2017, 32, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Minakuchi, T.; Nishimori, I.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Molecular cloning, characterization, and inhibition studies of the Rv1284 beta-carbonic anhydrase from Mycobacterium tuberculosis with sulfonamides and a sulfamate. J. Med. Chem. 2009, 52, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, I.; Minakuchi, T.; Vullo, D.; Scozzafava, A.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new beta-carbonic anhydrase from Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 3116–3120. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Maresca, A.; Covarrubias, A.S.; Mowbray, S.L.; Jones, T.A.; Supuran, C.T. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active beta-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg. Med. Chem. Lett. 2009, 19, 6649–6654. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, I.; Minakuchi, T.; Morimoto, K.; Sano, S.; Onishi, S.; Takeuchi, H.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J. Med. Chem. 2006, 49, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.J.; Bermudez, L.E. Identification of Bicarbonate as a Trigger and Genes Involved with Extracellular DNA Export in Mycobacterial Biofilms. MBio 2016, 7, e01597-16. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Colvin, C.J.; Needle, D.B.; Mba Medie, F.; Champion, P.A.; Abramovitch, R.B. The Carbonic Anhydrase Inhibitor Ethoxzolamide Inhibits the Mycobacterium tuberculosis PhoPR Regulon and Esx-1 Secretion and Attenuates Virulence. Antimicrob Agents Chemother. 2015, 59, 4436–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimori, I.; Minakuchi, T.; Maresca, A.; Carta, F.; Scozzafava, A.; Supuran, C.T. The beta-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr. Pharm. Des. 2010, 16, 3300–3309. [Google Scholar] [CrossRef] [PubMed]
- Güzel, O.; Maresca, A.; Scozzafava, A.; Salman, A.; Balaban, A.T.; Supuran, C.T. Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial beta-carbonic anhydrases Rv1284 and Rv3273. J. Med. Chem. 2009, 52, 4063–4067. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of the Rv1284 and Rv3273 beta-carbonic anhydrases from Mycobacterium tuberculosis with diazenylbenzenesulfonamides. Bioorg. Med. Chem. Lett. 2009, 19, 4929–4932. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Maresca, A.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Carbonic anhydrase inhibitors. Diazenylbenzenesulfonamides are potent and selective inhibitors of the tumor-associated isozymes IX and XII over the cytosolic isoforms I and II. Bioorg. Med. Chem. 2009, 17, 7093–7099. [Google Scholar] [CrossRef] [PubMed]
- Pacchiano, F.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Inhibition of beta-carbonic anhydrases with ureido-substituted benzenesulfonamides. Bioorg. Med. Chem. Lett. 2011, 21, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three beta-class carbonic anhydrases from Mycobacterium tuberculosis. J. Enzym. Inhib. Med. Chem. 2013, 28, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Ceruso, M.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Sulfonamides incorporating fluorine and 1,3,5-triazine moieties are effective inhibitors of three beta-class carbonic anhydrases from Mycobacterium tuberculosis. J. Enzym. Inhib. Med. Chem. 2014, 29, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Wani, T.V.; Bua, S.; Khude, P.S.; Chowdhary, A.H.; Supuran, C.T.; Toraskar, M.P. Evaluation of sulphonamide derivatives acting as inhibitors of human carbonic anhydrase isoforms I, II and Mycobacterium tuberculosis beta-class enzyme Rv3273. J. Enzym. Inhib. Med. Chem. 2018, 33, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Carta, F.; Vullo, D.; Supuran, C.T. Dithiocarbamates strongly inhibit the beta-class carbonic anhydrases from Mycobacterium tuberculosis. J. Enzym. Inhib. Med. Chem. 2013, 28, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Hofmann, A.; Osman, A.; Hall, R.A.; Mühlschlegel, F.A.; Vullo, D.; Innocenti, A.; Supuran, C.T.; Poulsen, S.A. Natural product-based phenols as novel probes for mycobacterial and fungal carbonic anhydrases. J. Med. Chem. 2011, 54, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Cau, Y.; Mori, M.; Supuran, C.T.; Botta, M. Mycobacterial carbonic anhydrase inhibition with phenolic acids and esters: Kinetic and computational investigations. Org. Biomol. Chem. 2016, 14, 8322–8330. [Google Scholar] [CrossRef] [PubMed]
- Buchieri, M.V.; Riafrecha, L.E.; Rodríguez, O.M.; Vullo, D.; Morbidoni, H.R.; Supuran, C.T.; Colinas, P.A. Inhibition of the beta-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: Identification of the first inhibitor with anti-mycobacterial activity. Bioorg. Med. Chem. Lett. 2013, 23, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Vullo, D.; Scozzafava, A.; Manole, G.; Supuran, C.T. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J. Enzym. Inhib. Med. Chem. 2013, 28, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Antimycobacterial activity of 9-sulfonylated/sulfenylated-6-mercaptopurine derivatives. Bioorg. Med. Chem. Lett. 2001, 11, 1675–1678. [Google Scholar] [CrossRef]
- Rose, S.J.; Babrak, L.M.; Bermudez, L.E. Mycobacterium avium Possesses Extracellular DNA that Contributes to Biofilm Formation, Structural Integrity, and Tolerance to Antibiotics. PLoS ONE 2015, 10, e0128772. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Abramovitch, R.B. Small Molecules That Sabotage Bacterial Virulence. Trends Pharmacol. Sci. 2017, 38, 339–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAs | Gene ID | Protein | Kcat (s−1) | Kcat/Km (M−1 s−1) | Activity | KI (nM) a | Reference |
|---|---|---|---|---|---|---|---|
| Mtb β-CA1 | Rv1284 | 163 aa | 3.9 × 105 | 3.7 × 107 | Moderate | 480 | [27] |
| Mtb β-CA2 | Rv3588c | 207 aa | 9.8 × 105 | 9.3 × 107 | High | 9.8 | [29] |
| Mtb β-CA3 | Rv3273 | b 215 aa | 4.3 × 105 | 4.0 × 107 | Moderate | 104 | [28] |
| hCAII | CA2 | 260 aa | 1.4 × 106 | 1.5 × 108 | Very high | 12 | [29] |
| CAIs | hCAII | β-CA1 | β-CA2 | β-CA3 | Reference |
|---|---|---|---|---|---|
| 3-bromosulfanilamide | 40 | 186 | NS | NS | [27] |
| Diazenylbenzenesulfonamides | 105 | - | 45–955 | - | [29] |
| 2-amino-pyrimidin-4-yl-sulfanilamide | 33 | - | NA | 91 | [28] |
| Sulfonylated sulfonamide | NS | - | - | 170 | [28] |
| prontosil | NS | 126 | 148 | [39] | |
| Halogenated benzolamides | NS | 120–580 | 410–450 | 170–340 | [42] |
| N-mono- and N,N-dithiocarbamates | 0.7–325 | 0.9–481 | NA | 0.91–431 | [45] |
| Ureido-sulfonamides | 2.1–226 | 5–560 | NA | 6.4–533 | [41] |
| Cinnamoyl glycosides | NS | 140 | 130–640 | - | [48] |
| Triazinyl Sulfonamides | 4.9–5.5 | 42–580 | 8.1–10 | 2.1–210 | [43] |
| a Acetazolamide (ATZ) | 12 | 481 | 9 | 104 | [29] |
| a Ethoxzolamide (EZA) | 8 | - | 594 | 27 | [29] |
| a Dorzolamide (DZA) | 9 | 744 | 99 | 137 | [29] |
| a Indisulam | 15 | 97 | NS | NS | [27] |
| Inhibitor | Bacilli | Concentration | Reference |
|---|---|---|---|
| (E)-1-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-4-(3-hydroxyphenyl) but-3-en-2-one | Mtb H37Rv | b 3.125–6.25 μg/mL | [48] |
| 100 μg/mL | [48] | ||
| DTCs (Fc14-584b and Fc14-594a) | M. marinum | 17–18 μg/mL | [26] |
| 9-sulfonylated/sulfenylated-6-mercaptopurines | Mtb H37Rv | 0.39–3.39 μg/mL | [50] |
| 9-sulfonylated/sulfenylated-6-mercaptopurines | a Mtb | 1 μg/mL | [50] |
| Inhibitor | Bacterium | Inhibitory Effect | References |
|---|---|---|---|
| EZA | M. tuberculosisa | Attenuates virulence, inhibits PhoPR | [36] |
| EZA | M. aviumb | Transport of eDNA and biofilm formation | [35] |
| Dithiocarbamate 12 | M. marinumb | c Impairs grow of bacterium in the larvae | [26] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspatwar, A.; Winum, J.-Y.; Carta, F.; Supuran, C.T.; Hammaren, M.; Parikka, M.; Parkkila, S. Carbonic Anhydrase Inhibitors as Novel Drugs against Mycobacterial β-Carbonic Anhydrases: An Update on In Vitro and In Vivo Studies. Molecules 2018, 23, 2911. https://doi.org/10.3390/molecules23112911
Aspatwar A, Winum J-Y, Carta F, Supuran CT, Hammaren M, Parikka M, Parkkila S. Carbonic Anhydrase Inhibitors as Novel Drugs against Mycobacterial β-Carbonic Anhydrases: An Update on In Vitro and In Vivo Studies. Molecules. 2018; 23(11):2911. https://doi.org/10.3390/molecules23112911
Chicago/Turabian StyleAspatwar, Ashok, Jean-Yves Winum, Fabrizio Carta, Claudiu T. Supuran, Milka Hammaren, Mataleena Parikka, and Seppo Parkkila. 2018. "Carbonic Anhydrase Inhibitors as Novel Drugs against Mycobacterial β-Carbonic Anhydrases: An Update on In Vitro and In Vivo Studies" Molecules 23, no. 11: 2911. https://doi.org/10.3390/molecules23112911