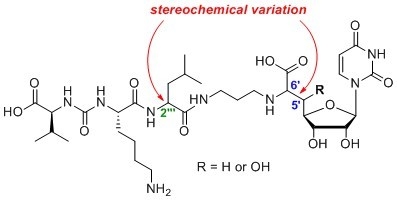
4.1. Synthesis of Muraymycin Epimers
General methods: All chemicals were purchased from standard suppliers. Reactions involving oxygen and/or moisture sensitive reagents were carried out under an atmosphere of argon using anhydrous solvents. Anhydrous solvents were obtained in the following manner. THF was dried over sodium/benzophenone and distilled, CH
2Cl
2 was dried over CaH
2 and distilled, MeOH was dried over activated molecular sieves (3 Å) and degassed. The obtained solvents were stored over molecular sieves (4 Å; in case of MeOH 3 Å). All other solvents were of technical quality and distilled prior to use, and deionised water was used throughout. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM, VWR, Darmstadt, Germany) under flash conditions except where indicated. TLC was performed on aluminium plates precoated with silica gel 60 F
254 (VWR, Darmstadt, Germany). Visualisation of the spots was carried out using UV light (254 nm) and/or staining under heating (H
2SO
4 staining solution: 4 g vanillin, 25 mL conc. H
2SO
4, 80 mL AcOH, and 680 mL MeOH; KMnO
4 staining solution: 1 g KMnO
4, 6 g K
2CO
3, and 1.5 mL 1.25 M NaOH solution, all dissolved in 100 mL H
2O; ninhydrin staining solution: 0.3 g ninhydrin, 3 mL AcOH, and 100 mL 1-butanol). Analytical HPLC was performed on a VWR-Hitachi system equipped with an L-2300 pump, an L-2200 autosampler, an L-2300 column oven (24 °C), an L-2455 Diode Array Detector (DAD), and a LiChroCart
TM column (4 × 125 mm) containing reversed phase silica gel Purospher
TM RP18e (5 μm) purchased from VWR (Darmstadt, Germany). The exact HPLC method is given in the
Supplementary Materials. Preparative HPLC was carried out on a Jasco system equipped with a DG-2080-53 degasser, two PU-2080 Plus pumps, a UV-2075 Plus UV/Vis detector (detection at 260 nm), and a column (21 × 250 mm) containing reversed phase silica gel Nucleodur™ 100-5 C18ec (5 μm) purchased from Macherey-Nagel (Düren, Germany). Method: eluent A water (+0.1% TFA), eluent B 80:20 MeCN:water (+0.1% TFA); 0–30 min gradient of B (10–60%), 30–32 min gradient of B (60–100%), 32–40 min 100% B, 40–42 min gradient of B (100–5%), 42–51 min 5% B; flow 10 mL/min. 300 MHz- and 600 MHz-
1H, and 75 MHz- and 126 MHz-
13C, as well as 282 MHz-
19F NMR spectra were recorded on Varian MERCURY 300, UNITY 300, and INOVA 600 spectrometers (Varian, Palo Alto, CA, USA). All
13C and
19F NMR spectra were
1H-decoupled. All spectra were recorded at room temperature except of samples in DMSO-
d6 and D
2O (standard 35 °C) and where indicated otherwise and were referenced internally to solvent reference frequencies wherever possible. Chemical shifts (δ) are quoted in ppm and coupling constants (
J) are reported in Hz. Assignment of signals was carried out using H,H-COSY, HSQC, and HMBC spectra obtained on the spectrometers mentioned above. The numbering of atoms of muraymycin target structure is depicted in the
Supplementary Material (Figure S20). Mass spectra of small molecules were measured on a Finnigan LCQ ion-trap mass spectrometer or on a Bruker microTOF spectrometer (Bremen, Germany). For ESI measurements in the negative mode, solutions of the compounds in pure MeOH were used, whereas for measurements in the positive mode, solutions in MeOH containing 0.1% formic acid were used. High resolution spectra were measured on a Bruker 7 Tesla Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker, Bremen, Germany). Melting points (mp) were measured on a Büchi instrument (Büchi, Essen, Germany) and are not corrected. Optical rotations were recorded on a PerkinElmer polarimeter 241 (Perkin-Elmer, Hamburg, Germany) with a Na source using a 10 cm cell. Solutions of the compounds (~10 mg) in CHCl
3, MeOH, or water (1 mL) were used, and concentrations are given in g/100 mL. Infrared spectroscopy (IR) was performed on a Jasco (Pfungstadt, Germany) FT/IR-4100 spectrometer equipped with an integrated ATR unit (GladiATR™, PIKE Technologies, Madison WI, USA). Wavenumbers (ν) are quoted in cm
−1. UV spectroscopy of small molecules was carried out on a PerkinElmer (Hamburg, Germany) Lambda 2 spectrometer. Measurements were performed with solutions of ~0.1 mg of the compound in 10 mL MeCN, MeOH, or water and in the range of 190 to 500 nm. Wavelengths of maximum absorption (λ
max) are reported in nm with the corresponding logarithmic molar extinction coefficient (log ε, ε/dm
3 mol
−1 cm
−1) given in parenthesis.
5′-deoxy-l-Leu muraymycin analogue (10) and 5′-deoxy-d-Leu muraymycin analogue (11): To a solution of the urea tripeptide 18 (10 mg, 0.018 mmol) in THF (2.5 mL), 1-hydroxybenzotriazole (HOBt, 2.4 mg, 0.018 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (ECD, 3.5 mg, 0.018 mmol), and N,N-diisopropylethylamine (DIPEA, 3.1 μL, 0.018 mmol) were added and the mixture was stirred at room temperature for 30 min. It was then added to a solution of amine 19 (15 mg, 0.023 mmol) in THF (2 mL) and stirred at room temperature for 20 h. EtOAc (30 mL) was added and the solution was washed with sat. NaHCO3 (30 mL). The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The protected analogues of 10 and 11 were obtained after column chromatography (98:2–95:5, CH2Cl2-MeOH) as colourless solids. This material was dissolved in TFA (80% in water, 2.7 mL) and stirred at room temperature for 24 h. Water (10 mL) was added and the solvent was removed under reduced pressure. The muraymycin analogues 10 (10 mg, 57%) and 11 (3.1 mg, 17%) were separated by preparative HPLC (10: tR = 17.9 min, 11: tR = 16.5 min) and obtained as colourless solids. 10: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.99 (d, J = 6.0 Hz, 3H, 5′′′-Ha), 1.04 (d, J = 6.0 Hz, 3H, 5′′′-Hb), 1.05 (d, J = 7.1 Hz, 3H, 4′′′′′-Ha), 1.08 (d, J = 6.6 Hz, 3H, 4′′′′′-Hb), 1.51–1.60 (m, 2H, 4′′′′-H), 1.66–1.83 (m, 6H, 3′′′-H, 4′′′-H, 3′′′′-Ha, 5′′′′-H), 1.88–1.94 (m, 1H, 3′′′′-Hb), 2.04 (dddd, J = 7.4, 7.2, 6.6, 6.4 Hz, 2H, 2′′-H), 2.28 (dqq, J = 7.1, 6.6, 6.2 Hz, 1H, 3′′′′′-H), 2.43 (ddd, J = 15.0, 9.7, 6.2 Hz, 1H, 5′-Ha), 2.59 (ddd, J = 15.0, 6.7, 2.3 Hz, 1H, 5′-Hb), 3.12 (dd, J = 7.6, 7.6 Hz, 2H, 6′′′′-H), 3.22 (dd, J = 7.4, 7.2 Hz, 2H, 1′′-H), 3.37 (ddd, J = 14.1, 6.6, 6.4 Hz, 1H, 3′′-Ha), 3.44 (ddd, J = 14.1, 6.6, 6.4 Hz, 1H, 3′′-Hb), 4.10 (dd, J = 6.7, 6.2 Hz, 1H, 6′-H), 4.20–4.23 (m, 2H, 3′-H, 2′′′′′-H), 4.24 (dd, J = 8.7, 5.4 Hz, 1H, 2′′′′-H), 4.29 (ddd, J = 9.7, 6.0, 2.3 Hz, 1H, 4′-H), 4.38 (dd, J = 10.8, 4.6 Hz, 1H, 2′′′-H), 4.56 (dd, J = 5.8, 4.0 Hz, 1H, 2′-H), 5.87 (d, J = 4.0 Hz, 1H, 1′-H), 6.01 (d, J = 8.1 Hz, 1H, 5-H), 7.76 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.69 (Ca-4′′′′′), 21.18 (Cb-4′′′′′), 23.37 (Ca-5′′′), 24.71 (C-4′′′′), 24.79 (Cb-5′′′), 27.09 (C-4′′′), 28.30 (C-2′′), 28.93 (C-5′′′′), 32.64 (C-3′′′′′), 33.41 (C-3′′′′), 35.29 (C-5′), 38.64 (C-3′′), 41.92 (C-6′′′′), 42.23 (C-3′′′), 46.93 (C-1′′), 55.20 (C-2′′′), 56.75 (C-2′′′′), 61.50 (C-2′′′′′), 61.73 (C-6′), 75.18 (C-2′), 75.55 (C-3′), 82.46 (C-4′), 94.41 (C-1′), 104.84 (C-5), 118.93 (q, 1JCF = 292.3 Hz, TFA-CF3), 145.32 (C-6), 153.89 (C-2), 161.95 (NC(=O)N), 165.36 (q, 2JCF = 35.3 Hz, TFA-COO), 168.58 (C-4), 173.96 (C-7′), 177.32 (C-1′′′), 177.79 (C-1′′′′), 179.05 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.86 (TFA-CF3). MS (ESI-): m/z = 741.4 [M − H]−. HRMS (ESI)−: calcd.: 741.3788 [M − H]−, found: 741.3792. IR (ATR): ν = 1638, 1551, 1198, 1182, 1131, 799, 720, 551, 519. UV (H2O): λmax (log ε) = 260 (4.01). Optical rotation: [α]D25 = −1.1 (c = 0.46, H2O). m.p. = 208 °C. 11: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.98 (d, J = 6.1 Hz, 3H, 5′′′-Ha), 1.04 (d, J = 5.9 Hz, 3H, 5′′′-Hb), 1.04 (d, J = 6.9 Hz, 3H, 4′′′′′-Ha), 1.08 (d, J = 6.7 Hz, 3H, 4′′′′′-Hb), 1.50–1.63 (m, 2H, 4′′′′-H), 1.66–1.85 (m, 6H, 3′′′-H, 4′′′-H, 3′′′′-Ha, 5′′′′-H), 1.87–1.93 (m, 1H, 3′′′′-Hb), 1.97–2.07 (m, 2H, 2′′-H), 2.27 (dqq, J = 6.9, 6.7, 5.7 Hz, 1H, 3′′′′′-H), 2.36 (ddd, J = 15.1, 10.0 Hz, 6.6 Hz, 1H, 5′-Ha), 2.54 (ddd, J = 15.1, 6.5 Hz, 3.0 Hz, 1H, 5′-Hb), 3.11 (dd, J = 7.6, 7.6 Hz, 2H, 6′′′′-H), 3.16 (ddd, J = 13.2, 8.8, 7.9 Hz, 1H, 1′′-Ha), 3.21 (ddd, J = 13.2, 8.0, 6.5 Hz, 1H, 1′′-Hb), 3.38 (ddd, J = 14.2, 6.5, 6.4 Hz, 1H, 3′′-Ha), 3.41 (ddd, J = 14.2, 7.0, 6.5 Hz, 1H, 3′′-Hb), 3.93 (dd, J = 6.6, 6.5 Hz, 1H, 6′-H), 4.17 (d, J = 5.7 Hz, 1H, 2′′′′′-H), 4.20 (dd, J = 6.4, 5.9 Hz, 1H, 3′-H), 4.24 (dd, J = 8.2, 6.0 Hz, 1H, 2′′′′-H), 4.28 (ddd, J = 10.0, 6.4, 3.0 Hz, 1H, 4′-H), 4.35 (dd, J = 10.4, 4.1 Hz, 1H, 2′′′-H), 4.54 (dd, J = 5.9, 3.8 Hz, 1H, 2′-H), 5.88 (d, J = 3.8 Hz, 1H, 1′-H), 6.01 (d, J = 8.1 Hz, 1H, 5-H), 7.77 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.74 (Ca-4′′′′′), 21.23 (Cb-4′′′′′), 22.99 (Ca-5′′′), 24.78 (C-4′′′′), 24.94 (Cb-5′′′), 27.13 (C-4′′′), 28.33 (C-2′′), 28.93 (C-5′′′′), 32.65 (C-3′′′′′), 33.50 (C-3′′′′), 35.77 (C-5′), 38.53 (C-3′′), 41.90 (C-6′′′′), 42.18 (C-3′′′), 46.87 (C-1′′), 55.21 (C-2′′′), 56.84 (C-2′′′′), 61.77 (C-2′′′′′), 62.91 (C-6′), 75.22 (C-2′), 75.51 (C-3′), 82.94 (C-4′), 94.32 (C-1′), 104.87 (C-5), 118.93 (q, 1JCF = 292.3 Hz, TFA-CF3), 145.32 (C-6), 153.91 (C-2), 161.82 (NC(=O)N), 165.41 (q, 2JCF = 34.0 Hz, TFA-COO), 168.59 (C-4), 174.75 (C-7′), 177.53 (C-1′′′), 178.10 (C-1′′′′), 179.42 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.87 (TFA-CF3). MS (ESI+): m/z = 743.5 [M + H]+. HRMS (ESI+): calcd.: 743.3934 [M + H]+, found: 743.3926. IR (ATR): ν [cm−1] = 1641, 1552, 1200, 1182, 1131, 799, 720, 550, 517. UV (H2O): λmax (log ε) = 261 (4.07). Optical rotation: [α]D25 = +31.4 (c = 0.14, H2O). m.p. = 211 °C.
5′-deoxy-6′-epi-l-Leu muraymycin analogue (12) and 5′-deoxy-6′-epi-d-Leu muraymycin analogue (13): To a solution of the urea tripeptide 18 (10 mg, 0.018 mmol) in THF (2.5 mL), HOBt (2.4 mg, 0.018 mmol), ECD (3.5 mg, 0.018 mmol), and DIPEA (3.1 μL, 0.018 mmol) were added and the mixture was stirred at room temperature for 30 min. It was then added to a solution of amine 20 (15 mg, 0.023 mmol) in THF (2 mL) and stirred at room temperature for 20 h. EtOAc (30 mL) was added and the solution was washed with sat. NaHCO3 (30 mL). The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The protected analogues of 12 and 13 were obtained after column chromatography (98:2–95:5, CH2Cl2-MeOH) as colourless solids. This material was dissolved in TFA (80% in water, 2.7 mL) and stirred at room temperature for 24 h. Water (10 mL) was added and the solvent was removed under reduced pressure. The muraymycin analogues 12 (10 mg, 57%) and 13 (3.3 mg, 19%) were separated by preparative HPLC (12: tR = 17.8 min, 13: tR = 16.4 min) and obtained as colourless solids. 12: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.99 (d, J = 5.8 Hz, 3H, 5′′′-Ha), 1.04 (d, J = 5.4 Hz, 3H, 5′′′-Hb), 1.05 (d, J = 6.7 Hz, 3H, 4′′′′′-Ha), 1.08 (d, J = 6.8 Hz, 3H, 4′′′′′-Hb), 1.51–1.61 (m, 2H, 4′′′′-H), 1.66–1.84 (m, 6H, 3′′′-H, 4′′′-H, 3′′′′-Ha, 5′′′′-H), 1.88–1.94 (m, 1H, 3′′′′-Hb), 1.96–2.04 (m, 2H, 2′′-H), 2.26 (dqq, J = 6.8, 6.7, 6.1 Hz, 1H, 3′′′′′-H), 2.47 (ddd, J = 15.5, 10.1, 4.8 Hz, 1H, 5′-Ha), 2.59 (ddd, J = 15.5, 5.3, 2.9 Hz, 1H, 5′-Hb), 3.12 (dd, J = 7.6, 7.5 Hz, 2H, 6′′′′-H), 3.16 (ddd, J = 12.2, 8.8, 7.6 Hz, 1H, 1′′-Ha), 3.21 (ddd, J = 12.2, 8.6, 7.3 Hz, 1H, 1′′-Hb), 3.35 (ddd, J = 14.3, 8.0, 6.7 Hz, 1H, 3′′-Ha), 3.40 (ddd, J = 14.3, 7.5, 6.8 Hz, 1H, 3′′-Hb), 4.08 (dd, J = 5.3, 4.8 Hz, 1H, 6′-H), 4.16 (ddd, J = 10.1, 7.9, 2.9 Hz, 1H, 4′-H), 4.21–4.25 (m, 3H, 3′-H, 2′′′′-H, 2′′′′′-H), 4.37 (dd, J = 9.7, 4.7 Hz, 1H, 2′′′-H), 4.56 (dd, J = 5.6, 3.8 Hz, 1H, 2′-H), 5.84 (d, J = 3.8 Hz, 1H, 1′-H), 6.00 (d, J = 8.1 Hz, 1H, 5-H), 7.78 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.68 (Ca-4′′′′′), 21.18 (Cb-4′′′′′), 23.37 (Ca-5′′′), 24.73 (C-4′′′′), 24.79 (Cb-5′′′), 27.09 (C-4′′′), 28.21 (C-2′′), 28.94 (C-5′′′′), 32.66 (C-3′′′′′), 33.42 (C-3′′′′), 34.63 (C-5′), 38.65 (C-3′′), 41.92 (C-6′′′′), 42.23 (C-3′′′), 47.29 (C-1′′), 55.21 (C-2′′′), 56.77 (C-2′′′′), 61.51 (C-2′′′′′), 62.56 (C-6′), 75.00 (C-2′), 75.39 (C-3′), 82.26 (C-4′), 94.93 (C-1′), 104.85 (C-5), 118.93 (q, 1JCF = 292.3 Hz, TFA-CF3), 145.71 (C-6), 153.92 (C-2), 161.94 (NC(=O)N), 165.37 (q, 2JCF = 35.3 Hz, TFA-COO), 168.53 (C-4), 174.09 (C-7′), 177.31 (C-1′′′), 177.82 (C-1′′′′), 179.08 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.86 (TFA-CF3). MS (ESI+): m/z = 743.5 [M + H]+. HRMS (ESI+): calcd.: 743.3934 [M + H]+, found: 743.3926. IR (ATR): ν [cm−1] = 1633, 1551, 1198, 1182, 1131, 1054, 799, 720, 551. UV (H2O): λmax (log ε) = 261 (4.00). Optical rotation: [α]D25 = −3.8 (c = 0.45, H2O). m.p. = 208 °C. 13: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.99 (d, J = 5.6 Hz, 3H, 5′′′-Ha), 1.05 (d, J = 6.1 Hz, 3H, 5′′′-Hb), 1.05 (d, J = 7.1 Hz, 3H, 4′′′′′-Ha), 1.08 (d, J = 6.6 Hz, 3H, 4′′′′′-Hb), 1.49–1.63 (m, 2H, 4′′′′-H), 1.68–1.93 (m, 7H, 3′′′-H, 4′′′-H, 3′′′′-H, 5′′′′-H), 1.97–2.02 (m, 2H, 2′′-H), 2.27 (dqq, J = 7.1, 6.6, 5.8 Hz, 1H, 3′′′′′-H), 2.47 (ddd, J = 15.3, 10.1, 4.7 Hz, 1H, 5′-Ha), 2.59 (ddd, J = 15.3, 5.3, 2.8 Hz, 1H, 5′-Hb), 3.11 (dd, J = 8.0, 7.8 Hz, 2H, 6′′′′-H), 3.14 (ddd, J = 12.7, 7.7, 7.3 Hz, 1H, 1′′-Ha), 3.20 (ddd, J = 12.3, 7.2, 7.0 Hz, 1H, 1′′-Hb), 3.38 (dd, J = 6.7, 6.6 Hz, 2H, 3′′-H), 4.03 (dd, J = 5.3, 4.7 Hz, 1H, 6′-H), 4.16 (ddd, J = 10.1, 7.1, 2.8 Hz, 1H, 4′-H), 4.18 (d, J = 5.8 Hz, 1H, 2′′′′′-H), 4.24 (dd, J = 9.2, 6.3 Hz, 1H, 3′-H), 4.24 (dd, J = 5.9, 2.1 Hz, 1H, 2′′′′-H), 4.36 (dd, J = 10.3, 4.0 Hz, 1H, 2′′′-H), 4.55 (dd, J = 9.2, 3.8 Hz, 1H, 2′-H), 5.85 (d, J = 3.8 Hz, 1H, 1′-H), 6.00 (d, J = 8.1 Hz, 1H, 5-H), 7.79 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.72 (Ca -4′′′′′), 21.19 (Cb-4′′′′′), 23.00 (Ca-5′′′), 24.79 (C-4′′′′), 24.93 (Cb-5′′′), 27.14 (C-4′′′), 28.28 (C-2′′), 28.94 (C-5′′′′), 32.61 (C-3′′′′′), 33.48 (C-3′′′′), 34.72 (C-5′), 38.58 (C-3′′), 41.90 (C-6′′′′), 42.16 (C-3′′′), 47.30 (C-1′′), 55.21 (C-2′′′), 56.87 (C-2′′′′), 61.63 (C-2′′′′′), 62.96 (C-6′), 75.00 (C-2′), 75.41 (C-3′), 82.35 (C-4′), 94.78 (C-1′), 104.88 (C-5), 118.94 (q, 1JCF = 291.1 Hz, TFA-CF3), 145.67 (C-6), 153.96 (C-2), 161.81 (NC(=O)N), 165.40 (q, 2JCF = 35.3 Hz, TFA-COO), 168.55 (C-4), 174.33 (C-7′), 177.55 (C-1′′′), 178.10 (C-1′′′′), 179.23 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.87 (TFA-CF3). MS (ESI+): m/z = 743.5 [M + H]+. HRMS (ESI+): calcd.: 743.3934 [M + H]+, found: 743.3928. IR (ATR): ν [cm−1] = 1633, 1552, 1198, 1181, 1130, 1053, 720, 550, 518. UV (H2O): λmax (log ε) = 260 (4.01). Optical rotation: [α]D25 = +28.7 (c = 0.15, H2O). m.p. = 209 °C.
6′-epi-l-Leu muraymycin analogue (14) and 6′-epi-d-Leu muraymycin analogue (15): To a solution of the urea tripeptide 18 (13 mg, 0.024 mmol) in THF (3 mL), HOBt (3.2 mg, 0.024 mmol), EDC (4.6 mg, 0.024 mmol), and DIPEA (4.2 μL, 0.024 mmol) were added and the mixture was stirred at room temperature for 45 min. It was then added to a solution of amine 21 (20 mg, 0.030 mmol) in THF (2 mL) and stirred at room temperature for 18 h. EtOAc (30 mL) was added and the solution was washed with sat. NaHCO3 (30 mL). The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The protected analogues of 14 and 15 were obtained after column chromatography (98:2–96:4, CH2Cl2-MeOH) as colourless solids. This material was dissolved in TFA (80% in water, 3.6 mL) and stirred at room temperature for 24 h. Water (10 mL) was added and the solvent was removed under reduced pressure. The muraymycin analogues 14 (14 mg, 59%) and 15 (5.6 mg, 24%) were separated by preparative HPLC (14: tR = 17.5 min, 15: tR = 16.1 min) and obtained as colourless solids. 14: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.99 (d, J = 5.8 Hz, 3H, 5′′′-Ha), 1.04 (d, J = 5.7 Hz, 3H, 5′′′-Hb), 1.05 (d, J = 6.6 Hz, 3H, 4′′′′′-Ha), 1.09 (d, J = 6.8 Hz, 3H, 4′′′′′-Hb), 1.51–1.60 (m, 2H, 4′′′′-H), 1.66–1.85 (m, 6H, 3′′′-H, 4′′′-H, 3′′′′-Ha, 5′′′′-H), 1.89–1.94 (m, 1H, 3′′′′-Hb), 2.04 (dddd, J = 7.4, 7.3, 6.7, 6.5 Hz, 2H, 2′′-H), 2.28 (dqq, J = 6.8, 6.6, 6.4 Hz, 1H, 3′′′′′-H), 3.12 (dd, J = 7.7, 7.6 Hz, 2H, 6′′′′-H), 3.18 (ddd, J = 12.9, 7.4, 7.3 Hz, 1H, 1′′-Ha), 3.25 (ddd, J = 12.9, 7.4, 7.3 Hz, 1H, 1′′-Hb), 3.38 (ddd, J = 14.3, 6.5, 6.5 Hz, 1H, 3′′-Ha), 3.45 (ddd, J = 14.3, 6.7, 6.7 Hz, 1H, 3′′-Hb), 4.10 (d, J = 4.5 Hz, 1H, 6′-H), 4.22–4.25 (m, 3H, 4′-H, 2′′′′-H, 2′′′′′-H), 4.37 (dd, J = 9.6, 4.9 Hz, 1H, 2′′′-H), 4.46 (dd, J = 5.8, 5.3 Hz, 1H, 3′-H), 4.48 (dd, J = 5.3, 4.0 Hz, 1H, 2′-H), 4.58 (dd, J = 4.5, 1.1 Hz, 1H, 5′-H), 5.99 (d, J = 8.1 Hz, 1H, 5-H), 6.00 (d, J = 4.0 Hz, 1H, 1′-H), 8.14 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.69 (Ca-4′′′′′), 21.18 (Cb-4′′′′′), 23.33 (Ca-5′′′), 24.72 (C-4′′′′), 24.80 (Cb-5′′′), 27.08 (C-4′′′), 27.81 (C-2′′), 28.93 (C-5′′′′), 32.69 (C-3′′′′′), 33.35 (C-3′′′′), 38.32 (C-3′′), 41.92 (C-6′′′′), 42.15 (C-3′′′), 48.22 (C-1′′), 55.23 (C-2′′′), 56.87 (C-2′′′′), 61.51 (C-2′′′′′), 68.51 (C-6′), 69.43 (C-5′), 72.48 (C-3′), 75.64 (C-2′), 85.67 (C-4′), 92.87 (C-1′), 104.74 (C-5), 118.93 (q, 1JCF = 292.3 Hz, TFA-CF3), 145.06 (C-6), 154.07 (C-2), 161.96 (NC(=O)N), 165.37 (q, 2JCF = 35.3 Hz, TFA-COO), 168.55 (C-4), 172.02 (C-7′), 177.43 (C-1′′′), 177.93 (C-1′′′′), 179.06 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.86 (TFA-CF3). MS (ESI+): m/z = 759.5 [M + H]+. HRMS (ESI+): calcd.: 759.3883 [M + H]+, found: 759.3886. IR (ATR): ν [cm-1] = 1632, 1549, 1198, 1182, 1128, 1049, 720, 560, 520. UV (H2O): λmax (log ε) = 262 (3.94). Optical rotation: [α]D25 = +5.4 (c = 0.54, H2O). m.p. = 215 °C. 15: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.98 (d, J = 5.3 Hz, 3H, 5′′′-Ha), 1.04 (d, J = 5.3 Hz, 3H, 5′′′-Hb), 1.04 (d, J = 6.5 Hz, 3H, 4′′′′′-Ha), 1.08 (d, J = 6.8 Hz, 3H, 4′′′′′-Hb), 1.49–1.54 (m, 1H, 4′′′′-Ha), 1.56–1.62 (m, 1H, 4′′′′-Hb), 1.66–1.92 (m, 7H, 3′′′-H, 4′′′-H, 3′′′′-H, 5′′′′-H), 1.98–2.07 (m, 2H, 2′′-H), 2.27 (dqq, J = 6.8, 6.5, 4.8 Hz, 1H, 3′′′′′-H), 3.11 (dd, J = 7.6, 7.6 Hz, 2H, 6′′′′-H), 3.18 (ddd, J = 13.7, 7.1, 6.0 Hz, 1H, 1′′-Ha), 3.24 (ddd, J = 13.7, 7.3, 7.2 Hz, 1H, 1′′-Hb), 3.37–3.43 (m, 2H, 3′′-H), 4.14 (d, J = 5.4 Hz, 1H, 6′-H), 4.19 (d, J = 4.8 Hz, 1H, 2′′′′′-H), 4.22 (dd, J = 6.1, 4.9 Hz, 1H, 2′′′′-H), 4.23 (dd, J = 5.8, 1.8 Hz, 1H, 4′-H), 4.35 (dd, J = 10.3, 4.1 Hz, 1H, 2′′′-H), 4.45 (dd, J = 5.8, 5.3 Hz, 1H, 3′-H), 4.48 (dd, J = 5.3, 4.1 Hz, 1H, 2′-H), 4.58 (dd, J = 5.4, 1.8 Hz, 1H, 5′-H), 5.98 (d, J = 8.1 Hz, 1H, 5-H), 5.99 (d, J = 4.1 Hz, 1H, 1′-H), 8.12 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.69 (Ca-4′′′′′), 21.18 (Cb-4′′′′′), 23.00 (Ca-5′′′), 24.79 (C-4′′′′), 24.93 (Cb-5′′′), 27.13 (C-4′′′), 27.87 (C-2′′), 28.94 (C-5′′′′), 32.61 (C-3′′′′′), 33.47 (C-3′′′′), 38.23 (C-3′′), 41.90 (C-6′′′′), 42.14 (C-3′′′), 48.20 (C-1′′), 55.22 (C-2′′′), 56.88 (C-2′′′′), 61.53 (C-2′′′′′), 68.51 (C-6′), 69.43 (C-5′), 72.51 (C-3′), 75.58 (C-2′), 85.77 (C-4′), 92.84 (C-1′), 104.79 (C-5), 118.93 (q, 1JCF = 292.3 Hz, TFA-CF3), 145.10 (C-6), 154.12 (C-2), 161.80 (NC(=O)N), 165.39 (q, 2JCF = 36.5 Hz, TFA-COO), 168.57 (C-4), 171.97 (C-7′), 177.63 (C-1′′′), 178.09 (C-1′′′′), 179.16 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.87 (TFA-CF3). MS (ESI+): m/z = 759.5 [M + H]+. HRMS (ESI+): calcd.: 759.3883 [M+H]+, found: 759.3884. IR (ATR): ν [cm−1] = 1686, 1637, 1552, 1200, 1131, 721, 636, 568, 554. UV (H2O): λmax (log ε) = 261 (4.08). Optical rotation: [α]D25 = +84.5 (c = 0.22, H2O). m.p. = 210 °C.
5′-epi-l-Leu muraymycin analogue (16) and 5′-epi-d-Leu muraymycin analogue (17): To a solution of the urea tripeptide 18 (6.7 mg, 0.012 mmol) in THF (1.5 mL), HOBt (1.6 mg, 0.012 mmol), ECD (2.3 mg, 0.012 mmol), and DIPEA (2.1 μL, 0.012 mmol) were added and the mixture was stirred at room temperature for 45 min. It was then added to a solution of amine 22 (10 mg, 0.015 mmol) in THF (3 mL) and stirred at room temperature for 18 h. EtOAc (30 mL) was added and the solution was washed with sat. NaHCO3 (30 mL). The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The protected analogues of 16 and 17 were obtained after column chromatography (98:2–96:4, CH2Cl2-MeOH) as colourless solids. This material was dissolved in TFA (80% in water, 1.8 mL) and stirred at room temperature for 24 h. Water (10 mL) was added and the solvent was removed under reduced pressure. The muraymycin analogues 16 (6.4 mg, 54%) and 17 (3.2 mg, 27%) were separated by preparative HPLC (16: tR = 17.9 min, 17: tR = 16.3 min) and obtained as colourless solids. 16: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.98 (d, J = 5.7 Hz, 3H, 5′′′-Ha), 1.03 (d, J = 5.5 Hz, 3H, 5′′′-Hb), 1.04 (d, J = 6.4 Hz, 3H, 4′′′′′-Ha), 1.08 (d, J = 7.0 Hz, 3H, 4′′′′′-Hb), 1.50–1.60 (m, 2H, 4′′′′-H), 1.66–1.83 (m, 6H, 3′′′-H, 4′′′-H, 3′′′′-Ha, 5′′′′-H), 1.87–1.93 (m, 1H, 3′′′′-Hb), 2.07 (dddd, J = 7.7, 7.6, 6.6, 6.5 Hz, 2H, 2′′-H), 2.28 (dqq, J = 7.0, 6.4, 5.7 Hz, 1H, 3′′′′′-H), 3.11 (dd, J = 7.7, 7.7 Hz, 2H, 6′′′′-H), 3.25 (dd, J = 7.7, 7.6 Hz, 2H, 1′′-H), 3.39 (ddd, J = 14.2, 6.6, 6.5 Hz, 1H, 3′′-Ha), 3.48 (ddd, J = 14.2, 6.6, 6.5 Hz, 1H, 3′′-Hb), 4.14 (d, J = 2.9 Hz, 1H, 6′-H), 4.22 (d, J = 5.7 Hz, 1H, 2′′′′′-H), 4.24 (dd, J = 8.7, 5.5 Hz, 1H, 2′′′′-H), 4.30 (dd, J = 8.4, 3.5 Hz, 1H, 4′-H), 4.39 (dd, J = 9.5, 4.7 Hz, 1H, 2′′′-H), 4.46 (dd, J = 8.4, 2.9 Hz, 1H, 5′-H), 4.49 (dd, J = 5.5, 3.5 Hz, 1H, 3′-H), 4.74 (dd, J = 5.9, 5.5 Hz, 1H, 2′-H), 5.84 (d, J = 5.9 Hz, 1H, 1′-H), 6.00 (d, J = 8.0 Hz, 1H, 5-H), 7.78 (d, J = 8.0 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.68 (Ca-4′′′′′), 21.18 (Cb-4′′′′′), 23.39 (Ca-5′′′), 24.69 (C-4′′′′), 24.76 (Cb-5′′′), 27.09 (C-4′′′), 28.12 (C-2′′), 28.93 (C-5′′′′), 32.66 (C-3′′′′′), 33.45 (C-3′′′′), 38.74 (C-3′′), 41.92 (C-6′′′′), 42.23 (C-3′′′), 47.50 (C-1′′), 55.25 (C-2′′′), 56.73 (C-2′′′′), 61.48 (C-2′′′′′), 65.70 (C-6′), 71.73 (C-5′), 73.67 (C-3′), 74.33 (C-2′), 86.08 (C-4′), 94.17 (C-1′), 104.86 (C-5), 118.93 (q, 1JCF = 291.1 Hz, TFA-CF3), 146.11 (C-6), 153.90 (C-2), 161.92 (NC(=O)N), 165.38 (q, 2JCF = 35.3 Hz, TFA-COO), 168.59 (C-4), 171.19 (C-7′), 177.37 (C-1′′′), 177.76 (C-1′′′′), 179.05 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = −72.88 (TFA-CF3). MS (ESI+): m/z = 759.5 [M + H]+. HRMS (ESI+): calcd.: 759.3883 [M + H]+, found: 759.3884. IR (ATR): ν [cm−1] = 1660, 1633, 1551, 1197, 1184, 1132, 720, 547, 511. UV (H2O): λmax (log ε) = 260 (3.98). Optical rotation: [α]D25 = -10.6 (c = 0.16, H2O). m.p. = 214 °C. 17: 1H NMR (600 MHz, D2O, 35 °C): δ [ppm] = 0.98 (d, J = 5.3 Hz, 3H, 5′′′-Ha), 1.00 (d, J = 6.8 Hz, 3H, 5′′′-Hb), 1.04 (d, J = 6.0 Hz, 3H, 4′′′′′-Ha), 1.05 (d, J = 6.8 Hz, 3H, 4′′′′′-Hb), 1.51–1.63 (m, 2H, 4′′′′-H), 1.69–1.87 (m, 6H, 3′′′-H, 4′′′-H, 3′′′′-Ha, 5′′′′-H), 1.88–1.94 (m, 1H, 3′′′′-Hb), 2.07 (dddd, J = 7.3, 7.2, 6.6, 6.3 Hz, 2H, 2′′-H), 2.21 (dqq, J = 6.8, 6.0, 5.9 Hz, 1H, 3′′′′′-H), 3.12 (dd, J = 7.6, 7.4 Hz, 2H, 6′′′′-H), 3.20–3.26 (m, 2H, 1′′-H), 3.39 (ddd, J = 14.3, 6.6, 6.3 Hz, 1H, 3′′-Ha), 3.47 (ddd, J = 14.3, 6.6, 6.3 Hz, 1H, 3′′-Hb), 3.95 (d, J = 3.0 Hz, 1H, 6′-H), 4.06 (d, J = 5.9 Hz, 1H, 2′′′′′-H), 4.23 (dd, J = 7.3, 7.1 Hz, 1H, 2′′′′-H), 4.29 (dd, J = 7.6, 4.4 Hz, 1H, 4′-H), 4.38 (dd, J = 9.5, 4.1 Hz, 1H, 2′′′-H), 4.44 (dd, J = 7.6, 3.0 Hz, 1H, 5′-H), 4.49 (dd, J = 5.3, 4.4 Hz, 1H, 3′-H), 4.65 (dd, J = 5.6, 5.3 Hz, 1H, 2′-H), 5.89 (d, J = 5.6 Hz, 1H, 1′-H), 6.01 (d, J = 8.1 Hz, 1H, 5-H), 7.83 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O, 35 °C): δ [ppm] = 19.77 (Ca-4′′′′′), 21.56 (Cb-4′′′′′), 22.98 (Ca-5′′′), 24.79 (C-4′′′′), 24.95 (Cb-5′′′), 27.12 (C-4′′′), 28.13 (C-2′′), 28.98 (C-5′′′′), 33.04 (C-3′′′′′), 33.38 (C-3′′′′), 38.77 (C-3′′), 41.92 (C-6′′′′), 42.17 (C-3′′′), 47.46 (C-1′′), 55.19 (C-2′′′), 56.84 (C-2′′′′), 63.09 (C-2′′′′′), 66.68 (C-6′), 71.79 (C-5′), 73.11 (C-3′), 74.83 (C-2′), 86.09 (C-4′), 94.45 (C-1′), 104.88 (C-5), 118.89 (q, 1JCF = 291.1 Hz, TFA-CF3), 145.66 (C-6), 154.01 (C-2), 161.81 (NC(=O)N), 165.39 (q, 2JCF = 37.8 Hz, TFA-COO), 168.63 (C-4), 171.91 (C-7′), 177.01 (C-1′′′), 177.54 (C-1′′′′), 178.33 (C-1′′′′′). 19F NMR (282 MHz, D2O, 35 °C): δ [ppm] = –72.88 (TFA-CF3). MS (ESI+): m/z = 759.5 [M + H]+. HRMS (ESI+): calcd.: 759.3883 [M + H]+, found: 759.3888. IR (ATR): ν [cm−1] = 1667, 1634, 1552, 1201, 1132, 1056, 800, 721, 547. UV (H2O): λmax (log ε) = 260 (3.95). optical rotation: [α]D25 = +13.1 (c = 0.13, H2O). m.p. = 218 °C.
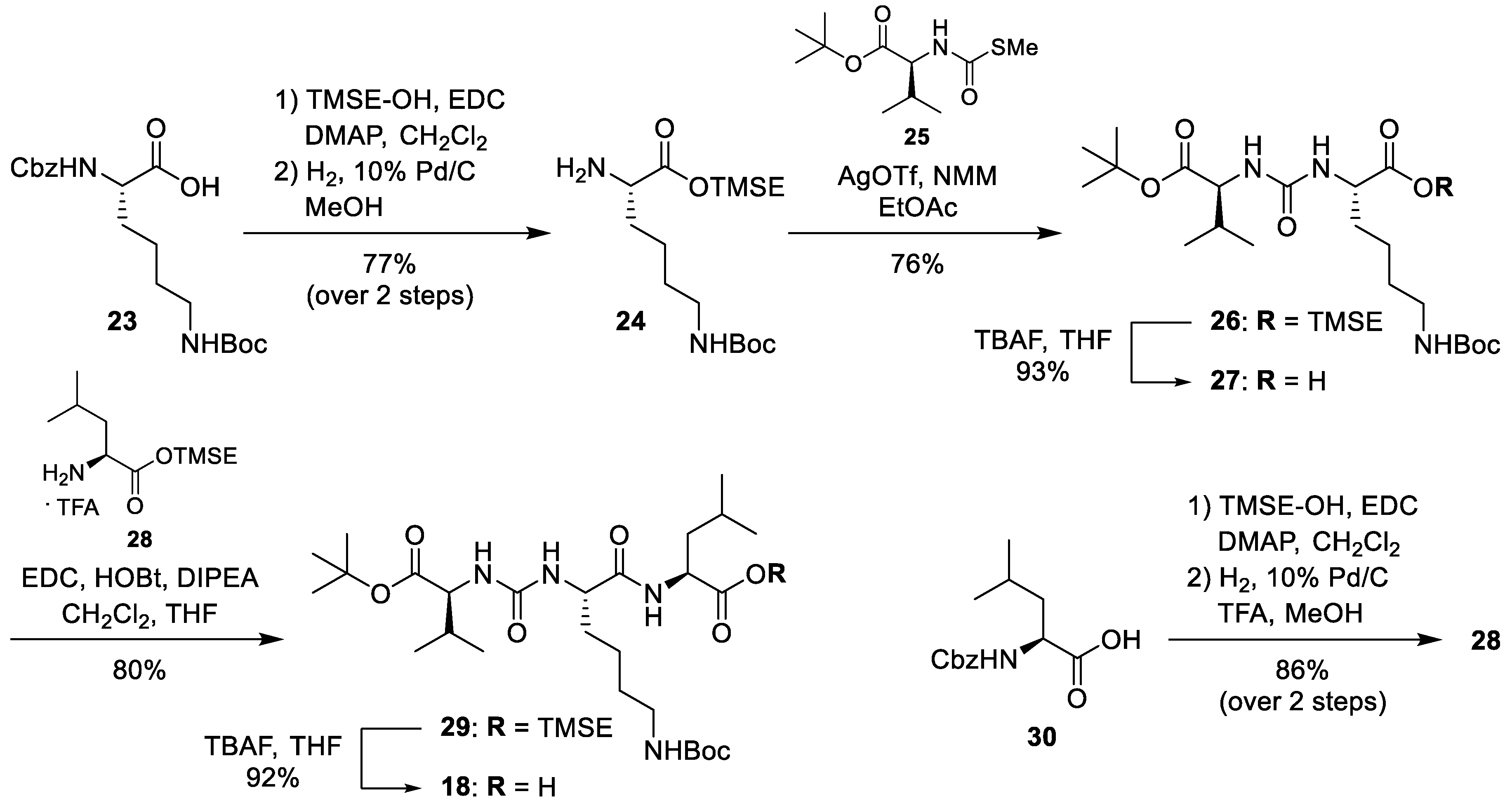
Protected urea tripeptide (18): To a solution of 29 (50 mg, 0.076 mmol) in THF (4 mL), tetrabutylammonium fluoride (TBAF, 1 m in THF, 91 μL, 0.091 mmol) was added at 0 °C and the mixture was stirred at 0 °C for 1 h. More TBAF (1 m in THF, 30 μL, 0.030 mmol) was added and the mixture was stirred at room temperature for 3 h. The solvent was removed under reduced pressure. The urea tripeptide 18 was obtained after column chromatography (95:4:1, CH2Cl2-MeOH-HOAc) as colourless solid (39 mg, 92%). 1H NMR (600 MHz, DMSO-d6, 35 °C): δ [ppm] = 0.83 (d, J = 6.9 Hz, 3H, 4-Ha), 0.83 (d, J = 6.5 Hz, 3H, 5′′-Ha), 0.85 (d, J = 6.8 Hz, 3H, 4-Hb), 0.88 (d, J = 6.6 Hz, 3H, 5′′-Hb), 1.22–1.28 (m, 2 H, 4′-H), 1.35–1.44 (m, 3H, 3′-Ha, 5′′-H), 1.38 (s, 9H, OC(CH3)3), 1.40 (s, 9H, OC(CH3)3), 1.52 (ddd, J = 9.1, 5.3, 5.0 Hz, 2H, 3′′-H), 1.53–1.58 (m, 1H, 3′-Hb), 1.57.1.65 (m, 1H, 4′′-H), 1.96 (dqq, J = 6.9, 6.8, 5.1 Hz, 1H, 3-H), 2.84–2.88 (m, 2H, 6′-NH), 3.92 (dd, J = 8.6, 5.1 Hz, 1H, 2-H), 4.14 (ddd, J = 8.2, 7.7, 5.3 Hz, 1H, 2′-H), 4.20 (ddd, J = 9.1, 8.0, 5.6 Hz, 1H, 2′′-H), 6.25 (d, J = 8.6 Hz, 1H, 2-NH), 6.27 (d, J = 7.7 Hz, 1H, 2′-NH), 6.66 (dd, J = 6.0, 5.8 Hz, 1H, 6′-NH), 8.01 (d, J = 8.0 Hz, 1H, 2′′-NH). 13C NMR (126 MHz, DMSO-d6, 35 °C): δ [ppm] = 17.59 (Ca-4), 18.92 (Cb-4), 21.33 (Ca-5′′), 22.24 (C-4′), 24.79 (Cb-5′′), 24.17 (C-4′′), 27.64 (OC(CH3)3), 28.20 (OC(CH3)3), 29.30 (C-5′), 30.43 (C-3), 32.97 (C-3′), 39.76 (C-6′), 39.92 (C-3′′), 50.09 (C-2′′), 52.41 (C-2′), 58.10 (C-2), 77.13 (OC(CH3)3), 80.04 (OC(CH3)3), 155.25 (NC(=O)O), 157.09 (NC(=O)N), 171.36 (C-1), 172.09 (C-1′), 173.64 (C-1′′). MS (ESI+): m/z = 581.3 [M + Na]+. HRMS (ESI+): calcd.: 581.3521 [M + Na]+, found: 581.3522. IR (ATR): ν [cm−1] = 1719, 1688, 1633, 1546, 1391, 1366, 1250, 1156, 665. Optical rotation: [α]D25 = −3.3 (c = 0.24, CHCl3). m.p. = 73 °C. TLC: Rf = 0.25 (94:5:1, CH2Cl2-MeOH-AcOH).
6′-epi nucleoside building block (21): To a solution of 33 (45 mg, 0.057 mmol) in MeOH (4 mL), Pd/C (10%, 10 mg, 9.4 μmol) and 1,4-cyclohexadiene (54 μL, 0.57 mmol) were added and the mixture was stirred at room temperature for 2 h. More Pd/C (10%, 5 mg, 5 μmol) and 1,4-cyclohexadiene (54 μL, 0.57 mmol) were added and the mixture was stirred at room temperature for 1 h. The mixture was filtered and the residue was washed with MeOH (3 × 4 mL). The solvent of the combined filtrates was removed under reduced pressure to give 21 as a colourless solid (37 mg, 99%). 1H NMR (600 MHz, CD3OD): δ [ppm] = 0.05 (s, 3H, SiCH3), 0.07 (s, 3H, SiCH3), 0.12 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.88 (s, 9H, SiC(CH3)3), 0.94 (s, 9H, SiC(CH3)3), 1.49 (s, 9H, OC(CH3)3), 1.66–1.75 (m, 2H, 2′′-H), 2.63 (ddd, J = 12.3, 6.2, 5.9 Hz, 1H, 1′′-Ha), 2.76 (ddd, J = 12.3, 7.2, 5.6 Hz, 1H, 1′′-Hb), 2.94–2.96 (m, 2H, 3′′-H), 3.38 (d, J = 7.4 Hz, 1 H, 6′-H), 3.87 (dd, J = 7.4, 1.2 Hz, 1H, 5′-H), 4.17 (dd, J = 4.2, 3.7 Hz, 1H, 3′-H), 4.20 (dd, J = 3.7, 1.2 Hz, 1H, 4′-H), 4.36 (dd, J = 5.3, 4.2 Hz, 1H, 2′-H), 5.72 (d, J = 8.1 Hz, 1H, 5-H), 5.88 (d, J = 5.3 Hz, 1H, 1′-H), 8.18 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, CD3OD): δ [ppm] = −4.58 (SiCH3), −4.49 (SiCH3), −4.49 (SiCH3), −4.22 (SiCH3), 18.76 (SiC(CH3)3), 18.85 (SiC(CH3)3), 26.24 (SiC(CH3)3), 26.31 (SiC(CH3)3), 28.29 (OC(CH3)3), 29.82 (C-2′′), 40.38 (C-3′′), 46.92 (C-1′′), 65.99 (C-6′), 71.28 (C-5′), 74.41 (C-3′), 76.01 (C-2′), 82.79 (OC(CH3)3), 86.15 (C-4′), 90.00 (C-1′), 102.72 (C-5), 142.68 (C-6), 152.45 (C-2), 166.11 (C-4), 174.08 (C-7′). MS (ESI+): m/z = 659.4 [M + H]+. HRMS (ESI+): calcd.: 659.3866 [M + H]+, found: 659.3867. IR (ATR): ν [cm−1] = 1686, 1253, 1153, 1113, 1051, 869, 834, 812, 773. UV (MeOH): λmax (log ε) = 207 (3.98), 262 (3.98). Optical rotation: [α]D25 = +24.0 (c = 0.30, MeOH). m.p. = 115 °C.
5′-epi nucleoside building block (22): To a solution of 34 (5.0 mg, 6.3 μmol) in MeOH (4 mL), Pd/C (10%, 5 mg, 5 μmol), and 1,4-cyclohexadiene (6.3 μL, 0.063 mmol) were added and the mixture was stirred at room temperature for 30 min. More Pd/C (10%, 5 mg, 5 μmol) was added and the mixture was stirred at room temperature for 30 min. The mixture was filtered and the residue was washed with MeOH (3 × 4 mL). The solvent of the combined filtrates was removed under reduced pressure to give 22 as a colourless solid (4.1 mg, 99%). 1H NMR (600 MHz, pyridine-d5, 35 °C): δ [ppm] = 0.14 (s, 3H, SiCH3), 0.16 (s, 3H, SiCH3), 0.26 (s, 3H, SiCH3), 0.35 (s, 3H, SiCH3), 0.94 (s, 9H, SiC(CH3)3), 1.06 (s, 9H, SiC(CH3)3), 1.59 (s, 9H, OC(CH3)3), 2.11–2.16 (m, 1H, 2′′-Ha), 2.21–2.26 (m, 1H, 2′′-Hb), 3.09 (ddd, J = 12.3, 7.2, 5.6 Hz, 1H, 1′′-Ha), 3.22 (ddd, J = 12.3, 6.1, 6.1 Hz, 1H, 1′′-Hb), 3.45 (ddd, J = 12.5, 6.8, 6.7 Hz, 1H, 3′′-Ha), 3.50 (ddd, J = 12.5, 6.6 Hz, 6.5 Hz, 1H, 3′′-Hb), 4.06 (d, J = 3.8 Hz, 1H, 6′-H), 4.68 (d, J = 7.9 Hz, 1H, 4′-H), 4.79 (d, J = 4.3 Hz, 1H, 3′-H), 4.88 (dd, J = 7.9, 3.8 Hz, 1H, 5′-H), 5.12 (dd, J = 7.9, 4.3 Hz, 1H, 2′-H), 6.03 (d, J = 8.1 Hz, 1H, 5-H), 6.88 (d, J = 7.9 Hz, 1H, 1′-H), 8.41 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, pyridine-d5, 35 °C): δ [ppm] = −4.46 (SiCH3), −4.05 (SiCH3), −4.00 (SiCH3), −3.95 (SiCH3), 18.32 (SiC(CH3)3), 18.51 (SiC(CH3)3), 26.16 (SiC(CH3)3), 26.26 (SiC(CH3)3), 27.75 (C-2′′), 28.41 (OC(CH3)3), 39.51 (C-3′′), 46.86 (C-1′′), 64.60 (C-6′), 73.58 (C-5′), 73.85 (C-2′), 74.03 (C-3′), 81.72 (OC(CH3)3), 87.19 (C-4′), 87.19 (C-1′), 103.29 (C-5), 142.71 (C-6), 152.37 (C-2), 164.15 (C-4), 171.56 (C-7′). MS (ESI+): m/z = 659.4 [M + H]+. HRMS (ESI+): calcd.: 659.3866 [M + H]+, found: 659.3871. IR (ATR): ν [cm−1] = 1678, 1252, 1155, 1057, 865, 833, 813, 776, 542. UV (MeOH): λmax (log ε) = 205 (3.98), 260 (3.84). Optical rotation: [α]D25 = −32.1 (c = 0.42, MeOH). m.p. = 184 °C.
Nε-tert-butyloxycarbonyl-l-lysine trimethylsilylethyl ester (24): To a solution of Nα-benzyloxycarbonyl-Nε-tert-butyloxycarbonyl-l-lysine 23 (667 mg, 1.75 mmol), ECD (673 mg, 3.51 mmol), and 4-(dimethylamino)pyridine (DMAP, 175 mg, 1.43 mmol) in CH2Cl2 (8.9 mL), 2-(trimethylsilyl)ethanol (380 μL, 310 mg, 2.62 mmol) was added and the mixture was stirred at room temperature for 25 h. It was then washed with sat. NaHCO3 (3 × 30 mL), brine (3 × 30 mL), and water (60 mL). The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The resultant crude product was purified by column chromatography (6:4, petroleum ether-CH2Cl2) to give the Nα-protected lysine ester as a colourless oil (653 mg, 77%). This material (600 mg, 1.25 mmol) was dissolved in MeOH (2 mL), Pd/C (10%, 190 mg, 0.179 mmol) was added and the mixture was stirred under a hydrogen atmosphere (1 bar) at room temperature for 3 h. It was then filtered over celiterTM, the residue was washed with MeOH and the solvent of the combined filtrates was removed under reduced pressure to give 24 as a colourless oil (422 mg, 99%, 77% over 2 steps from 23). 1H NMR (300 MHz, CD2Cl4, 100 °C): δ [ppm] = 4.53–4.44 (m, 1H, NεH), 4.26–4.20 (m, 2H, H-1′), 3.39–3.35 (m, 1H, H-2), 3.14–3.07 (m, 2H, H-6), 1.80–1.68 (m, 1H, H-3a), 1.59–1.39 (m, 5H, H-3b, H-4, H-5), 1.45 (s, 9H, Boc-CH3), 1.05–1.00 (m, 2H, H-2′), 0.08 (s, 9H, Si(CH3)3). 13C-NMR (75 MHz, CD2Cl4, 100 °C): δ [ppm] = 175.38 (C-1), 155.57 (Boc-C=O), 78.67 (Boc-C), 62.70 (C-1′), 54.33 (C-2), 40.48 (C-6), 34.25 (C-3), 29.64 (C-5), 28.23 (Boc-CH3), 22.71 (C-4), 17.34 (C-2′), -1.77 (Si(CH3)3). MS (ESI+): m/z = 347.2 [M + H]+. HRMS (ESI+): calcd.: 347.2361 [M + H]+, found: 347.2364. IR (ATR): ν [cm−1] = 3372, 2952, 2364, 1713, 1520, 1365, 1250, 1171, 838. Optical rotation: [α]D25 = +5.5 (c = 0.58, CHCl3). TLC: Rf (95:5, CH2Cl2-MeOH) = 0.80.
Protected urea dipeptide trimethylsilylethyl ester (26): To a solution of 24 (10 mg, 29 μmol) and N-(S-methylthiocarbonyl)-l-valine tert-butyl ester 25 (7.9 mg, 32 μmol) in EtOAc (1 mL), N-methylmorpholine (NMM, 9.5 μL, 8.7 mg, 86 μmol) and silver(I)-trifluoromethanesulfonate (AgOTf, 11 mg, 43 μmol) were added and the mixture was stirred at room temperature for 17 h. The solvent was removed under reduced pressure, and the resultant crude product was purified by column chromatography (3:1, petroleum ether-EtOAc) to give 26 as colourless oil (12 mg, 76%). 1H NMR (300 MHz, CD2Cl4, 100 °C): δ [ppm] = 4.98 (d, J = 8.0 Hz, 1H, Lys-NαH), 4.95 (d, J = 8.8 Hz, 1H, Val-NH), 4.63–4.48 (m, 1H, Lys-NεH), 4.38 (ddd, J = 8.0, 7.6, 5.5 Hz, 1H, Lys-H-2), 4.31–4.14 (m, 3H, Val-H-2, H-1), 3.10 (dd, J = 13.0, 6.7 Hz, 2H, Lys-H-6), 2.18–2.03 (m, 1H, Val-H-3), 1.89–1.75 (m, 1H, Lys-H-3a), 1.75–1.59 (m, 1H, Lys-H-3b), 1.59–1.32 (m, 4H, Lys-H-4, Lys-H-5), 1.49 (s, 9H, t-Bu-CH3) 1.46 (s, 9H, Boc-CH3), 1.08–0.99 (m, 2H, H-2), 0.97 (d, J = 9.5 Hz, 3H, Val-H-4), 0.94 (d, J = 9.5 Hz, 3H, Val-H-4), 0.08 (s, 9H, Si(CH3)3). 13C NMR (75 MHz, CD2Cl4, 100 °C): δ [ppm] = 172.89 (Lys-C-1), 171.56 (Val-C-1), 156.78 (Boc-C=O), 155.69 (urea-C=O), 81.42 (t-Bu-C), 78.71 (Boc-C), 63.24 (C-1), 58.68 (Val-C-2), 53.13 (Lys-C-2), 40.35 (Lys-C-6), 32.33 (Lys-C-3), 31.16 (Val-C-3), 29.46 (Lys-C-5), 28.26 (t-Bu-CH3), 27.90 (Boc-CH3), 22.35 (Lys-C-4), 18.55 (Val-C-4), 17.60 (Val-C-4), 17.32 (C-2), -1.80 (Si(CH3)3). MS (ESI+): m/z = 568.3 [M + Na]+. HRMS (ESI)+: calcd.: 568.3388 [M + Na]+, found: 568.3391. IR (ATR): ν [cm−1] = 3355, 2961, 1715, 1644, 1550, 1365, 1249, 1164, 836. Optical rotation: [α]D25 = 7.7 (c = 0.38, CHCl3). TLC: Rf (3:2, petroleum ether-EtOAc) = 0.47.
Protected urea dipeptide (27): To a solution of 26 (380 mg, 0.696 mmol) in THF (8.7 mL), tetrabutylammoniumfluoride solution (TBAF, 1 m in THF, 840 μL, 0.840 mmol) was added at 0 °C and the mixture was stirred at room temperature for 5 h. The solvent was removed under reduced pressure, and the resultant crude product was purified by column chromatography (95:5:1, CH2Cl2-MeOH-HOAc) to give 27 as colourless oil (287 mg, 93%). 1H NMR (300 MHz, CD2Cl4, 100 °C): δ [ppm] = 5.69–5.37 (m, 2H, Lys-NαH, Val-NαH). 4.95–4.83 (m, 1H, Lys-NεH), 4.36–4.28 (m, 1H, Lys-H-2), 4.25–4.18 (m, 1H, Val-H-2), 3.16–3.06 (m, 2H, Lys-H-6), 2.23–2.02 (m, 1H, Val-H-3), 1.95–1.80 (m, 1H, Lys-H-3a), 1.80–1.66 (m, 1H, Lys-H-3b), 1.51–1.48 (m, 4H, Lys-H-4, Lys-H-5), 1.48 (s, 9H, t-Bu-CH3) 1.47 (s, 9H, Boc-CH3), 0.97 (d, J = 10.5 Hz, 3H, Val-H-4), 0.94 (d, J = 10.5 Hz, 3H, Val-H-4). 13C NMR (75 MHz, CD2Cl4, 100 °C): δ [ppm] = 175.12 (Lys-C-1) 171.99 (Val-C-1), 158.34 (Boc-C=O), 156.21 (urea-C=O), 81.98 (t-Bu-C), 81.74 (Boc-C), 58.46 (Val-C-2), 53.38 (Lys-C-2), 40.04 (Lys-C-6), 31.16 (Lys-C-3), 31.11 (Val-C-3), 29.29 (Lys-C-5), 28.26 (t-Bu-CH3), 27.85 (Boc-CH3), 22.25 (Lys-C-4), 18.69 (Val-C-4), 17.43 (Val-C-4). MS (ESI+): m/z = 468.3 [M + Na]+. HRMS (ESI+): calcd.: 468.2680 [M + Na]+, found: 468.2681. IR (ATR): ν [cm−1] = 3359, 2967, 1715, 1640, 1552, 1366, 1159, 847. Optical rotation: [α]D25 = 19.9 (c = 0.80, CHCl3). TLC: Rf (7:3:1, CH2Cl2:MeOH:HOAc) = 0.41.
l-leucine trimethylsilylethyl ester trifluoroacetate (28): To a solution of N-benzyloxycarbonyl-l-leucine 30 (10.0 g, 37.8 mmol) in CH2Cl2 (385 mL), EDC (9.21 g, 48 mmol), DMAP (922 mg, 7.55 mmol), and 2-(trimethylsilyl)ethanol (6.87 mL, 5.67 g, 48.0 mmol) were added and the mixture was stirred at room temperature for 14 h. It was then washed with HCl (1 m, 3 × 600 mL), sat. NaHCO3 (3 × 600 mL), and brine (3 × 600 mL). The organic layer was dried over NaSO4 and the solvent was removed under reduced pressure. N-benzyloxycarbonyl-l-leucine trimethylsilylethyl ester was isolated after column chromatography as a colourless oil (11.8 g, 86%). To a solution of this material (366 mg, 1.00 mmol) in MeOH (40 mL), TFA (80 μL, 0.12 g, 1.0 mmol) and Pd/C (10%, 146 mg, 0.130 mmol) were added and the mixture was stirred under hydrogen atmosphere (1 bar) at room temperature for 4 h. It was then filtered over celite™ and the residue was washed with MeOH. The solvent of the combined filtrates was removed under reduced pressure to give 28 as a colourless solid (345 mg, quant., 86% over 2 steps from 30). 1H NMR (300 MHz, CDCl3): δ [ppm] = 4.29–4.23 (m, 2H, H-1′), 3.90 (dd, J = 7.0, 7.0 Hz, 1H, H-2), 1.87–1.73 (m, 3H, H-3, H-4), 1.05–0.99 (m, 2H, H-2′), 0.97 (d, J = 6.1 Hz, 3H, H-5), 0.95 (d, J = 6.1 Hz, 3H, H-5), 0.04 (s, 9H, Si(CH3)3). 13C NMR (75 MHz, CDCl3): δ [ppm] = 170.12 (C-1), 162.73–161.77 (m, TFA-COO), 116.32–114.45 (m, TFA-CF3), 65.12 (C-1′), 51.54 (C-2), 39.61 (C-3), 24.29 (C-4), 22.09 (C-5), 21.74 (C-5), 17.19 (C-2′), −1.68 (Si(CH3)3). 19F NMR (282 MHz, CDCl3): δ [ppm] = −75.98 (TFA-CF3). MS (ESI+): m/z = 232.2 [M − TFA]+. HRMS (ESI+): calcd.: 232.1727 [M − TFA]+, found: 232.1724. IR (ATR): ν [cm−1] = 1733, 1665, 1250, 1201, 1174, 1137, 1042, 930, 834. Optical rotation: [α]D25 = 3.6 (c = 1.0, CHCl3). m.p. = 85 °C. TLC: Rf (9:1, petroleum ether:EtOAc) = 0.76.
Protected urea tripeptide trimethylsilylethyl ester (29): To a solution of 27 (15 mg, 35 μmol) in THF (1 mL), HOBt (4.7 mg, 35 μmol), and EDC (6.7 mg, 35 μmol) were added and the mixture was stirred at room temperature for 30 min. A solution of 28 (12 mg, 35 μmol) and DIPEA (12 μL, 9.0 mg, 70 μmol) in CH2Cl2 (1 mL) was added and the mixture was stirred at room temperature for 18 h. The solvent was removed under reduced pressure, and the resultant crude product was purified by column chromatography (99:1, CH2Cl2-MeOH) to give 29 as a colourless solid (18 mg, 80%). 1H NMR (300 MHz, CD2Cl4, 100 °C): δ [ppm] = 6.59 (d, J = 8.1 Hz, 1H, Lys-NαH), 5.21 (d, J = 7.8 Hz, 1H, Leu-NH), 5.14 (d, J = 8.7 Hz, 1H, Val-NH), 4.76–4.64 (m, 1H, Lys-NεH), 4.53 (ddd, J = 8.2, 8.1, 5.2 Hz, 1H, Lys-H-2), 4.31–4.12 (m, 4H, Val-H-2, Leu-H-2, H-1), 3.10 (dd, J = 13.1, 6.3 Hz, 2H, Lys-H-6), 2.20–2.02 (m, 1H, Val-H-3), 1.93–1.76 (m, 1H, Lys-H-3a), 1.77–1.34 (m, 8H, Lys-H-3b, Lys-H-4, Lys-H-5, Leu-H-3, Leu-H-4), 1.46 (s, 9H, Boc-CH3), 1.49 (s, 9H, t-Bu-CH3), 1.10–1.00 (m, 2H, H-2), 1.01–0.87 (m, 12H, 2 × Val-H-4, 2 × Leu-H-5), 0.07 (s, 9H, Si(CH3)3). 13C NMR (75 MHz, CD2Cl4, 100 °C): δ [ppm] = 172.41 (Lys-C-1), 172.36 (Val-C-1), 172.06 (Leu-C-1), 157.52 (Boc-C=O), 155.97 (urea-C=O), 81.55 (t-Bu-C), 78.78 (Boc-C), 63.24 (C-1), 58.34 (Val-C-2), 53.51 (Lys-C-2), 50.77 (Leu-C-2), 41.06 (Lys-C-6), 39.90 (Leu-C-3), 31.51 (Lys-C-3), 31.03 (Val-C-3), 29.47 (Lys-C-5), 28.29 (t-Bu-CH3), 27.88 (Boc-CH3), 24.52 (Leu-C-4), 22.57 (Lys-C-4), 22.39 (Val-C-4), 21.79 (Val-C-4), 18.80 (Leu-C-5), 17.43 (Leu-C-5), 17.15 (C-2), −1.73 (Si(CH3)3). MS (ESI+): m/z = 681.50 [M + Na]+. HRMS (ESI+): calcd.: 681.4229 [M + Na]+, found: 681.4233. IR (ATR): ν [cm−1] = 3339, 2958, 1731, 1688, 1633, 1546, 1249, 1153, 837. Optical rotation: [α]D25 = −14.1 (c = 0.91, CHCl3). m.p. = 108 °C. TLC: Rf (98:2, CH2Cl2:MeOH) = 0.48.
Cbz-protected 6′-epi nucleoside building block (33): To a solution of the uridine-derived epoxy tert-butyl ester 31 (60 mg, 0.10 mmol) in i-PrOH (4 mL), N-benzyloxycarbonyl-1,3-diaminopropane 32 (31 mg, 0.15 mmol) was added and the mixture was stirred under reflux for 4 d. The solvent was removed under reduced pressure, and the resultant crude product was purified by column chromatography (1:1, petroleum ether-EtOAc) to give 33 as a colourless solid (61 mg, 77%). 1H NMR (600 MHz, CDCl3): δ [ppm] = 0.05 (s, 3H, SiCH3), 0.05 (s, 3H, SiCH3), 0.08 (s, 3H, SiCH3), 0.10 (s, 3H, SiCH3), 0.86 (s, 9H, SiC(CH3)3), 0.91 (s, 9H, SiC(CH3)3), 1.47 (s, 9H, OC(CH3)3), 1.60–1.65 (m, 2H, 2′′-H), 2.39 (ddd, J = 11.6, 7.1, 6.9 Hz, 1H, 1′′-Ha), 2.84 (ddd, J = 11.6, 5.8, 5.8 Hz, 1H, 1′′-Hb), 3.26–3.36 (m, 3H, 6′-H, 3′′-H), 3.84 (dd, J = 7.2, 1.1 Hz, 1H, 5′-H), 4.07 (dd, 4.6, 1.1 Hz, 1H, 4′-H), 4.12 (dd, J = 4.6, 4.4 Hz, 1H, 3′-H), 4.36 (dd, J = 4.5, 4.4 Hz, 1H, 2′-H), 5.03 (dd, J = 6.2, 6.1 Hz, 1H, 3′′-NH), 5.08 (d, J = 12.2 Hz, 1H, 1′′′-Ha), 5.10 (d, J = 12.2 Hz, 1H, 1′′′-Hb), 5.52 (d, J = 4.5 Hz, 1H, 1′-H), 5.70 (d, J = 8.1 Hz, 1H, 5-H), 7.29–7.35 (m, 5H, aryl-H), 7.59 (d, J = 8.1 Hz, 1H, 6-H), 8.69 (s, 1H, 3-H). 13C NMR (126 MHz, CDCl3): δ [ppm] = −4.71 (SiCH3), −4.67 (SiCH3), −4.62 (SiCH3), −4.23 (SiCH3), 17.99 (SiC(CH3)3), 18.11 (SiC(CH3)3), 25.82 (SiC(CH3)3), 25.90 (SiC(CH3)3), 28.13 (OC(CH3)3), 30.13 (C-2′′), 38.70 (C-3′′), 45.65 (C-1′′), 64.55 (C-6′), 66.67 (C-1′′′), 68.96 (C-5′), 72.18 (C-3′), 73.77 (C-2′), 82.19 (OC(CH3)3), 84.82 (C-4′), 92.31 (C-1′), 102.03 (C-5), 128.00, 128.08, 128.43 (aryl-C), 136.54 (C-2′′′), 141.81 (C-6), 149.94 (C-2), 156.38 (Cbz-C=O), 162.74 (C-4), 172.04 (C-7′). MS (ESI+): m/z = 793.5 [M + H]+. HRMS (ESI+): calcd.: 793.4234 [M + H]+, found: 793.4234. IR (ATR): ν [cm−1] = 1682, 1252, 1154, 1121, 867, 834, 813, 776, 735. UV (MeCN): λmax (log ε) = 204 (4.24), 261 (3.99). Optical rotation: [α]D25 = +18.0 (c = 0.55, CHCl3). m.p. = 62 °C. TLC: Rf = 0.13 (2:3, petroleum ether:EtOAc).
Cbz-protected 5′-epi nucleoside building block (35): To a solution of uridine-derived epoxy tert-butyl ester 34 (10 mg, 0.017 mmol) in i-PrOH (4 mL), N-benzyloxycarbonyl-1,3-diaminopropane 32 (5.4 mg, 0.026 mmol) was added and the mixture was stirred under reflux for 3 d. The solvent was removed under reduced pressure, and the resultant crude product was purified by column chromatography (97:3, CH2Cl2-MeOH) to give 35 as a colourless solid (12 mg, 89%). 1H NMR (600 MHz, DMSO-d6, 35 °C): δ [ppm] = −0.07 (s, 3H, SiCH3), 0.01 (s, 3H, SiCH3), 0.08 (s, 3H, SiCH3), 0.11 (s, 3H, SiCH3), 0.81 (s, 9H, SiC(CH3)3), 0.89 (s, 9H, SiC(CH3)3), 1.42 (s, 9H, OC(CH3)3), 1.49–1.54 (m, 2H, 2′′-H), 2.04 (s, 1H, 6′-NH), 2.34 (ddd, J = 10.6, 8.5, 8.2 Hz, 1H, 1′′-Ha), 2.54 (ddd, J = 10.6, 7.1, 6.8 Hz, 1H, 1′′-Hb), 2.99–3.06 (m, 3H, 6′-H, 3′′-H), 3.70 (ddd, J = 6.3, 6.1, 5.4 Hz, 1H, 5′-H), 4.10 (d, J = 5.4 Hz, 1H, 4′-H), 4.23 (d, J = 4.4 Hz, 1H, 3′-H), 4.32 (dd, J = 7.7, 4.4 Hz, 1H, 2′-H), 5.00 (s, 2H, 1′′′-H), 5.58 (d, J = 6.3 Hz, 1H, OH), 5.67 (d, J = 8.1 Hz, 1H, 5-H), 5.87 (d, J = 7.7 Hz, 1H, 1′-H), 7.16 (dd, J = 5.9, 5.8 Hz, 1H, 3′′-NH), 7.33–7.36 (m, 5H, aryl-H), 7.73 (d, J = 8.1 Hz, 1H, 6-H), 11.31 (s, 1H, 3-H). 13C NMR (126 MHz, DMSO-d6, 35 °C): δ [ppm] = −5.15 (SiCH3), −4.88 (SiCH3), −4.63 (SiCH3), −4.53 (SiCH3), 17.54 (SiC(CH3)3), 17.67 (SiC(CH3)3), 25.54 (SiC(CH3)3), 25.62 (SiC(CH3)3), 27.76 (OC(CH3)3), 30.07 (C-2′′), 38.44 (C-3′′), 44.96 (C-1′′), 64.21 (C-6′), 64.97 (C-1′′′), 71.89 (C-3′), 72.22 (C-5′), 73.76 (C-2′), 80.11 (OC(CH3)3), 85.66 (C-1′), 86.04 (C-4′), 102.09 (C-5), 127.43, 127.47, 128.07 (aryl-C), 137.02 (C-2′′′), 140.51 (C-6), 150.64 (C-2), 155.81 (Cbz-C=O), 162.52 (C-4), 171.66 (C-7′). MS (ESI+): m/z = 793.5 [M + H]+. HRMS (ESI+): calcd.: 793.4234 [M + H]+, found: 793.4239. IR (ATR): ν [cm−1] = 1689, 1252, 1153, 1057, 833, 813, 775, 735, 697. UV (MeCN): λmax (log ε) = 204 (4.13), 259 (3.77). Optical rotation: [α]D25 = −30.0 (c = 0.24, CHCl3). m.p. = 78 °C. TLC: Rf = 0.15 (96:4, CH2Cl2:MeOH).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
