Failure of the Anti-Inflammatory Parasitic Worm Product ES-62 to Provide Protection in Mouse Models of Type I Diabetes, Multiple Sclerosis, and Inflammatory Bowel Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. ES-62 and SMAs
2.2. Type I Diabetes Model
2.3. Experimental Autoimmune Encephalomyelitis
2.4. Induction and Assessment of Chronic Colitis
2.5. T Cell Transfer Model
3. Results
3.1. Type I Diabetes
3.2. Multiple Sclerosis
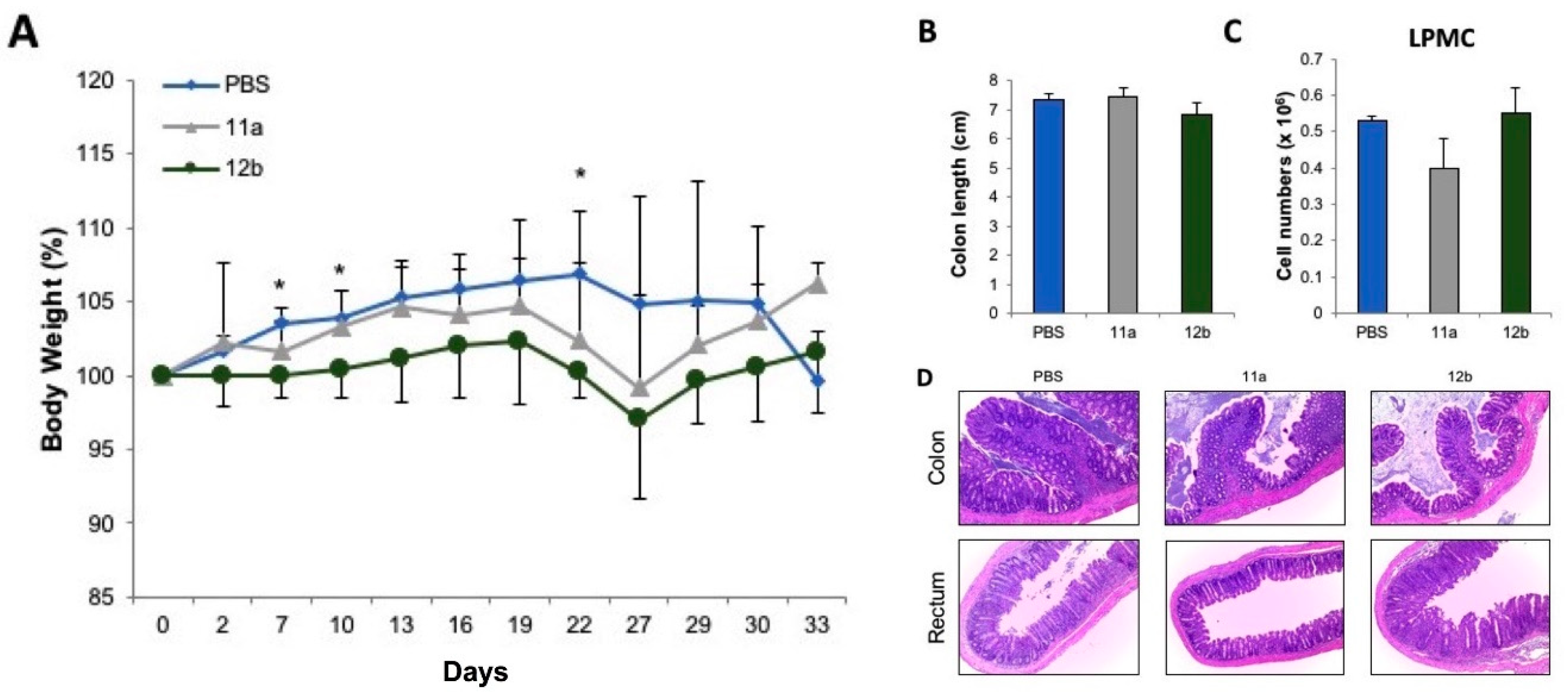
3.3. Inflammatory Bowel Disease
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maizels, R.M.; McSorley, H.J. Regulation of the host immune system by helminth parasites. J. Allergy Clin. Immunol. 2016, 138, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.A.; Lumb, F.; Harnett, M.M.; Harnett, W. ES-62, a therapeutic anti-inflammatory agent evolved by the filarial nematode Acanthocheilonema viteae. Mol. Biochem. Parasitol. 2014, 194, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.T.; McGrath, M.A.; Pineda, M.A.; Al-Riyami, L.; Rzepecka, J.; Lumb, F.; Harnett, W.; Harnett, M.M. The parasitic worm product, ES-62 targets MyD88-dependent effector mechanisms to suppress ANA production and proteinuria in MRL/Lpr mice. Arthritis Rheumatol. 2015, 67, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, J.; Siebeke, I.; Coltherd, J.C.; Kean, D.E.; Steiger, C.N.; Al-Riyami, L.; McSharry, C.; Harnett, M.M.; Harnett, W. The helminth product, ES-62, protects against airway inflammation by resetting the Th cell phenotype. Int. J. Parasitol. 2013, 43, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Coltherd, J.C.; Rodgers, D.T.; Lawrie, R.E.; Al-Riyami, L.; Suckling, C.J.; Harnett, W.; Harnett, M.M. The parasitic worm-derived immunomodulator, ES-62 and its drug-like small molecule analogues exhibit therapeutic potential in a model of chronic asthma. Sci. Rep. 2016, 6, 19224. [Google Scholar] [CrossRef] [PubMed]
- Al-Riyami, L.; Pineda, M.A.; Rzepecka, J.; Huggan, J.K.; Khalaf, A.I.; Suckling, C.J.; Scott, F.J.; Rodgers, D.T.; Harnett, M.M.; Harnett, W. Designing anti-inflammatory drugs from parasitic worms: A synthetic small molecule analogue of the Acanthocheilonema viteae product ES-62 prevents development of collagen-induced arthritis. J. Med. Chem. 2013, 56, 9982–10002. [Google Scholar] [CrossRef] [PubMed]
- Al-Riyami, L.; Rodgers, D.; Rzepecka, J.; Pineda, M.A.; Suckling, C.J.; Harnett, M.M.; Harnett, W. Protective effect of small molecule analogues of the Acanthocheilonema viteae secreted product ES-62 on oxazolone-induced ear inflammation. Exp. Parasitol. 2015, 158, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Janicova, L.; Rzepecka, J.; Rodgers, D.T.; Doonan, J.; Bell, K.S.; Lumb, F.E.; Suckling, C.J.; Harnett, M.M.; Harnett, W. Testing small molecule analogues of the Acanthocheilonema viteae immunomodulator ES-62 against clinically relevant allergens. Parasite Immunol. 2016, 38, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, J.; Coates, M.L.; Saggar, M.; Al-Riyami, L.; Coltherd, J.; Tay, H.K.; Huggan, J.K.; Janicova, L.; Khalaf, A.I.; Siebeke, I.; et al. Small molecule analogues of the immunomodulatory parasitic helminth product ES-62 have anti-allergy properties. Int. J. Parasitol. 2014, 44, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, J.; Pineda, M.A.; Al-Riyami, L.; Rodgers, D.T.; Huggan, J.K.; Lumb, F.E.; Khalaf, A.I.; Meakin, P.J.; Corbet, M.; Ashford, M.L.; et al. Prophylactic and therapeutic treatment with a synthetic analogue of a parasitic worm product prevents experimental arthritis and inhibits IL-1β production via NRF2-mediated counter-regulation of the inflammasome. J. Autoimmun. 2015, 60, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Lumb, F.E.; Doonan, J.; Bell, K.S.; Pineda, M.A.; Corbet, M.; Suckling, C.J.; Harnett, M.M.; Harnett, W. Dendritic cells provide a therapeutic target for synthetic small molecule analogues of the parasitic worm product, ES-62. Sci. Rep. 2017, 7, 1704. [Google Scholar] [CrossRef] [PubMed]
- Doonan, J.; Lumb, F.E.; Pineda, M.A.; Tarafdar, A.; Crowe, J.; Khan, A.M.; Suckling, C.J.; Harnett, M.M.; Harnett, W. Protection Against Arthritis by the Parasitic Worm Product ES-62, and Its Drug-Like Small Molecule Analogues, Is Associated with Inhibition of Osteoclastogenesis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, J.; Harnett, W. Can the study of helminths be fruitful for human diseases? In Helminth Infections and Their Impact on Global Public Health; Bruschi, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 479–502. [Google Scholar]
- Zaccone, P.; Fehérvári, Z.; Jones, F.M.; Sidobre, S.; Kronenberg, M.; Dunne, D.W.; Cooke, A. Schistosoma mansoni antigens modulate the activity of the innate immune response and prevent onset of type 1 diabetes. Eur. J. Immunol. 2003, 33, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, P.; Burton, O.; Miller, N.; Jones, F.M.; Dunne, D.W.; Cooke, A. Schistosoma mansoni egg antigens induce Treg that participate in diabetes prevention in NOD mice. Eur. J. Immunol. 2009, 39, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.E.; O’Brien, B.A.; Hutchinson, A.T.; Robinson, M.W.; Simpson, A.M.; Dalton, J.P.; Donnelly, S. Secreted Proteins from the Helminth Fasciola hepatica Inhibit the Initiation of Autoreactive T Cell Responses and Prevent Diabetes in the NOD Mouse. PLoS ONE 2014, 9, e86289. [Google Scholar] [CrossRef] [PubMed]
- Hübner, M.P.; Stocker, J.T.; Mitre, E. Inhibition of type 1 diabetes in filaria-infected non-obese diabetic mice is associated with a T helper type 2 shift and induction of FoxP3+ regulatory T cells. Immunology 2009, 127, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Sewell, D.; Qing, Z.; Reinke, E.; Elliot, D.; Weinstock, J.; Sandor, M.; Fabry, Z. Immunomodulation of experimental autoimmune encephalomyelitis by helminth ova immunization. Int. Immunol. 2003, 15, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hu, X.; Zhou, G.; Lu, Z.; Qiu, W.; Bao, J.; Dai, Y. Soluble egg antigen from Schistosoma japonicum modulates the progression of chronic progressive experimental autoimmune encephalomyelitis via Th2-shift response. J. Neuroimmunol. 2008, 194, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.P.; Brady, M.T.; Finlay, C.M.; Boon, L.; Mills, K.H.G. Infection with a Helminth Parasite Attenuates Autoimmunity through TGF-β-Mediated Suppression of Th17 and Th1 Responses. J. Immunol. 2009, 183, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Donskow-Łysoniewska, K.; Krawczak, K.; Doligalska, M. Heligmosomoides polygyrus: EAE remission is correlated with different systemic cytokine profiles provoked by L4 and adult nematodes. Exp. Parasitol. 2012, 132, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Taylor, M.D.; O’Gorman, M.T.; Balic, A.; Barr, T.A.; Filbey, K.; Anderton, S.M.; Maizels, R.M. Helminth-induced CD19+CD23hi B cells modulate experimental allergic and autoimmune inflammation. Eur. J. Immunol. 2010, 40, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Hasby, E.A.; Hasby Saad, M.A.; Shohieb, Z.; El Noby, K. FoxP3+ T regulatory cells and immunomodulation after Schistosoma mansoni egg antigen immunization in experimental model of inflammatory bowel disease. Cell. Immunol. 2015, 295, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Smyth, D.; Gaze, S.; Aziz, A.; Giacomin, P.; Ruyssers, N.; Artis, D.; Laha, T.; Navarro, S.; Loukas, A.; et al. Hookworm Excretory/Secretory Products Induce Interleukin-4 (IL-4)+IL-10+ CD4+ T Cell Responses and Suppress Pathology in a Mouse Model of Colitis. Infect. Immun. 2013, 81, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.H.; Deehan, M.R.; Katz, E.; Brown, K.S.; Houston, K.M.; O’Grady, J.; Harnett, M.M.; Harnett, W. Hyporesponsiveness of murine B lymphocytes exposed to the filarial nematode secreted product ES-62 in vivo. Immunology 2003, 109, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.D.; Wang, B.; Haskins, K.; Benoist, C.; Mathis, D. Following a dibetogenic T cell from genesis through pathogenesis. Cell 1993, 74, 1089–1100. [Google Scholar] [CrossRef]
- Takedatsu, H.; Michelsen, K.S.; Wei, B.; Landers, C.J.; Thomas, L.S.; Dhall, D.; Braun, J.; Targan, S.R. TL1A (TNFS15) regulates the development of chronic colitis by modulating both T-helper and T-helper 17 activation. Gastroenterology 2008, 135, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Weigmann, B.; Tubbe, I.; Seidel, D.; Nicolaev, A.; Bewcker, C.; Neurath, M.F. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat. Protocol. 2007, 2, 2307–2311. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, P.; RAine, T.; Sidobre, S.; Kronenberg, M.; Mastroeni, P.; Cooke, A. Salmonella typhimurium infection halts development of type 1 diabetes in NOD mice. Eur. J. Immunol. 2004, 34, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.A.; McGrath, M.A.; Smith, P.C.; Al-Riyami, L.; Rzepecka, J.; Gracie, J.A.; Harnett, W.; Harnett, M.M. The parasitic helminth product ES-62 suppresses pathogenesis in collagen-induced arthritis by targeting the interleukin-17-producing cellular network at multiple sites. Arthritis Rheum. 2012, 64, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.A.; Rodgers, D.T.; Al-Riyami, L.; Harnett, W.; Harnett, M.M. ES-62 protects against collagen-induced arthritis by resetting interleukin-22 toward resolution of inflammation in the joints. Arthritis Rheumatol. Hoboken 2014, 66, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Finney, C.A.M.; Taylor, M.D.; Wilson, M.S.; Maizels, R.M. Expansion and activation of CD4+CD25+ regulatory T cells in Heligmosomoides polygyrus infection. Eur. J. Immunol. 2007, 37, 1874–1886. [Google Scholar] [CrossRef] [PubMed]
- Hang, L.; Blum, A.M.; Setiawan, T.; Urban, J.P.; Stoyanoff, K.M.; Weinstock, J.V. Heligmosomoides polygyrus bakeri infection activates colonic Foxp3+ T cells enhancing their capacity to prevent colitis. J. Immunol. Baltim. 2013, 191, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.T.; Pineda, M.A.; McGrath, M.A.; Al-Riyami, L.; Harnett, W.; Harnett, M.M. Protection against collagen-induced arthritis in mice afforded by the parasitic worm product ES-62 is associated with restoration of the levels of interleukin 10-producing B cells and reduced plasma cell infiltration of the joints. Immunology 2013, 141, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Dittel, B.N. Mechanisms of regulatory B cell function in autoimmune and inflammatory diseases beyond IL-10. J. Clin. Med. 2017, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Oleinika, K.; Tonon, S.; Doyle, R.; Bosma, A.; Carter, N.A.; Harris, K.A.; Jones, S.A.; Klein, N.; Mauri, C.; et al. Cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat. Med. 2014, 20, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Burrows, M.P.; Volchkov, P.; Kobayashi, K.S.; Chervonsky, A.V. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 9973–9977. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Wang, Y.; Begum-Haque, S.; Dasgupta, S.; Kasper, D.L.; Kasper, L.H. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 2010, 3, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, Y.; Hu, Y.; Zhao, L.; Zhang, C. Initial gut microbiota structure affects sensitivity to DSS-induced colitis in a mouse model. Sci. China Life Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Smits, H.H.; Hiemstra, P.S.; Prazeres da Costa, C.; Ege, M.; Edwards, M.; Garn, H.; Howarth, P.H.; Jartti, T.; de Jong, E.C.; Maizels, R.M. Microbes and asthma: Opportunities for intervention. J. Allergy Clin. Immunol. 2016, 137, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Kyburz, A.; Muller, A. The Gastrointestinal Tract Microbiota and Allergic Diseases. Dig. Dis. 2016, 34, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Roodsaz, S.; Abramson, S.B.; Scher, J.U. The metabolic role of the gut microbiota in health and rheumatic disease: Mechanisms and interventions. Nat. Rev. Rheumatol. 2016, 12, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-K.; Lee, C.-G.; So, J.-S.; Chae, C.-S.; Hwang, J.-S.; Sahoo, A.; Nam, J.H.; Rhee, J.H.; Hwang, K.-C.; Im, S.-H. Generation of regulatory dendritic cells and CD4+Foxp3+ T cells by probiotics administration suppresses immune disorders. Proc. Natl. Acad. Sci. USA 2010, 107, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A.C.; Davis, J.W.; Spollen, W.; Bivens, N.; Givan, S.; Hagan, C.E.; McIntosh, M.; Franklin, C.L. Effects of Vendor and Genetic Background on the Composition of the Fecal Microbiota of Inbred Mice. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacteria microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.B.; Sonnenberg, G.F. Host-Microbiota Interactions Shape Local and Systemic Inflammatory Diseases. J. Immunol. 2017, 198, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Ball, D.H.; Tay, H.K.; Bell, K.S.; Coates, M.L.; Al-Riyami, L.; Rzepecka, J.; Harnett, W.; Harnett, M.M. Mast cell subsets and their functional modulation by the Acanthocheilonema viteae product ES-62. J. Parasitol. Res. 2013, 2013, 961268. [Google Scholar] [CrossRef] [PubMed]
- Eason, R.J.; Bell, K.S.; Marshall, F.A.; Rodgers, D.T.; Pineda, M.A.; Steiger, C.N.; Al-Riyami, L.; Harnett, W.; Harnett, M.M. The helminth product ES-62 modulates dendritic cell responses by inducing the selective autophagolysosomal degradation of TLR transducers, as exemplified by PKCδ. Sci. Rep. 2016, 6, 37276. [Google Scholar] [CrossRef] [PubMed]
- Suckling, C.J.; Alam, S.; Olson, M.A.; Saikh, K.U.; Harnett, M.M.; Harnett, W. Small molecule analogues of the parasitic worm product ES-62 interact with the TIR domain of MyD88 to inhibit pro-inflammatory signaling. Sci. Rep. 2018, 8, 2123. [Google Scholar] [CrossRef] [PubMed]
- Marta, M.; Andersson, A.; Isaksson, M.; Kampe, O.; Lobell, A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2008, 38, 565–575. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds ES-62, 11a and 12b are available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doonan, J.; Thomas, D.; Wong, M.H.; Ramage, H.J.; Al-Riyami, L.; Lumb, F.E.; Bell, K.S.; Fairlie-Clarke, K.J.; Suckling, C.J.; Michelsen, K.S.; et al. Failure of the Anti-Inflammatory Parasitic Worm Product ES-62 to Provide Protection in Mouse Models of Type I Diabetes, Multiple Sclerosis, and Inflammatory Bowel Disease. Molecules 2018, 23, 2669. https://doi.org/10.3390/molecules23102669
Doonan J, Thomas D, Wong MH, Ramage HJ, Al-Riyami L, Lumb FE, Bell KS, Fairlie-Clarke KJ, Suckling CJ, Michelsen KS, et al. Failure of the Anti-Inflammatory Parasitic Worm Product ES-62 to Provide Protection in Mouse Models of Type I Diabetes, Multiple Sclerosis, and Inflammatory Bowel Disease. Molecules. 2018; 23(10):2669. https://doi.org/10.3390/molecules23102669
Chicago/Turabian StyleDoonan, James, David Thomas, Michelle H. Wong, Hazel J. Ramage, Lamyaa Al-Riyami, Felicity E. Lumb, Kara S. Bell, Karen J. Fairlie-Clarke, Colin J. Suckling, Kathrin S. Michelsen, and et al. 2018. "Failure of the Anti-Inflammatory Parasitic Worm Product ES-62 to Provide Protection in Mouse Models of Type I Diabetes, Multiple Sclerosis, and Inflammatory Bowel Disease" Molecules 23, no. 10: 2669. https://doi.org/10.3390/molecules23102669
APA StyleDoonan, J., Thomas, D., Wong, M. H., Ramage, H. J., Al-Riyami, L., Lumb, F. E., Bell, K. S., Fairlie-Clarke, K. J., Suckling, C. J., Michelsen, K. S., Jiang, H.-R., Cooke, A., Harnett, M. M., & Harnett, W. (2018). Failure of the Anti-Inflammatory Parasitic Worm Product ES-62 to Provide Protection in Mouse Models of Type I Diabetes, Multiple Sclerosis, and Inflammatory Bowel Disease. Molecules, 23(10), 2669. https://doi.org/10.3390/molecules23102669