Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy: Part 1. Oligofluorenes, Oligophenylenes, Binaphthyls and Fused Aromatics



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
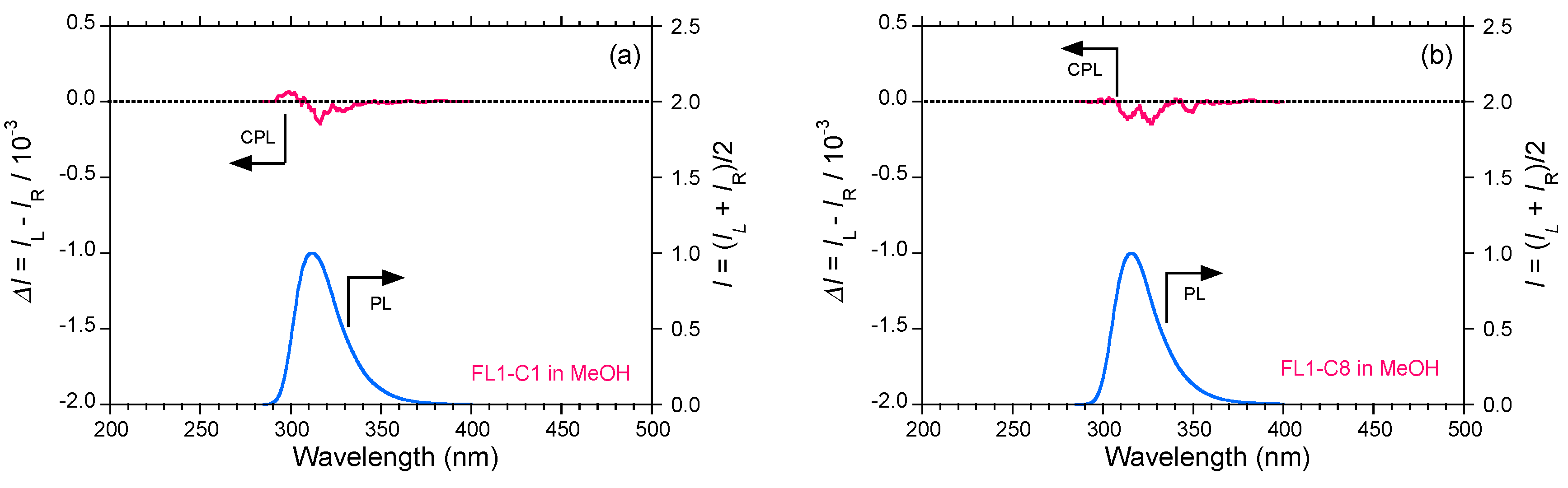{kind=link}
{kind=link}
{kind=link}
{kind=link}
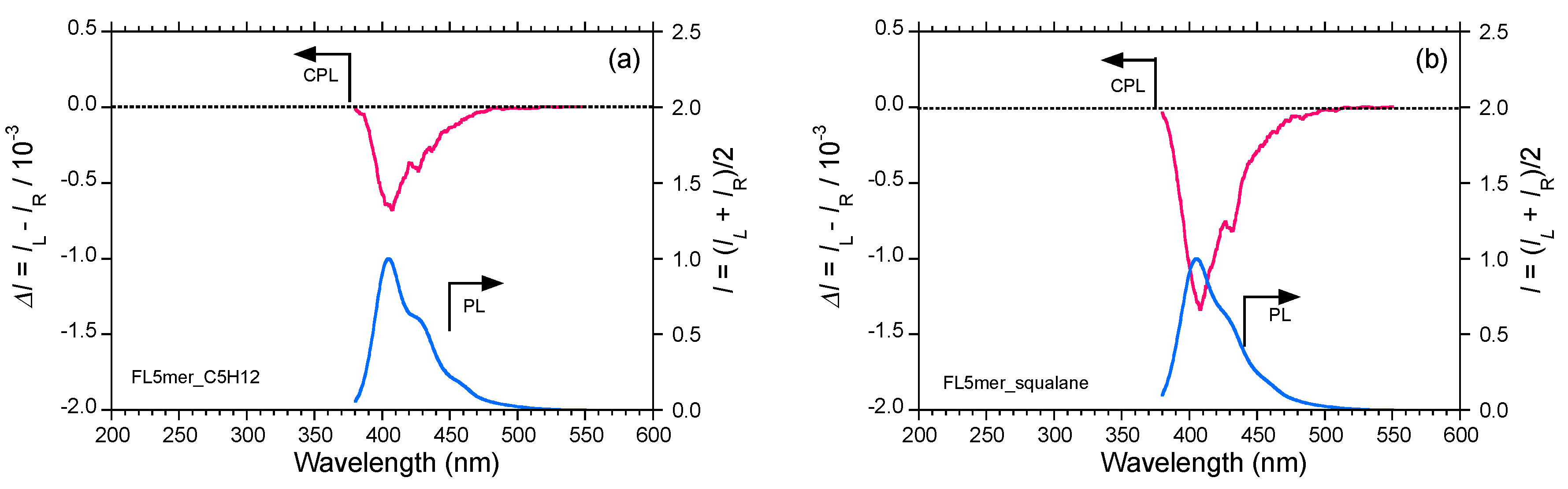{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
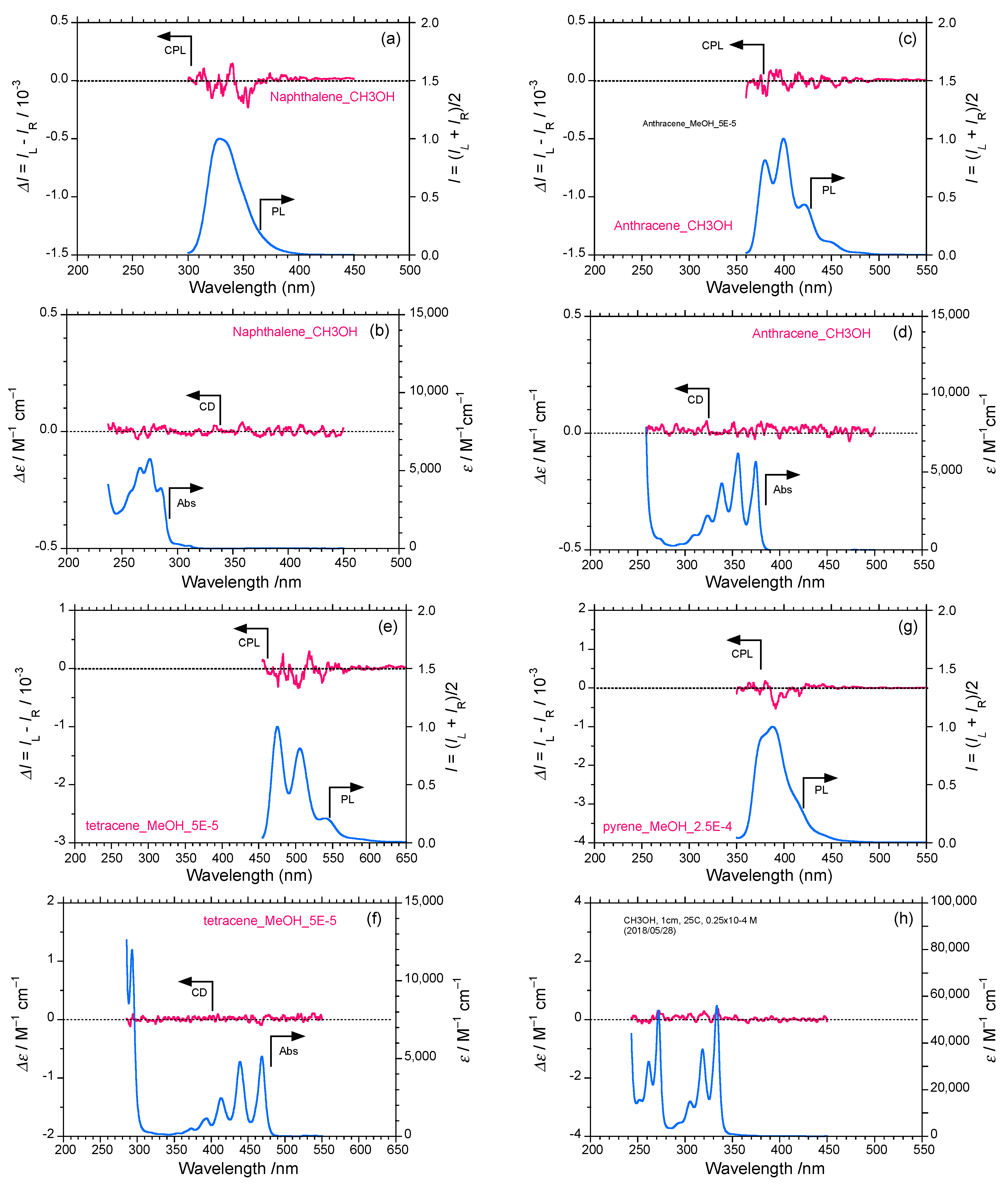
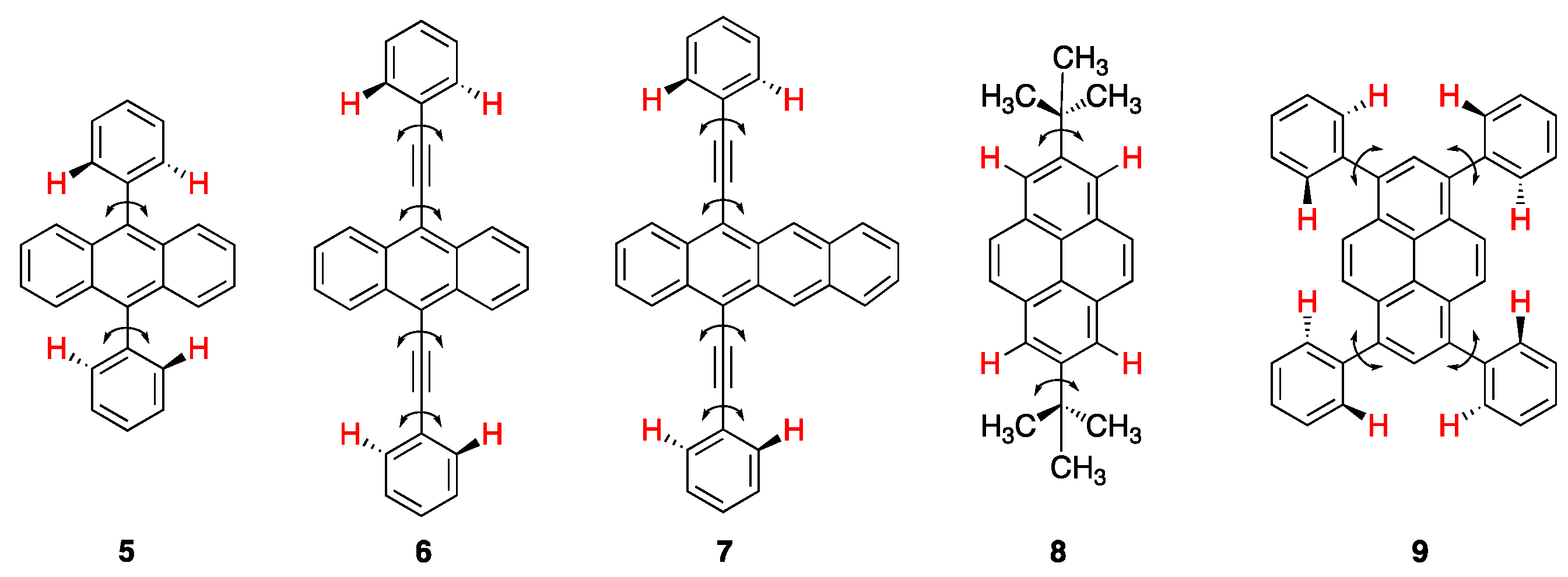
2.1. Rigid Achiral Planar π-Conjugated Luminophores
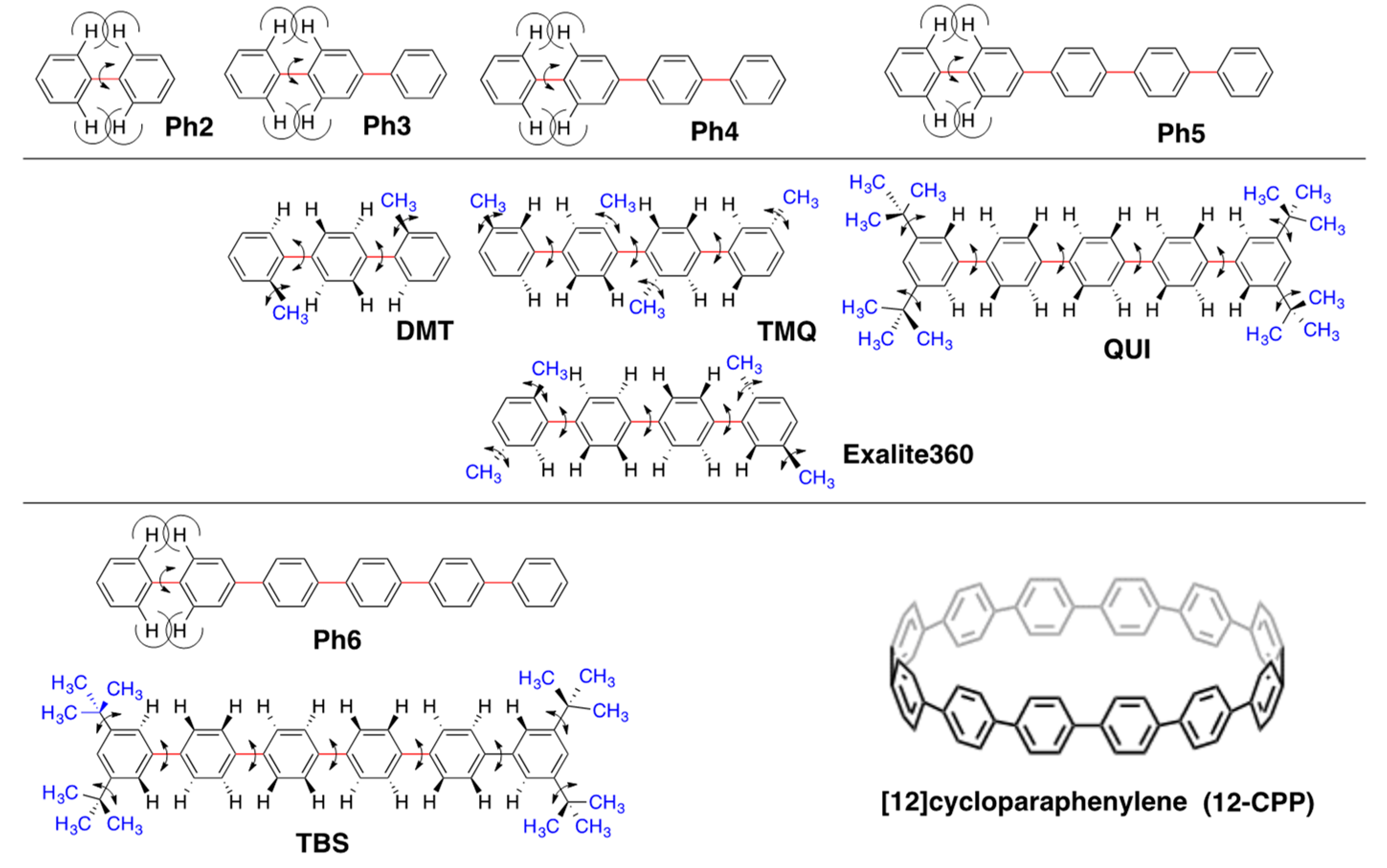
2.2. Rigid Planar π-Conjugated Luminophores Bearing Phenyl and Phenylethynyl Rotors
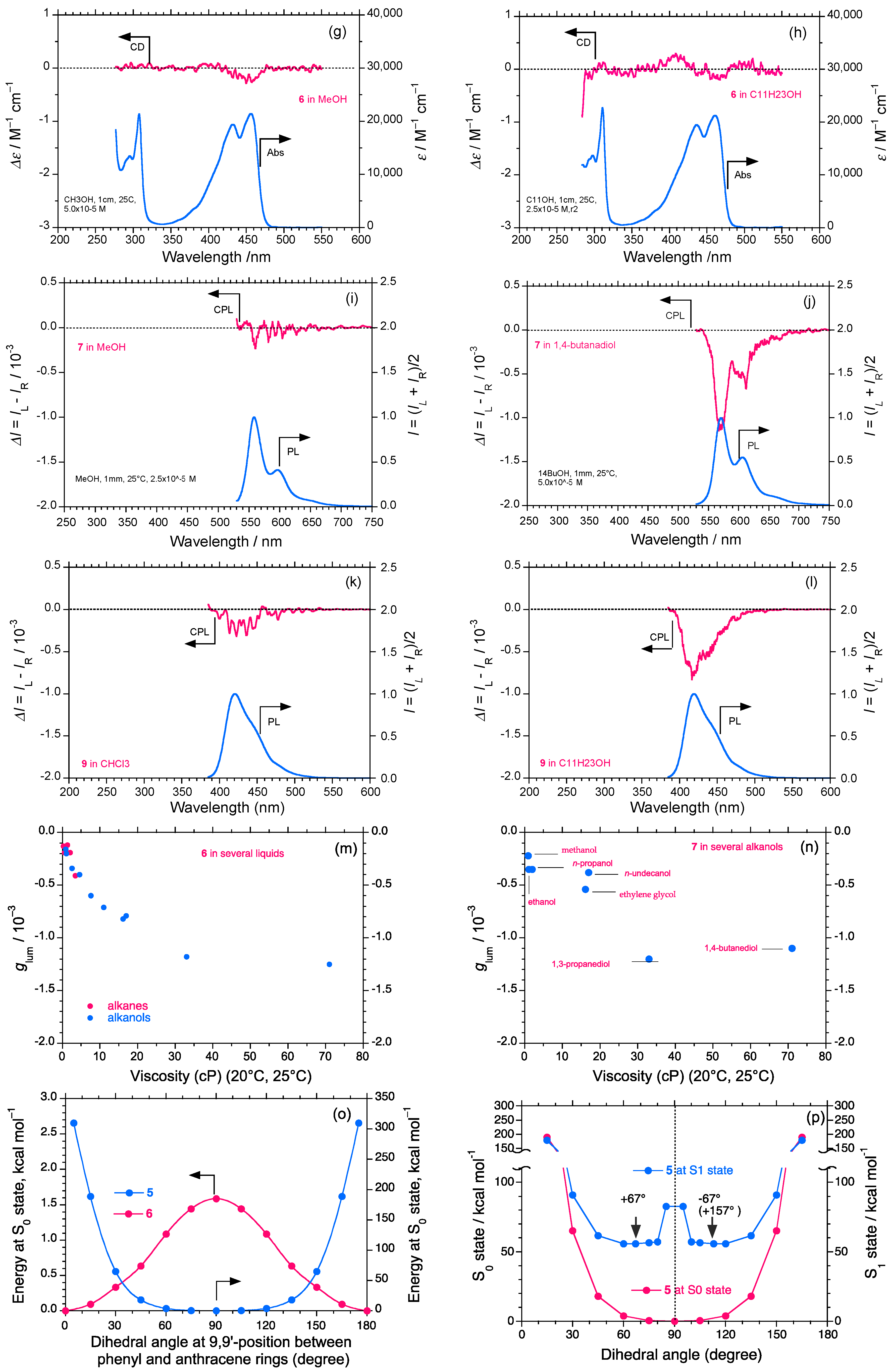
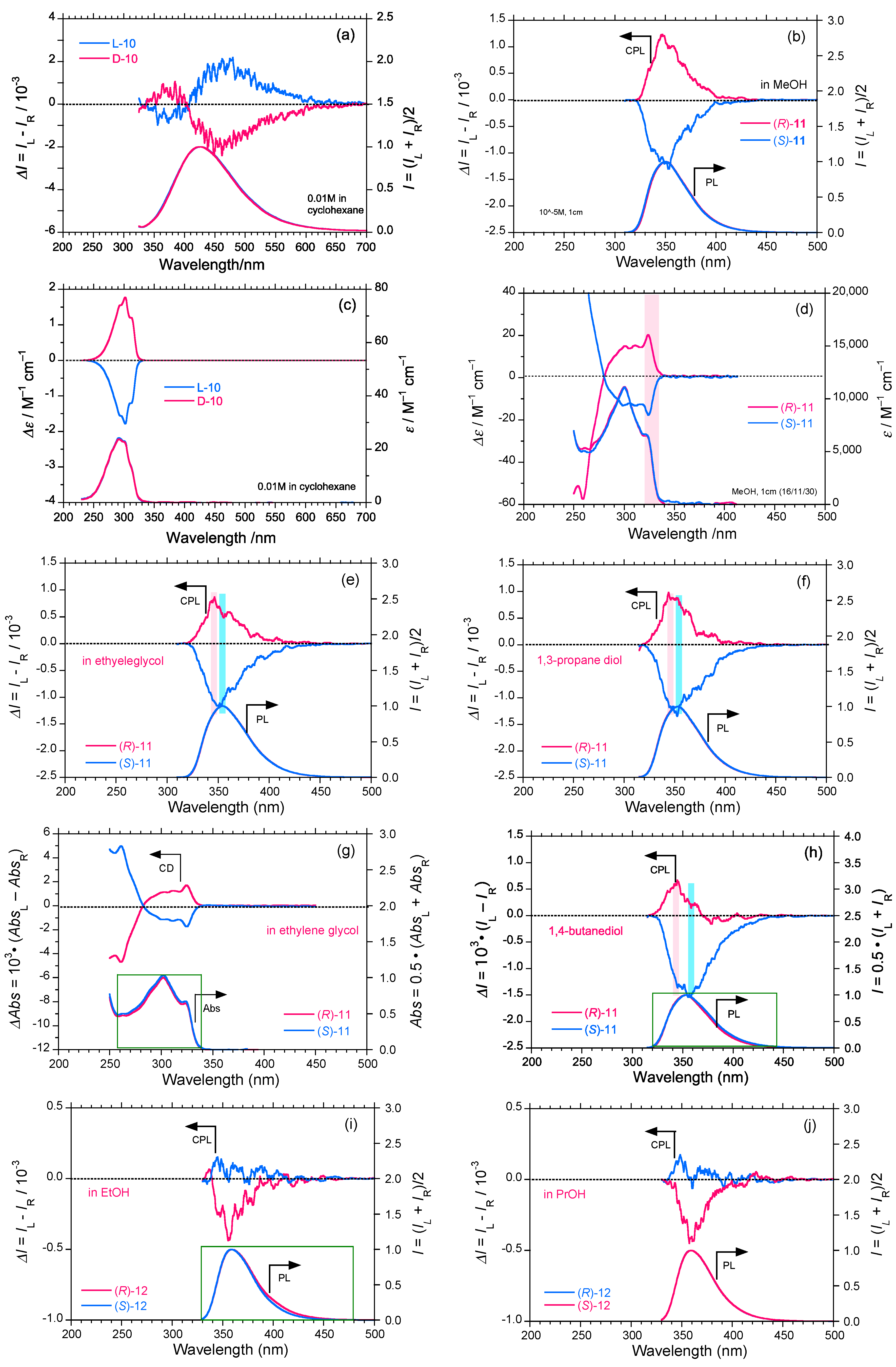
2.3. Rigid and Non-Rigid Chiral Luminophores
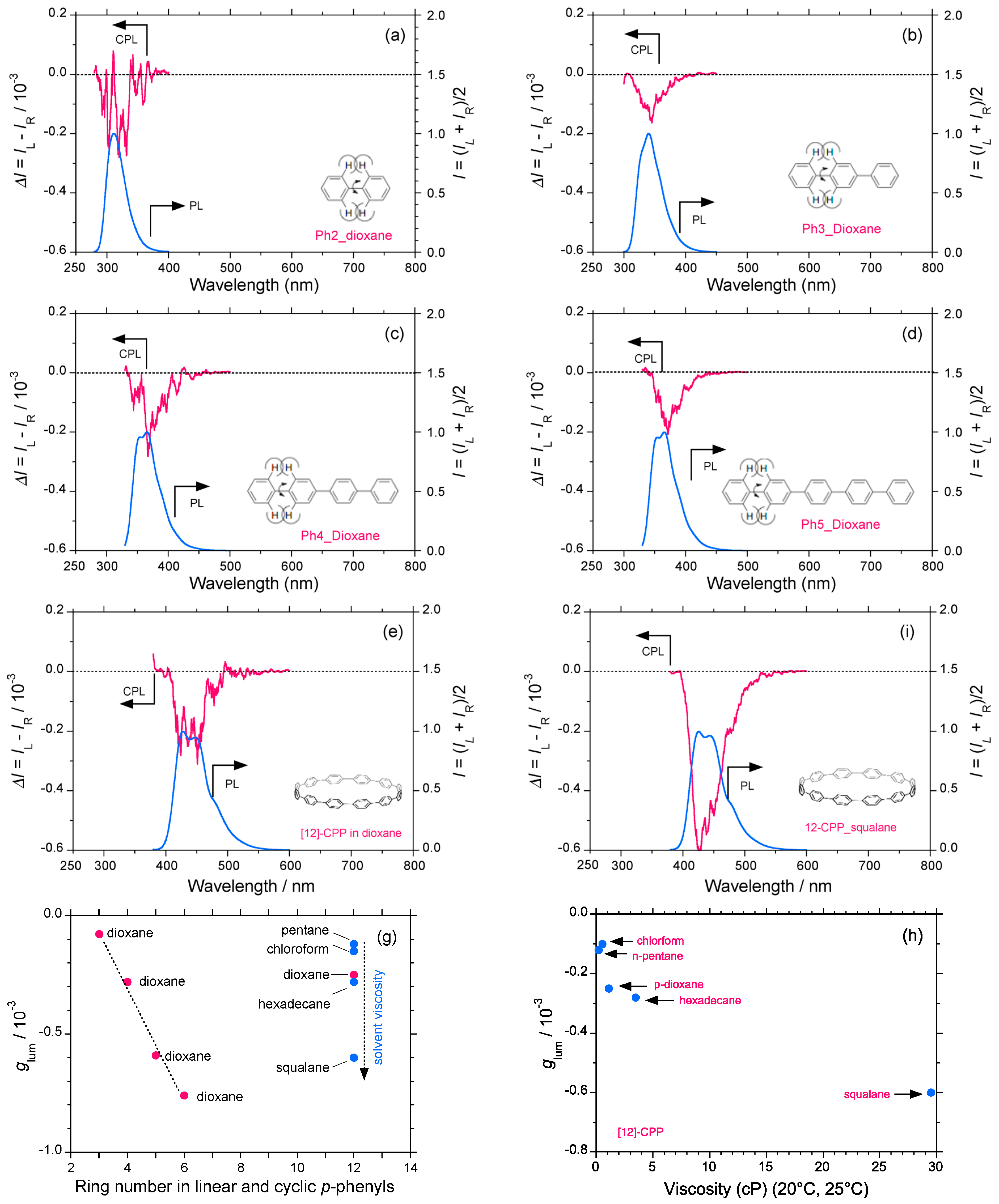
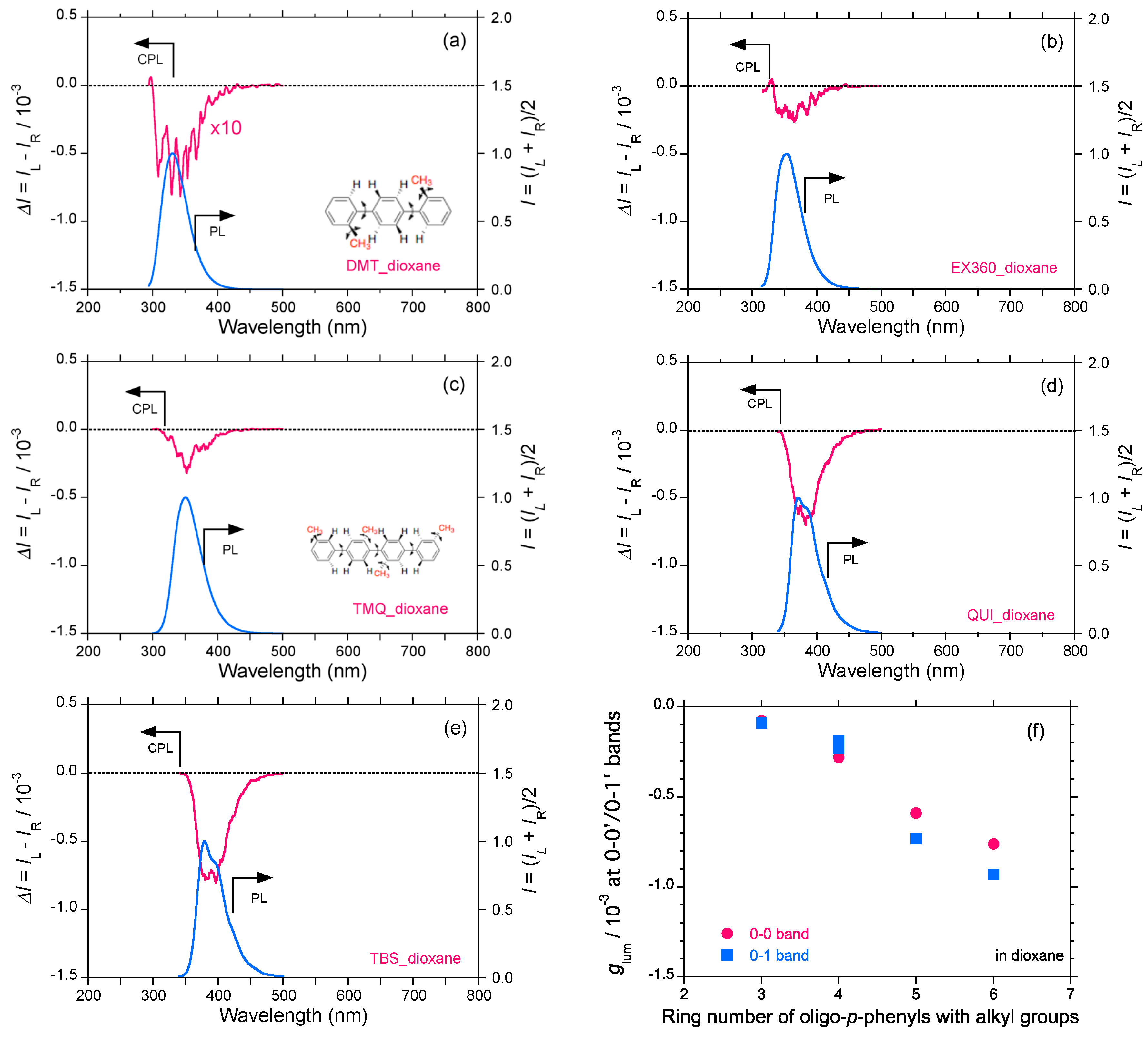
2.4. Linear and Cyclic Oligo-p-Phenyl Derivatives without and with Rotatable Alkyl Substituents
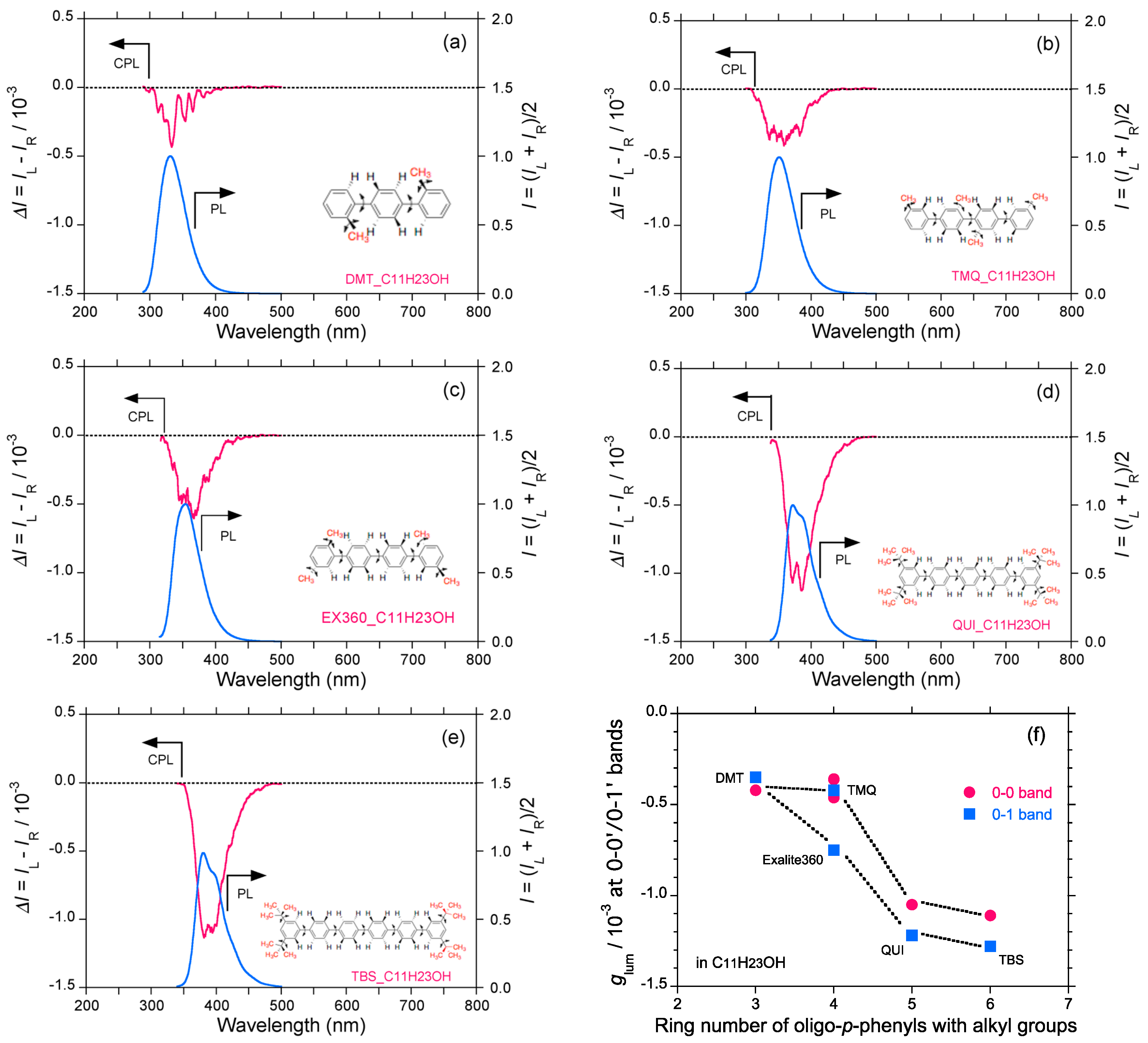
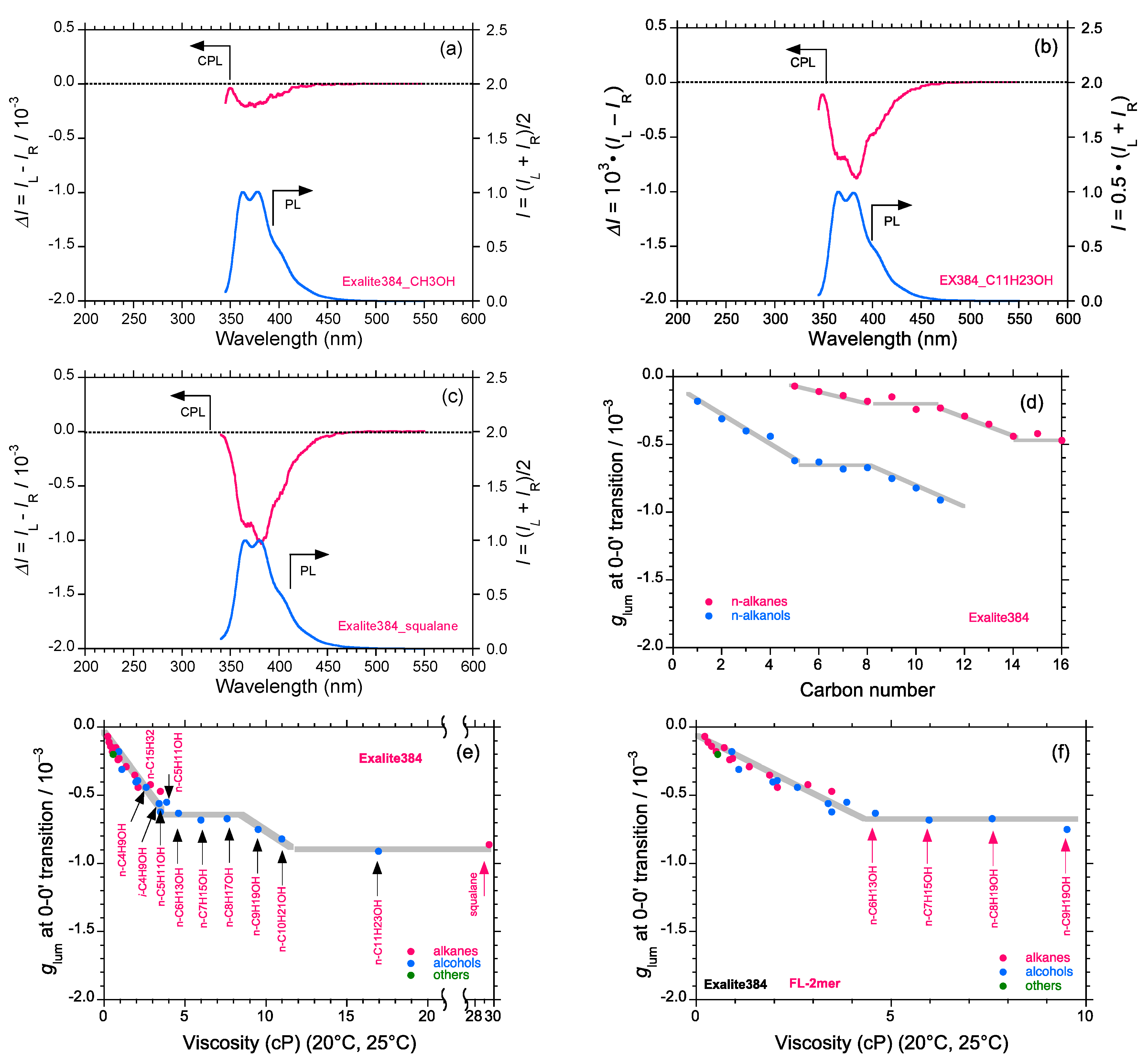
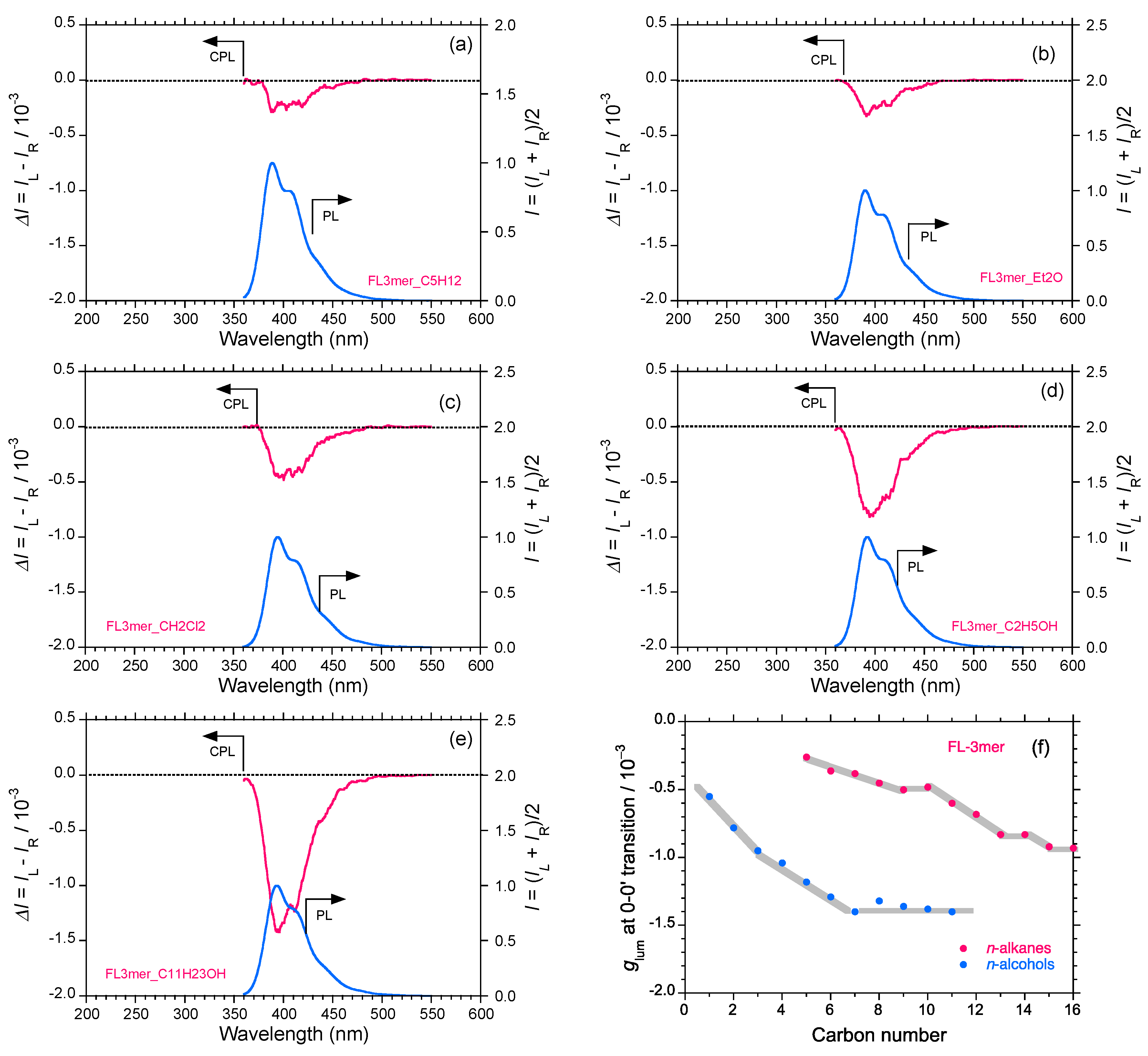
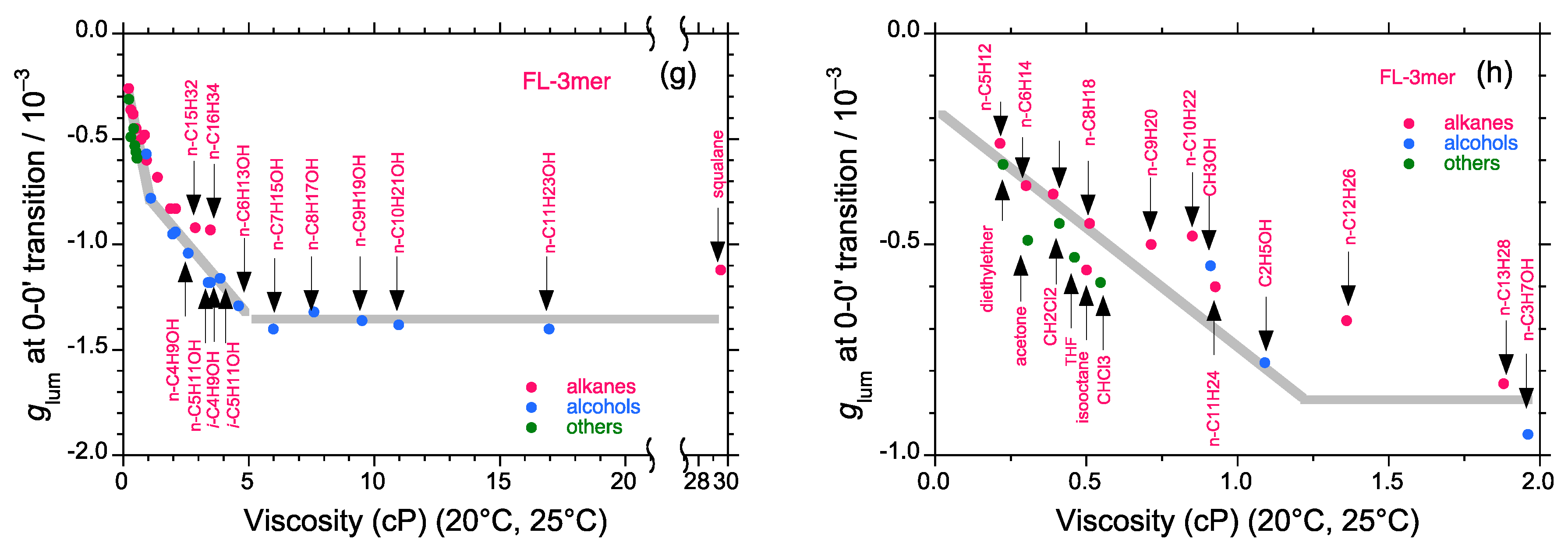
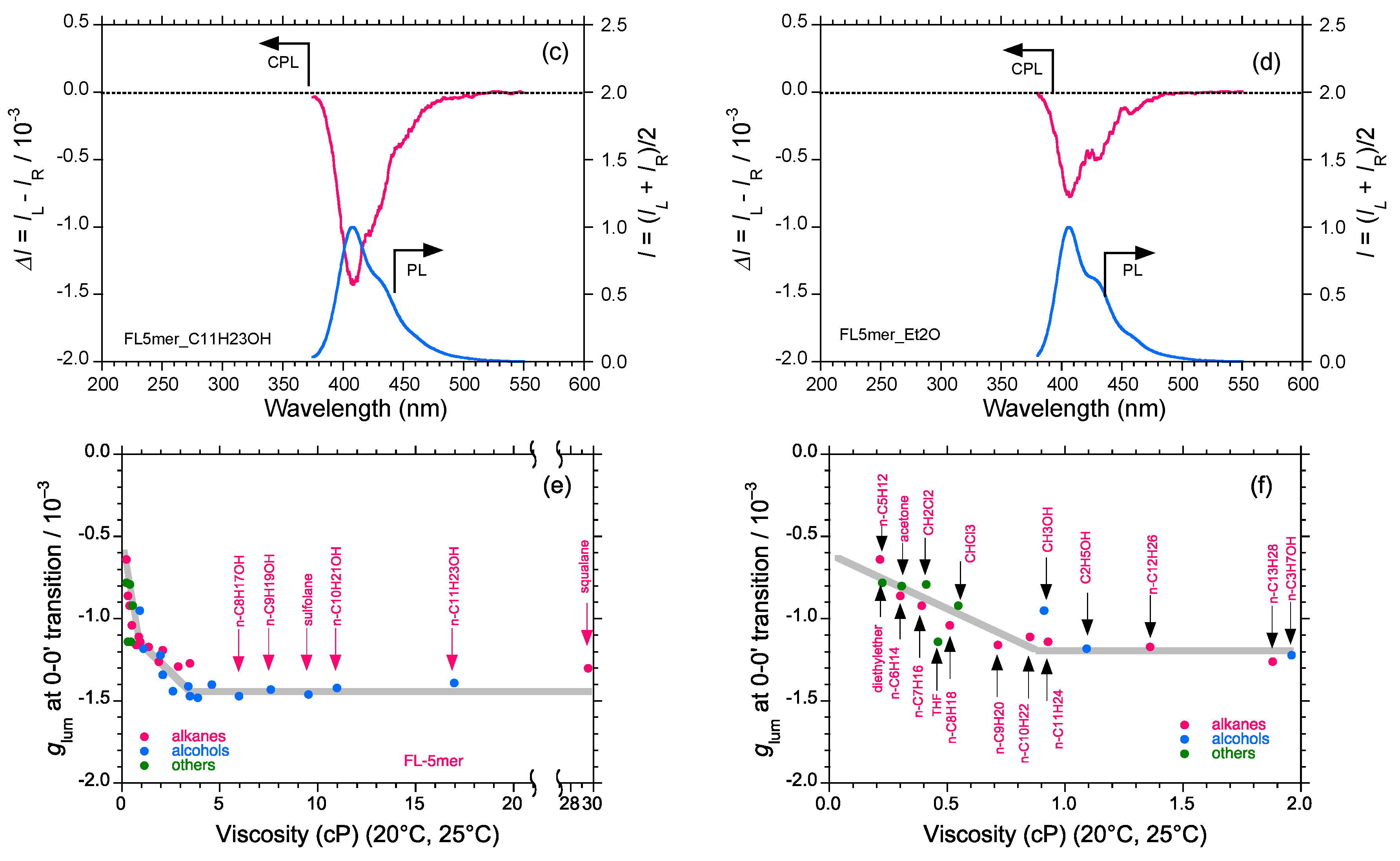
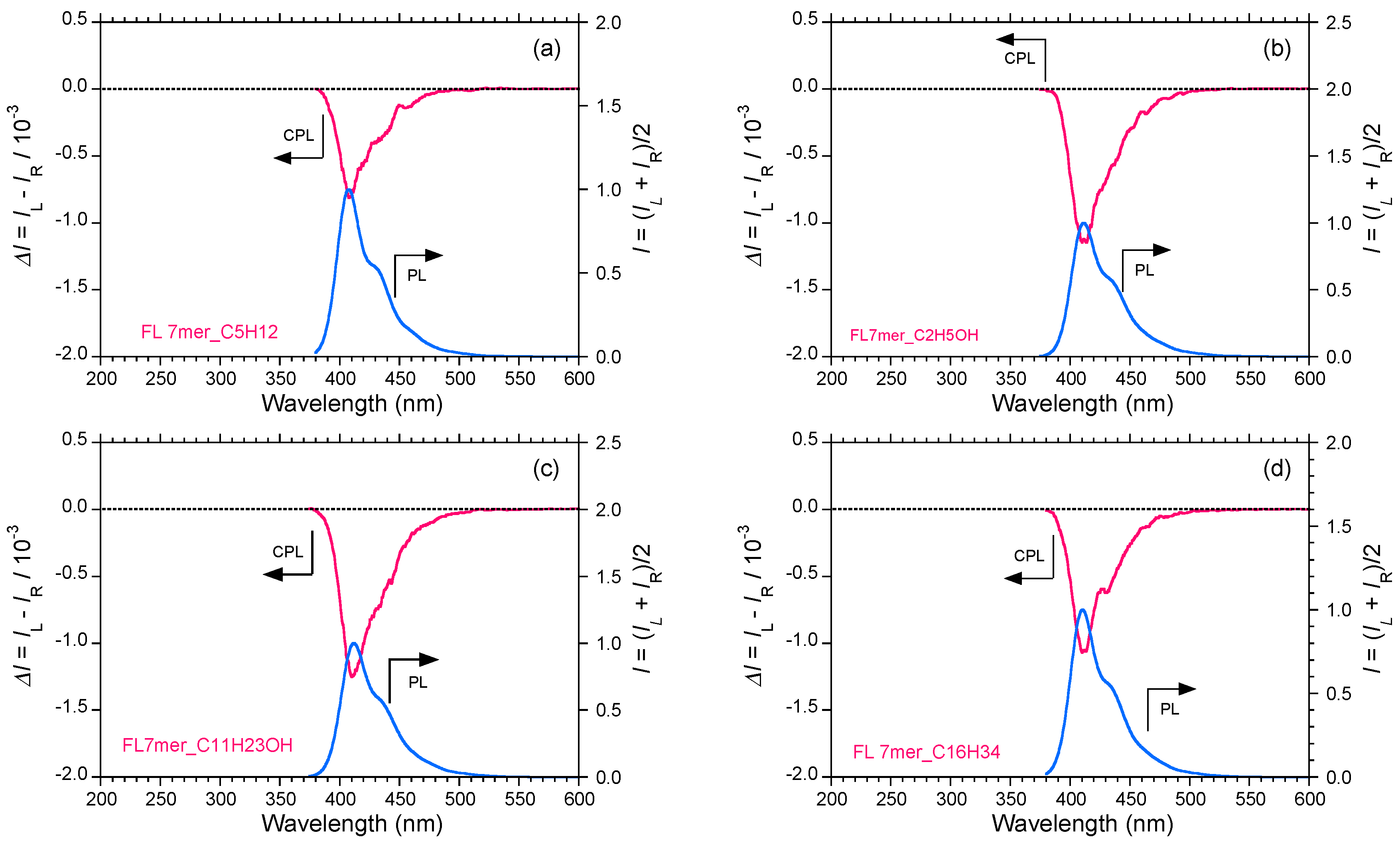
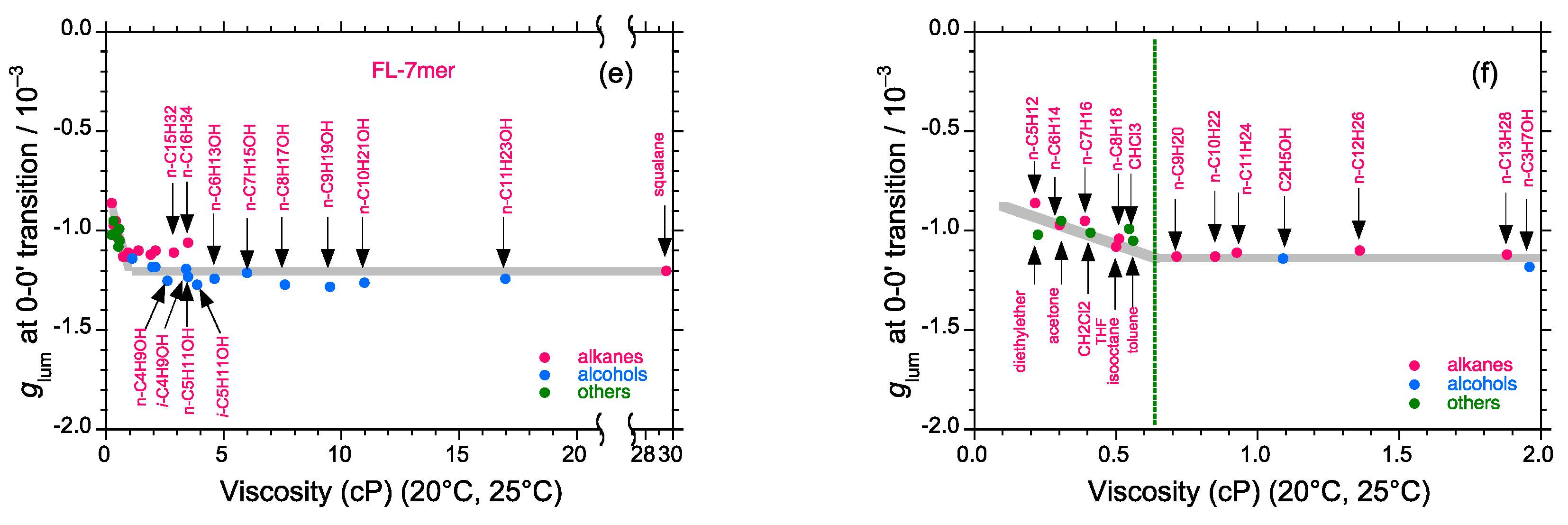
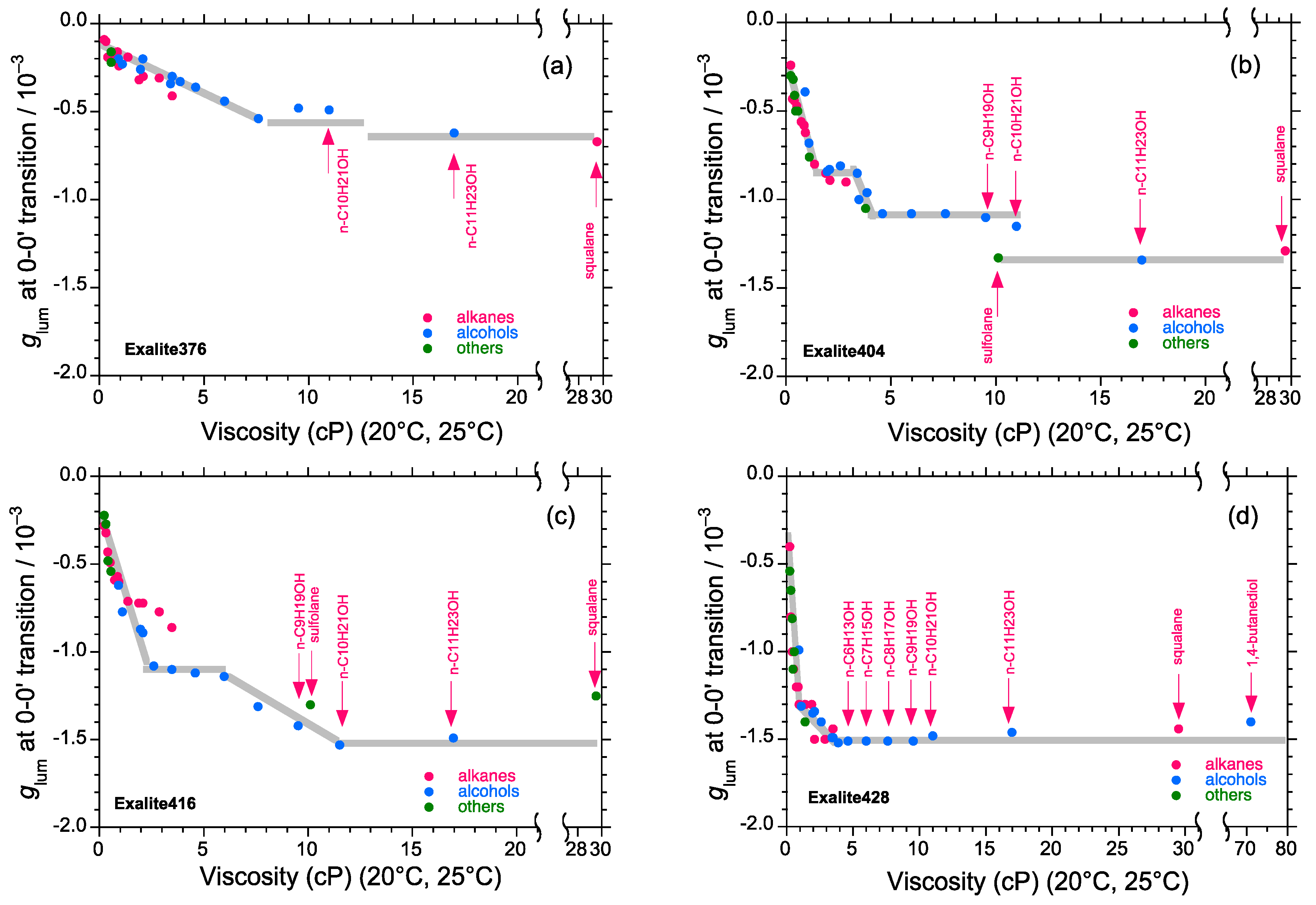
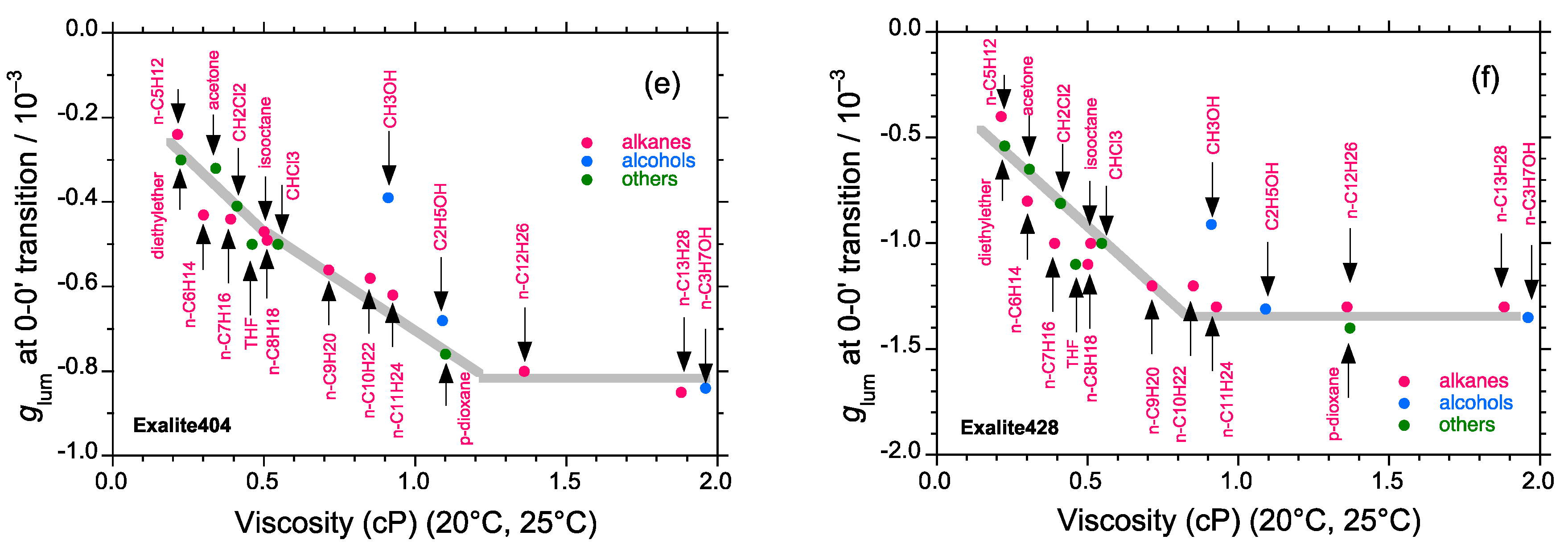
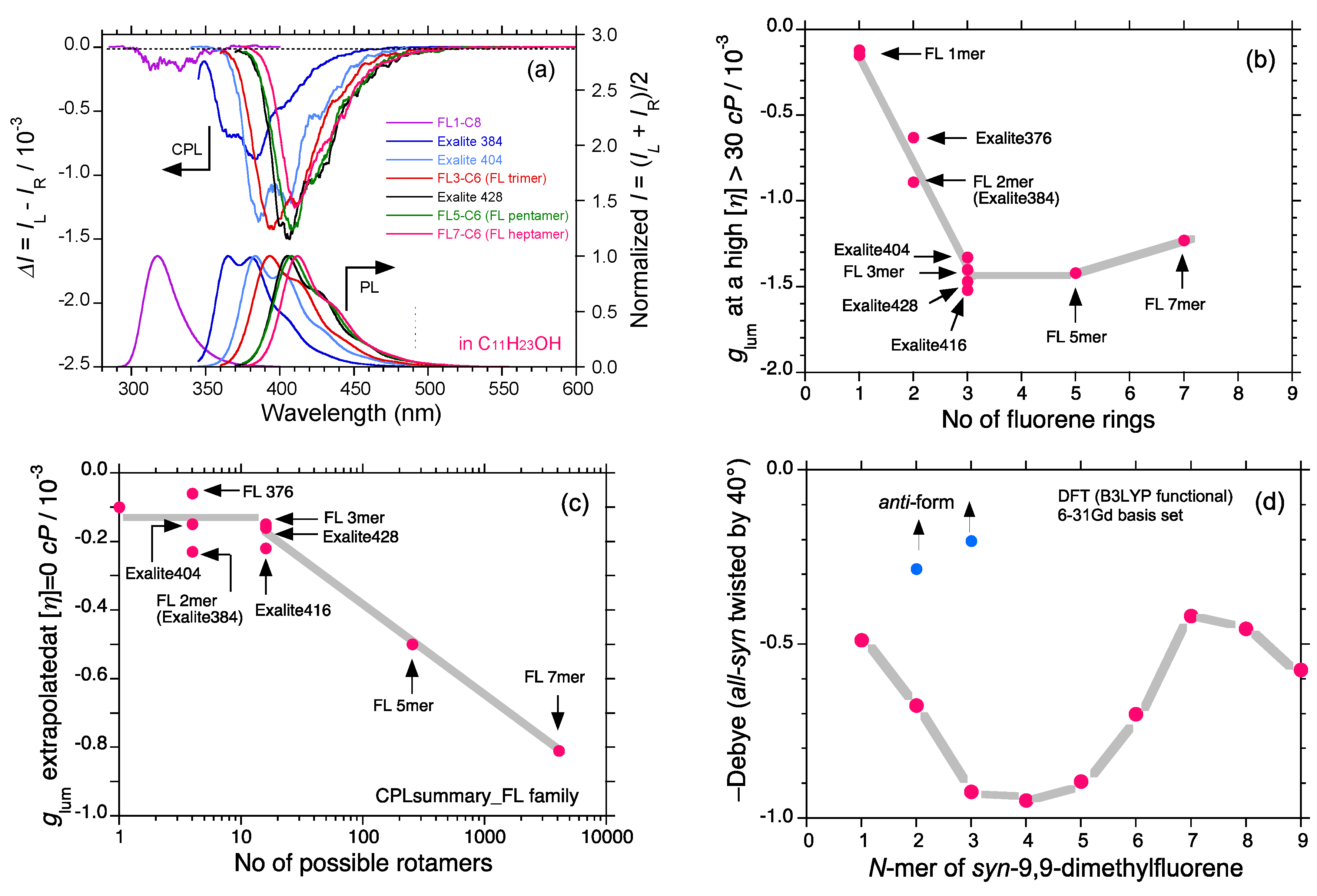
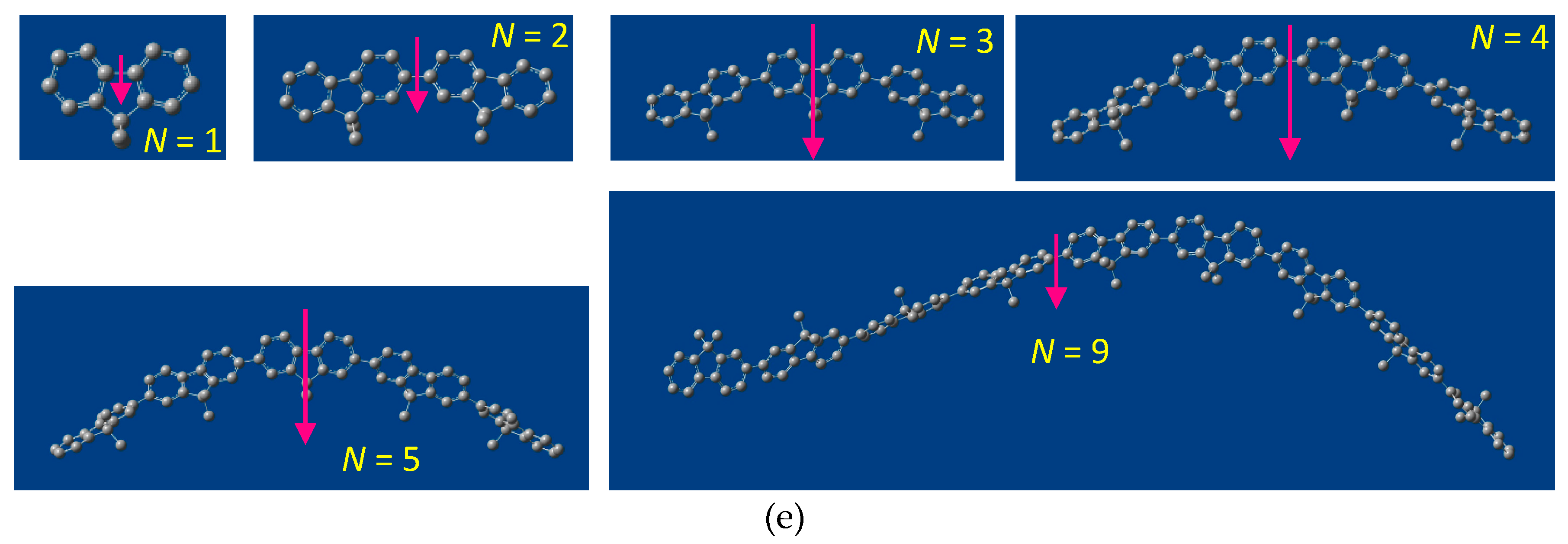
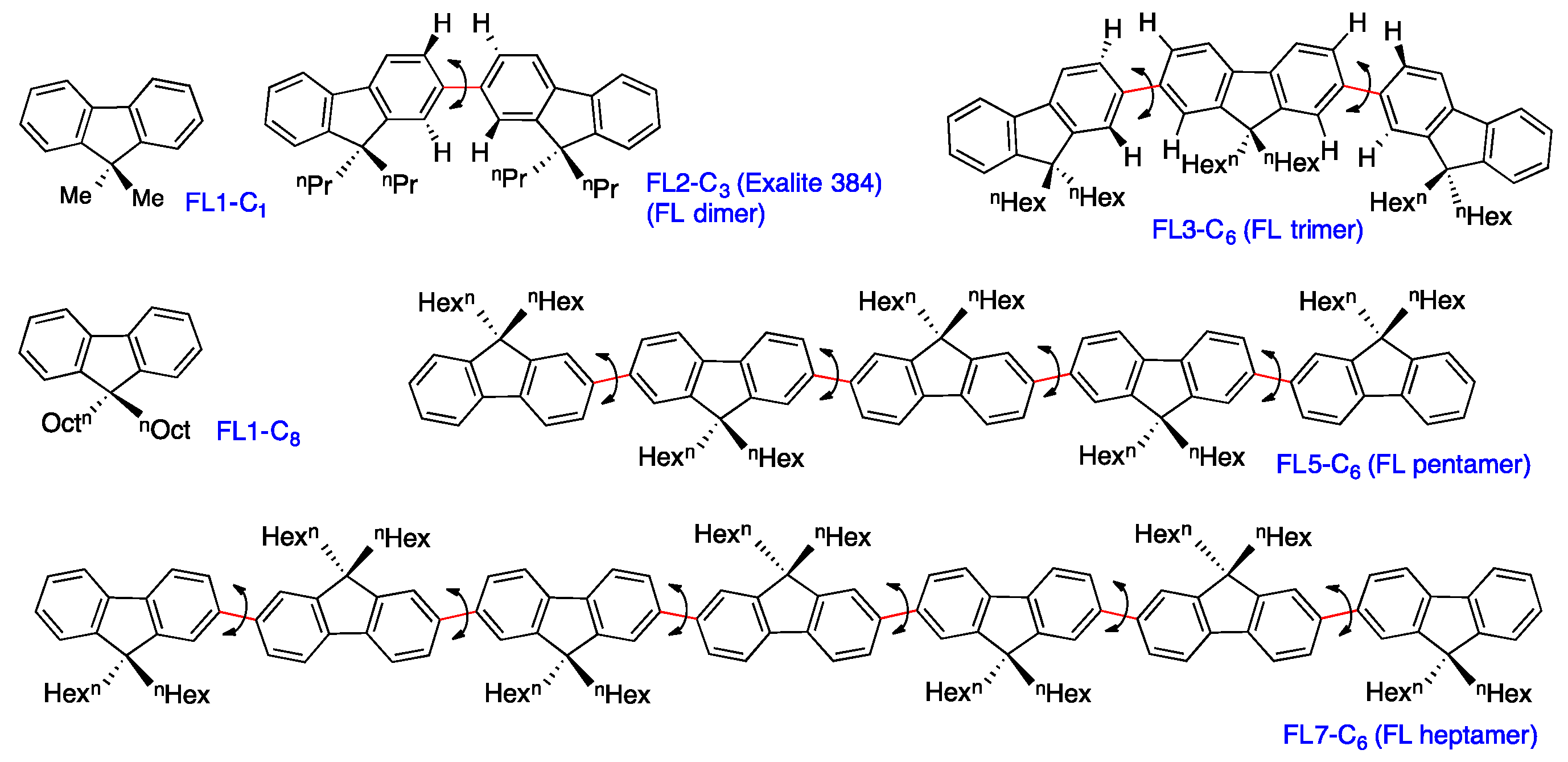
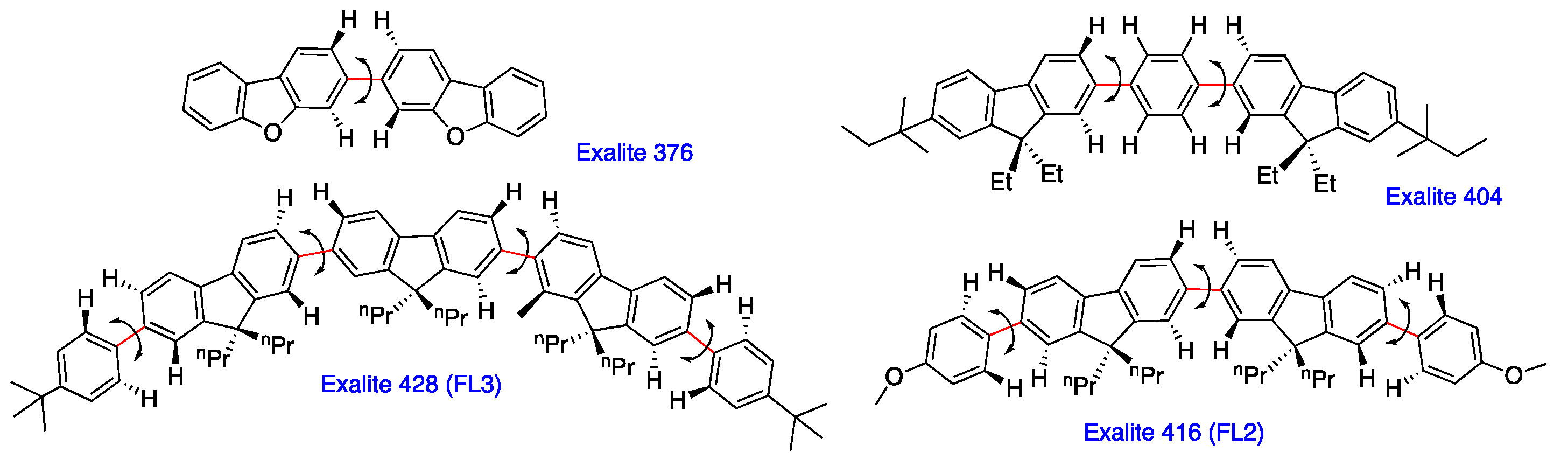
2.5. Chain-Like Non-Rigid Oligofluorene Derivatives Bearing Alkyl Groups
3. Discussion
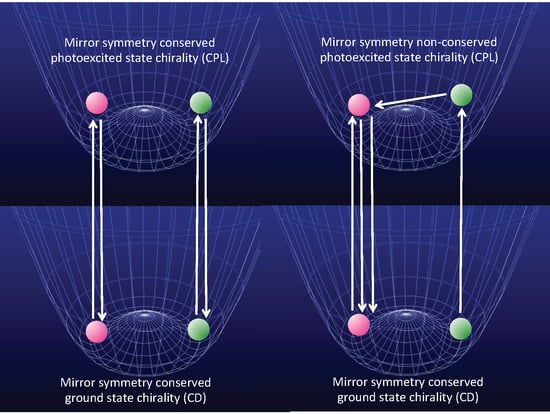
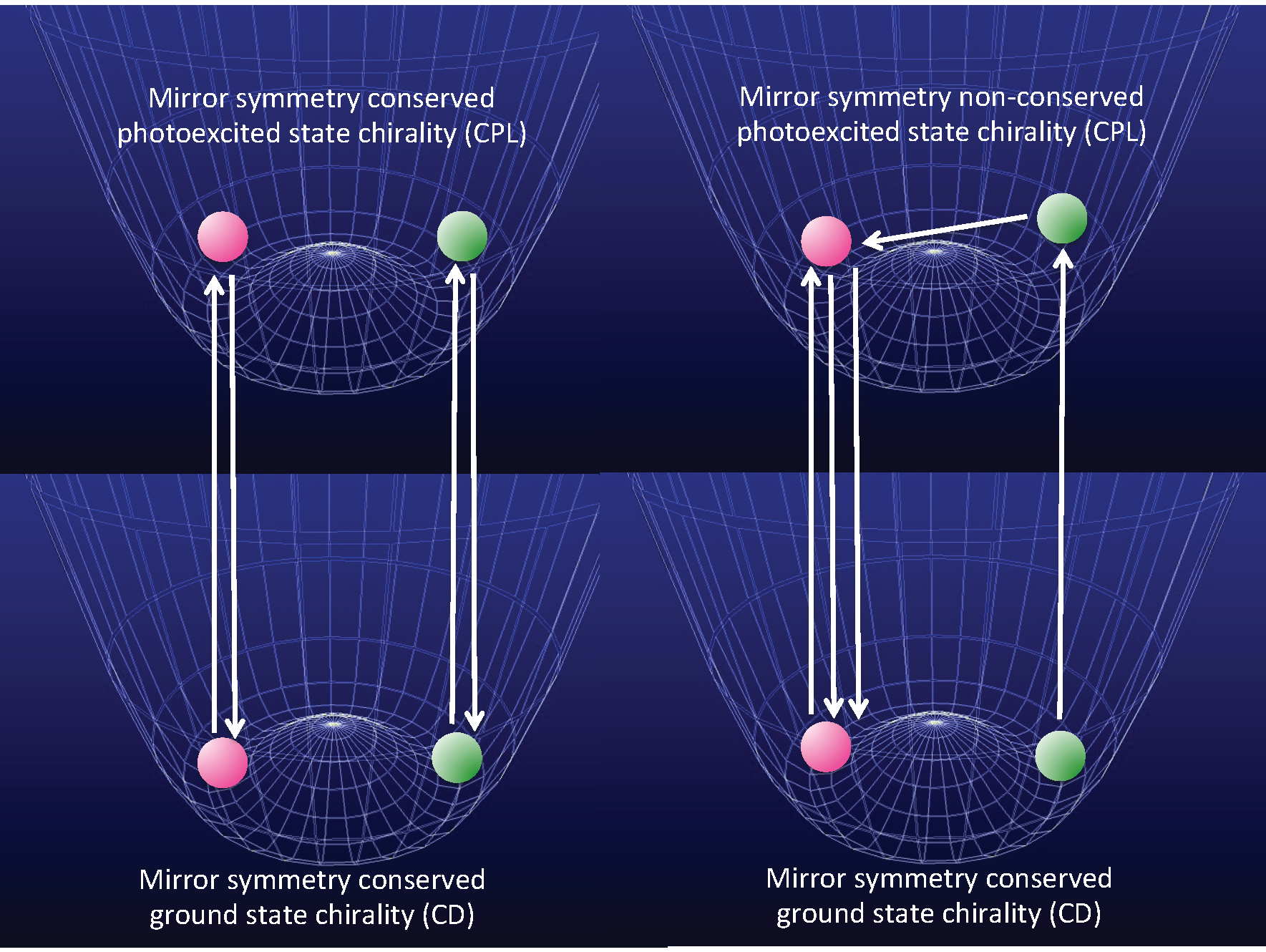
- Case 1.
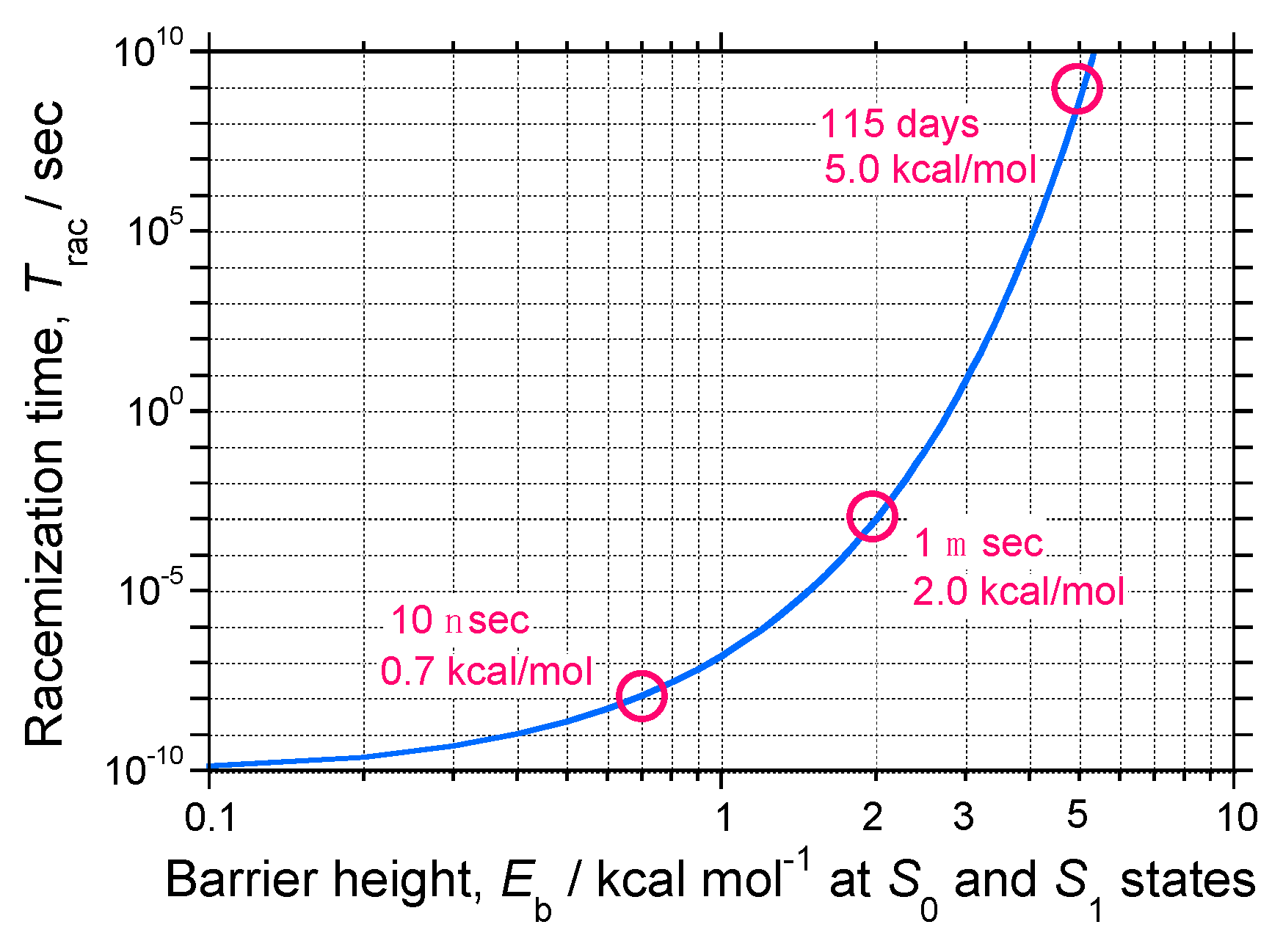
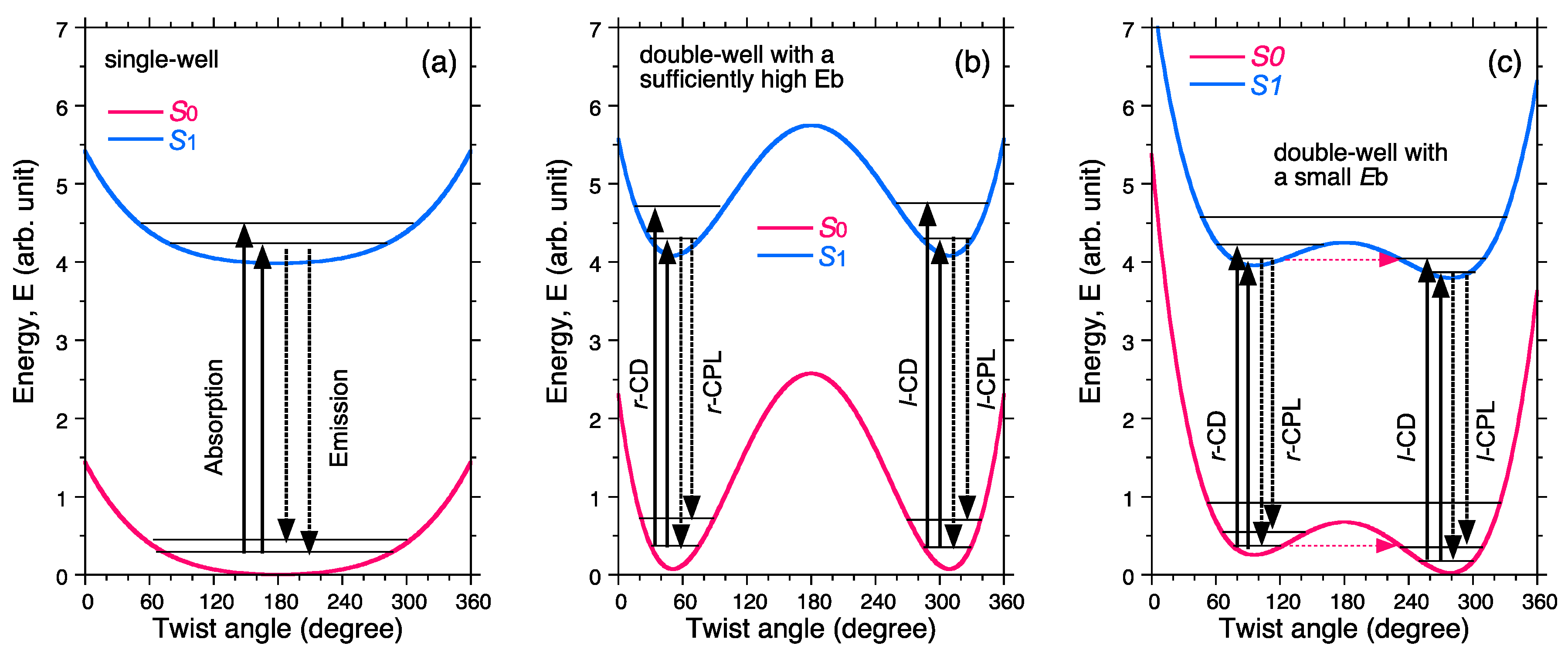
- Molecules in a single-well; no CD and no CPL signals are observable due to inherently achiral geometry at the S0/S1 states (Figure 15a).
- Case 2.
- Rigid chiral molecules enforced by a stereocenter in a DW with sufficiently high Eb at the S0/S1 states; for these, if a single (S)-enantiomer were obtained, the CD and CPL signals would be mirror-images of the (R)-enantiomer due to the inherent chiral geometry of the S0/S1 states (Figure 15b). The value of glum is similar to that of gabs and no change in sign between CPL and CD is expected. The stereocenter is responsible for the parity-conserving electromagnetic force.
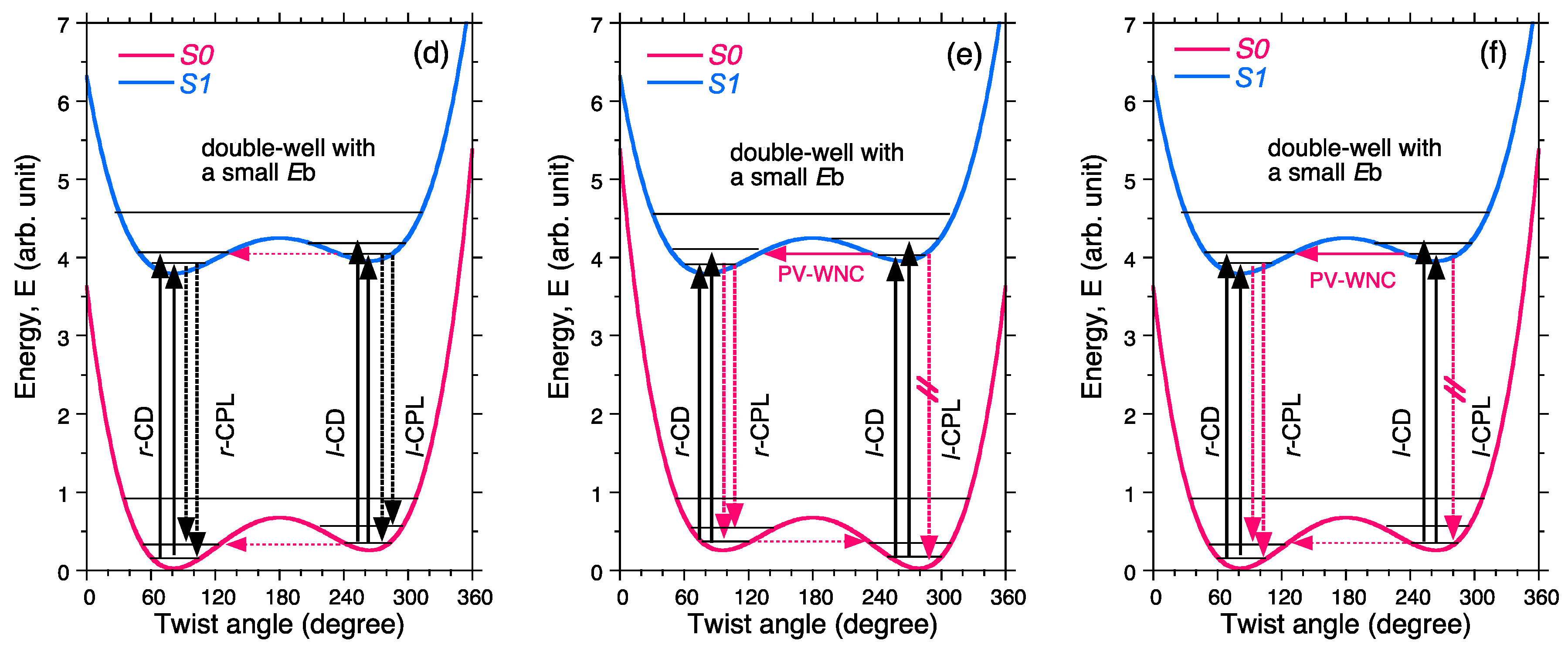
- Case 3.
- Non-rigid chiral molecules in a DW with sufficiently small Eb at the S0/S1 states; because the (S)-enantiomer is difficult to isolate due to rapid racemization by quantum tunneling, it is easy to be convinced that no CD or CPL signals at the S0/S1 states can be detected, and this is the conventional wisdom among chemists and described in textbooks on stereochemistry (Figure 14d and Figure 15c). The non-rigid chiral molecules are thus called CD-silent/CPL-silent molecules. The silence arises from quantum tunneling at the S0/S1 states. However, if the tunneling at the S1 state is a one-way event due to the PV-WNC, one can observe (–)-CPL (right-handed CPL only) (Figure 15e,f). In this case, the rapid left-right oscillation at the S1 state by quantum tunneling results in no detectable CD signals.
4. Materials and Methods
4.1. Instrumentation
4.2. Lists of Materials
4.2.1. Luminophores (vendor)
4.2.2. Solvents [Vender, Viscosity in cP (Temperature in °C)
4.3. Preparation of Sample Solutions
4.4. Chiroptical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janoschek, R. Theories on the origin of biomolecular Homochirality. In Chirality—From Weak Bosons to the α-Helix; Janoschek, R., Ed.; Springer: Berlin, Germany, 1991; ISBN 978-3-642-76569-8. [Google Scholar]
- Quack, M. Structure and dynamics of chiral molecules. Angew. Chem. Int. Ed. 1989, 28, 571–586. [Google Scholar] [CrossRef]
- Hund, F. Symmetriecharaktere yon termen bei systemen mit gleichen partikeln in der quantenmechanik. Z. Phys. 1927, 43, 788–803. [Google Scholar] [CrossRef]
- Bunker, P.R.; Jensen, P. The symmetry groups of non-rigid molecules. In Fundamentals of Molecular Symmetry; Institute of Physics Publishing: Philadelphia, PA, USA, 2005; pp. 274–290. ISBN 0750309415. [Google Scholar]
- Lee, T.D.; Yang, C.N. Question of parity conservation in weak interactions. Phys. Rev. 1956, 104, 254–258. [Google Scholar] [CrossRef]
- Wu, C.S.; Ambler, E.; Hayword, R.W.; Hoppes, D.D.; Hudson, R.P. Experimental test of parity conservation on beta decay. Phys. Rev. 1957, 105, 1413–1415. [Google Scholar] [CrossRef]
- Schopper, H. Circular polarization of γ-Rays: Further proof for parity failure in β Decay. Philos. Mag. 1957, 2, 710–713. [Google Scholar] [CrossRef]
- Goldhaber, M.; Grodzins, L.; Sunyar, A. Helicity of neutrinos. Phys. Rev. 1958, 109, 1015–1017. [Google Scholar] [CrossRef]
- Fagg, L.W.; Hanna, S.S. Polarization mmeasurements on nnuclear gamma rays. Rev. Mod. Phys. 1959, 31, 711–758. [Google Scholar] [CrossRef]
- Wu, C.S. Parity experiments in beta decay. Rev. Mod. Phys. 1959, 31, 783–790. [Google Scholar] [CrossRef]
- Electroweak Interaction. Available online: https://en.wikipedia:wiki/Electroweak_interaction (accessed on 28 July 2018).
- Walgate, R. What will come after the Z0? Nature 1983, 303, 473. [Google Scholar] [CrossRef]
- van der Meer, S. Stochastic cooling and the Accumulation of Antiprotons. Rev. Mod. Phys. 1985, 57, 689–698. [Google Scholar] [CrossRef]
- Rubbia, C. Experimental observation of the intermediate vector bosons W+, W− and Z0. Rev. Mod. Phys. 1985, 57, 699–722. [Google Scholar] [CrossRef]
- ENSDF Decay Data in the MIRD (Medical Internal Radiation Dose) Format for 60Co. Available online: https://www.orau:ptp/ptp%20library/library/doe/bnl/nuclidedata/mirco60.htm (accessed on 8 October 2018).
- ENSDF Decay Data in the MIRD (Medical Internal Radiation Dose) Format for 58Co. Available online: https://www.orau:ptp/ptp%20library/library/doe/bnl/nuclidedata/mirco58.htm (accessed on 8 October 2018).
- Bouchiat, M.A.; Bouchiat, C.C. Weak neutral currents in atomic physics. Phys. Lett. B 1974, 48, 111–114. [Google Scholar] [CrossRef]
- Close, E.E. Parity violation in atoms? Nature 1976, 264, 505–506. [Google Scholar] [CrossRef]
- Baied, P.E.G.; Brimcombe, M.W.S.; Roberts, G.J.; Sandars, P.G.H.; Soreide, D.C.; Fortson, E.N.; Lewis, L.L.; Lindahl, E.G.; Soreide, D.C. Search for parity non-conserving optical rotation in atomic bismuth. Nature 1976, 264, 528–529. [Google Scholar] [CrossRef]
- Bucksbaum, P.H.; Commins, E.D.; Hunter, L.R. Observations of parity nonconservation in atomic thallium. Phys. Rev. D 1981, 24, 1134–1148. [Google Scholar] [CrossRef] [Green Version]
- Emmons, T.P.; Reeves, J.M.; Fortson, E.N. Parity-nonconserving optical rotation in atomic lead. Phys. Rev. Lett. 1983, 51, 2089–2091. [Google Scholar] [CrossRef]
- Bouchiat, M.-A.; Pottier, L. Optical experiments and weak interactions. Science 1986, 234, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Parity Violation in Atoms and Polarized Electron Scattering; Bernard, F.; Bouchiat, M.-A. (Eds.) World Scientific: Singapore, 1999; ISBN 9810237316. [Google Scholar]
- Guéna, J.; Lintz, M.; Bouchiat, M.-A. Atomic parity violation: Principles, recent results, present motivations. Mod. Phys. Lett. A 2005, 20, 375–390. [Google Scholar] [CrossRef]
- Okun, L.B. Mirror particles and mirror matter: 50 years of speculation and search. Phys. Usp. 2007, 50, 380–389. [Google Scholar] [CrossRef]
- Tsigutkin, K.; Dounas-Frazer, D.; Family, A.; Stalnaker, J.E.; Yashchuk, V.V.; Budker, D. Observation of a large atomic parity violation effect in ytterbium. Phys. Rev. Lett. 2009, 103, 071601. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Heckel, B.R.; Ramsey, N.F.; Green, K.; Greene, G.L.; Byrne, J.; Pendlebury, J.M. First measurement of parity-nonconserving neutron-spin rotation: The tin isotopes. Phys. Rev. Lett. 1980, 45, 2088–2091. [Google Scholar] [CrossRef]
- Yamagata, Y. A hypothesis for the asymmetric appearance of biomolecules on earth. J. Theoret. Biol. 1966, 11, 495–498. [Google Scholar] [CrossRef]
- Rein, D.W. Some remarks on parity violating effects of intramolecular interactions. J. Mol. Evol. 1974, 4, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Letokhov, V.S. On difference of energy levels of left and right molecules due to weak interactions. Phys. Lett. A 1975, 53, 275–276. [Google Scholar] [CrossRef]
- Keszthelyi, L. Chemical evolution: Effect of high energy radiation. Orig. Life 1976, 7, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Zel’Dovich, Y.B.; Saakyan, D.B.; Sobe’Man, I.I. Energy difference between right-hand and left-hand molecules due to parity nonconservation in weak interactions of electrons with nuclei. JETP Lett. 1977, 25, 94–97. [Google Scholar]
- Keszthelyi, L. Origin of the asymmetry of biomolecules and weak interaction. Orig. Life 1977, 8, 299–340. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Stodolsky, L. Quantum beats in optical activity and weak interactions. Phys. Lett. B 1978, 78, 313–317. [Google Scholar] [CrossRef]
- Hegstrom, R.A.; Rein, D.W.; Sandars, P.G.H. Calculation of the parity nonconserving energy difference between mirror-image molecules. J. Chem. Phys. 1980, 73, 2329–2341. [Google Scholar] [CrossRef]
- Mason, S.F.; Tranter, G.E. Energy inequivalence of peptide enantiomers from parity non-conservation. Chem. Commun. 1983, 117–119. [Google Scholar] [CrossRef]
- Hegstrom, R.A. Parity Nonconservation and the origin of biological chirality: Theoretical calculations. Orig. Life 1984, 14, 1–4. [Google Scholar] [CrossRef]
- Mason, S.F. Origins of biomolecular handedness. Nature 1984, 311, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.F.; Tranter, G.E. The electroweak origin of biomolecular handedness. Proc. R. Soc. Lond. A 1985, 397, 45–65. [Google Scholar] [CrossRef]
- Hegstrom, R.A.; Kondepudi, D.K. The handedness of the universe. Sci. Am. 1990, 262, 108–115. [Google Scholar] [CrossRef]
- Salam, A. The role of chirality in the origin of life. J. Mol. Evol. 1991, 33, 105–113. [Google Scholar] [CrossRef]
- Macdermott, A.J. Electroweak enantioselection and the origin of life. Orig. Life Evol. Biosph. 1995, 25, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, O.; Kiyonaga, H. Parity-violating energy shift of helical n-alkanes. J. Mol. Struct. (Theochem.) 1994, 312, 271–274. [Google Scholar] [CrossRef]
- Bonner, W.A. Enantioselective autocatalysis. IV. implications for parity violation effects. Orig. Life Evol. Biosph. 1996, 26, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Szabó-Nagy, A.; Keszthelyi, L. Demonstration of the parity-violating energy difference between enantiomers. Proc. Natl. Acad. Sci. USA 1999, 96, 4252–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottselig, M.; Luckhaus, D.; Quack, M.; Stohner, J.; Willeke, M. Mode selective stereomutation and parity violation in disulfane isotopomers H2S2, D2S2, T2S2. Helv. Chim. Acta 2001, 84, 1846–1861. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Gierlich, J.; Bollwein, T. Large parity-violation eeffects in heavy-metal-containing chiral compounds. Angew. Chem. Int. Ed. 2003, 42, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- MacDermott, A.J.; Hegstrom, R.A. A proposed experiment to measure the parity-violating energy difference between enantiomers from the optical rotation of chiral ammonia-like “Cat” molecules. Chem. Phys. 2004, 305, 55–68. [Google Scholar] [CrossRef]
- Quack, M.; Stohner, J.; Willeke, M. High-resolution spectroscopic studies and theory of parity violation in chiral molecules. Annu. Rev. Phys. Chem. 2008, 59, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Darquié, B.; Stoeffler, C.; Shelkovnikov, A.; Daussy, C.; Amy-Klein, A.; Chardonnet, C.; Zrig, S.; Guy, L.; Crassous, J.; Soulard, P.; et al. Progress toward the first observation of parity violation in chiral molecules by high-resolution laser spectroscopy. Chirality 2010, 22, 870–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorta-Urra, A.; Peñate-Rodríguez, H.C.; Bargueño, P.; Rojas-Lorenzo, G.; Miret-Artés, S. Dissipative geometric phase and decoherence in parity-violating chiral molecules. J. Chem. Phys. 2012, 136, 174505, (6 pages). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, S.; Arn, F.; Bolotova, I.; Chen, Z.; Fábri, C.; Grassi, G.; Lerch, P.; Quack, M.; Seyfang, G.; Wokaun, A.; et al. Synchrotron-based highest resolution terahertz spectroscopy of the ν24 band system of 1,2-dithiine (C4H4S2): A candidate for measuring the parity violating energy difference between enantiomers of chiral molecules. J. Phys. Chem. Lett. 2016, 7, 3847–3853. [Google Scholar] [CrossRef] [PubMed]
- Daussy, C.; Marrel, T.; Amy-Klein, A.; Nguyen, C.T.; Bordé, C.J.; Chardonnet, C. Limit on the parity nonconserving energy difference between the enantiomers of a chiral molecule by laser spectroscopy. Phys. Rev. Lett. 1999, 83, 1554–1557. [Google Scholar] [CrossRef]
- Viedma, C. Selective chiral symmetry breaking during crystallization: Parity violation or cryptochiral environment in control? Cryst. Growth Des. 2007, 7, 553–556. [Google Scholar] [CrossRef]
- Wang, W.; Yi, F.; Ni, Y.; Zhao, Z.; Jin, X.; Tang, Y. Parity violation of electroweak force in phase transitions of single crystals of d- and l-alanine and valine. J. Biol. Phys. 2000, 26, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Scolnik, T.; Portnaya, I.; Cogan, U.; Tal, S.; Haimovitz, R.; Fridkin, M.; Elitzur, A.C.; Deamer, D.W.; Shinitzky, M. Subtle differences in structural transitions between poly-l- and poly-d-amino acids of equal length in water. Phys. Chem. Chem. Phys. 2006, 8, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kodona, E.K.; Alexopoulos, C.; Panou-Pomonis, E.; Pomonis, P.J. Chirality and helix stability of polyglutamic acid enantiomers. J. Colloid Interface Sci. 2008, 319, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, M. Experimental tests of parity violation at helical polysilylene level. Macromol. Rapid Commun. 2001, 22, 669–674. [Google Scholar] [CrossRef]
- Fujiki, M. Mirror symmetry breaking in helical polysilanes: Ppreference between left and right of chemical and physical origin. Symmetry 2010, 2, 1625–1652. [Google Scholar] [CrossRef]
- Fujiki, M.; Kawagoe, Y.; Nakano, Y.; Nakao, A. Mirror-symmetry-breaking in poly[(9,9-di-n-octylfluorenyl-2,7-diyl)-alt-biphenyl] (PF8P2) is susceptible to terpene chirality, achiral solvents and mechanical stirring. Molecules 2013, 18, 7035–7057. [Google Scholar] [CrossRef] [PubMed]
- Gabuda, S.P.; Kozlova, S.G. Chirality-related interactions and a mirror symmetry violation in handed nano structures. J. Chem. Phys. 2014, 141, 044701. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, S.G.; Gabuda, S.P. Thermal properties of Zn2(C8H4O4)2•C6H12N2 Metal-organic framework compound and mirror symmetry violation of dabco molecules. Sci. Rep. 2017, 7, 11505. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. Handbook of Organic Solvents; CRC Press: Boca Raton, FL, USA, 1994; ISBN 0849389305. [Google Scholar]
- Viswanath, D.S.; Ghosh, T.; Prasad, D.H.L.; Dutt, N.V.K.; Rani, K.Y. Viscosity of Liquids; Theory, Estimation, Experiment and Data. Springer: Berlin, Germany, 2007; ISBN 9048173787. [Google Scholar]
- Properties of Organic Solvents. Available online: http://murov.info/orgsolvents.htm. (accessed on 12 June 2018).
- Turro, N.J. Modern Molecular Photochemistry; University Science Books: Sausalito, CA, USA, 1991; ISBN 0935702717. [Google Scholar]
- Calvert, J.G.; Pitts, J.N. Photochemistry; John Wiley & Sons: Hoboken, NJ, USA, 1973; ISBN 0471130907. [Google Scholar]
- Shindo, Y.; Nakagawa, M. On the artifacts in circularly polarized emission spectroscopy. Appl. Spector. 1985, 39, 32–38. [Google Scholar] [CrossRef]
- Blok, P.M.L.; Dekkers, H.P.J.M. Measurement of the circular polarization of the luminescence of photoselected samples under artifact-free conditions. Appl. Spector. 1990, 44, 305–309. [Google Scholar] [CrossRef]
- Longhi, G.; Castiglioni, E.; Abbate, S.; Lebon, F.; Lightner, D.A. Experimental and calculated CPL spectra and related spectroscopic data of camphor and other simple chiral bicyclic ketones. Chirality 2013, 25, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Thi Duong, S.; Fujiki, M. The origin of bisignate circularly polarized luminescence (CPL) spectra from chiral Polymer aggregates and molecular camphor: Anti-kasha’s rule revealed by CPL excitation (CPLE) spectra. Polym. Chem. 2017, 8, 4673–4679. [Google Scholar] [CrossRef]
- Kinuta, T.; Sato, T.; Nakano, Y.; Harada, T.; Tajima, N.; Fujiki, M.; Kuroda, R.; Matsubara, Y.; Imai, Y. Solid-state chiral optical properties of axially chiral binaphthyl acid derivatives. J. Photochem. Photobiol. A Chem. 2011, 220, 134–138. [Google Scholar] [CrossRef]
- Kimoto, T.; Tajima, N.; Fujiki, M.; Imai, Y. Control of circularly polarized luminescence by using open- and closed-type binaphthyl derivatives with the same axial chirality. Chem.–Asian J. 2012, 7, 2836–2841. [Google Scholar] [CrossRef] [PubMed]
- Kinuta, T.; Tajima, N.; Fujiki, M.; Miyazawa, M.; Imai, Y. Control of circularly polarized photoluminescent property via dihedral angle of binaphthyl derivatives. Tetrahedron 2012, 68, 4791–4796. [Google Scholar] [CrossRef]
- Amako, T.; Kimoto, T.; Tajima, N.; Fujiki, M.; Imai, Y. A comparison of circularly polarized luminescence (CPL) and circular dichroism (CD) characteristics of four axially chiral binaphthyl-2,2′-diyl hydrogen phosphate derivatives. Tetrahedron 2013, 69, 2753–2757. [Google Scholar] [CrossRef]
- Schneider, D.J.; Landis, D.A.; Fleitz, P.A.; Seliskar, C.J.; Kauffman, J.M.; Steppel, R.N. Characterization of new excimer pumped UV laser dyes I. p-terphenyls, Laser Chem. 1991, 11, 49–62. [Google Scholar] [CrossRef]
- Fleitz, P.A.; Seliskar, C.J.; Steppel, R.N.; Kauffman, J.M.; Kelley, C.J.; Ghiorghis, A. Characterization of new excimer pumped UV laser dyes 2. p-quaterphenyls, Laser Chem. 1991, 11, 99–107. [Google Scholar] [CrossRef]
- Seliskar, C.J.; Landis, D.A.; Kauffman, J.M.; Aziz, M.A.; Steppel, R.N.; Kelley, C.J.; Qin, Y.; Ghiorghis, A. Characterization of new excimer pumped UV laser dyes 3. p-quinqui-, sexi-, octi- and deciphenyls. Laser Chem. 1993, 13, 19–28. [Google Scholar] [CrossRef]
- Omachi, H.; Segawa, Y.; Itami, K. Synthesis of cycloparaphenylenes and related carbon nanorings: A step toward the controlled synthesis of carbon nanotubes. Acc. Chem. Res. 2012, 45, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Omachi, H.; Nakayama, T.; Takahashi, E.; Segawa, Y.; Itami, K. Initiation of carbon nanotube growth by well-defined carbon nanorings. Nat. Chem. 2013, 5, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Monkman, A.; Rothe, C.; King, S.; Dias, F. Polyfluorene photophysics. Adv. Polym. Sci. 2008, 212, 187–225. [Google Scholar] [CrossRef]
- Klaerner, G.; Miller, R.D. Polyfluorene derivatives: Effective conjugation lengths from well-defined oligomers. Macromolecules 1998, 31, 2007–2009. [Google Scholar] [CrossRef]
- Shiraki, T.; Shindome, S.; Toshimitsu, F.; Fujigaya, T.; Nakashima, N. Strong main-chain length-dependence for the β-phase formation of oligofluorenes. Polym. Chem. 2015, 6, 5103–5109. [Google Scholar] [CrossRef]
- Kondepudi, D. Parity violations and the origin of bimolecular handedness. In Entropy, Information and Evolution: New Perspective on Physical and Biological Evolution; Weber, B.H., Depew, D.J., Smith, J.D., Eds.; MIT Press: Cambridge, MA, USA, 1988; ISBN 0262731681. [Google Scholar]
- Shinitzky, M. Configurational entropy in chiral solutions—Negative entropy of solvent envelopes. Entropy 2009, 11, 667–674. [Google Scholar] [CrossRef]
- Shinitzky, M. Space asymmetry as a possible global feature. Chirality 2013, 25, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Suzuki, N.; Liu, J.; Matsuda, T.; Rahim, N.A.A.; Zhang, W.; Fujiki, M.; Zhang, Z.; Zhou, N.; Zhu, X. Limonene induced chiroptical generation and inversion during aggregation of achiral polyfluorene analogs: Structure-dependence and mechanism. Polym. Chem. 2014, 5, 5920–5927. [Google Scholar] [CrossRef]
- Category: Proton-Proton Chain Reaction. Available online: https://en.wikipedia:wiki/Proton–proton_chain_reaction (accessed on 28 August 2018).
- Arrow of Time. Available online: https://en.wikipedia:wiki/Arrow_of_time (accessed on 15 September 2018).
- Franck-Condon-Principle. Available online: https://en.wikipedia:wiki/Franck–Condon_principle (accessed on 29 June 2018).
- Guijarro, A.; Yus, M. Origin of Chirality in the Molecules of life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem; RSC Publishing: Cambridge, UK, 2008; ISBN 978-0-85404-156-5. [Google Scholar]
- Chronology of the Universe. Available online: https://en.wikipedia:wiki/Chronology_of_the_universe (accessed on 6 September 2018).
- Freedman, R.; Geller, R.; Kaufmann, W.J. Universe, 10th ed.; Freeman, W.H., Ed.; W.H. Freeman and Company: London, UK, 2015; ISBN 1319042384. [Google Scholar]
- Story of the Universe. Available online: http://cms.web.cern.ch/content/story-universe (accessed on 7 December 2011).
- Superconductivity. Available online: https://en.wikipedia:wiki/Superconductivity (accessed on 24 August 2018).
- Allford, C.P.; Legg, R.E.; O’Donnell, R.A.; Dawson, P.; Missous, M.; Buckle, P.D. Thermally activated resonant tunnelling in GaAs/AlGaAs triple barrier heterostructures. Semicond. Sci. Technol. 2015, 30, 105035. [Google Scholar] [CrossRef] [Green Version]
- Avetisov, V.; Goldanskii, V. Mirror symmetry- breaking at the molecular level. Proc. Natl. Acad. Sci. USA 1996, 93, 11435–11442. [Google Scholar] [CrossRef] [PubMed]
- Soai, K.; Kawasaki, T. Asymmetric autocatalysis with amplification of chirality. Top. Curr. Chem. 2008, 284, 1–33. [Google Scholar] [CrossRef]
- Green, M.M.; Jain, V. Homochirality in life: Two equal runners, one tripped. Orig. Life Evol. Biosph. 2010, 40, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Sandars, P.G.H. A toy model for the generation of Homochirality during polymerization, Orig. Life Evol. Biosph. 2003, 33, 575–587. [Google Scholar] [CrossRef]
- Fujiki, M. Mirror symmetry breaking of silicon polymers—From weak bosons to artificial helix. Chem. Rec. 2009, 9, 271–298. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.D. Symmetry and molecular chirality. Chem. Soc. Rev. 1986, 15, 189–223. [Google Scholar] [CrossRef]
- Famiano, M.A.; Boyd, R.N.; Kajino, T.; Onaka, T.; Mo, Y. Amino acid chiral selection via weak interactions in stellar environments: Implications for the origin of life. Sci. Rep. 2018, 8, 8833. [Google Scholar] [CrossRef] [PubMed]
- Bargueño, P.; de Tudela, R.P. The role of supernova neutrinos on molecular homochirality, Orig. Life Evol. Bosh. 2007, 37, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Marcianò, A.; Smolin, L. Gravitational origin of the weak interaction’s chirality. Phys. Rev. D 2014, 89, 065017. [Google Scholar] [CrossRef]
- Bargueño, P. Gravitational origin parity violation. Chirality 2015, 27, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Ribó, J.M.; Blanco, C.; Crusats, J.; El-Hachemi, Z.; Hochberg, D.; Moyano, A. Absolute asymmetric synthesis in enantioselective autocatalytic reaction networks: Theoretical games, speculations on chemical evolution and perhaps a synthetic option. Chem. Eur. J. 2014, 20, 17250–17271. [Google Scholar] [CrossRef] [PubMed]
- Alez, G. Circular Polarization: General Description, Circular Dichroism, Circularly Polarized Luminescence, Antennas, Circular Polarization of Nature; Webster’s Digital Services: Wahroonga, NSW, Australia, 2012; ISBN 127618848X. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |





























© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiki, M.; Koe, J.R.; Mori, T.; Kimura, Y. Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy: Part 1. Oligofluorenes, Oligophenylenes, Binaphthyls and Fused Aromatics. Molecules 2018, 23, 2606. https://doi.org/10.3390/molecules23102606
Fujiki M, Koe JR, Mori T, Kimura Y. Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy: Part 1. Oligofluorenes, Oligophenylenes, Binaphthyls and Fused Aromatics. Molecules. 2018; 23(10):2606. https://doi.org/10.3390/molecules23102606
Chicago/Turabian StyleFujiki, Michiya, Julian R. Koe, Takashi Mori, and Yoshihiro Kimura. 2018. "Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy: Part 1. Oligofluorenes, Oligophenylenes, Binaphthyls and Fused Aromatics" Molecules 23, no. 10: 2606. https://doi.org/10.3390/molecules23102606
APA StyleFujiki, M., Koe, J. R., Mori, T., & Kimura, Y. (2018). Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy: Part 1. Oligofluorenes, Oligophenylenes, Binaphthyls and Fused Aromatics. Molecules, 23(10), 2606. https://doi.org/10.3390/molecules23102606