A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing

 ,
,
Abstract
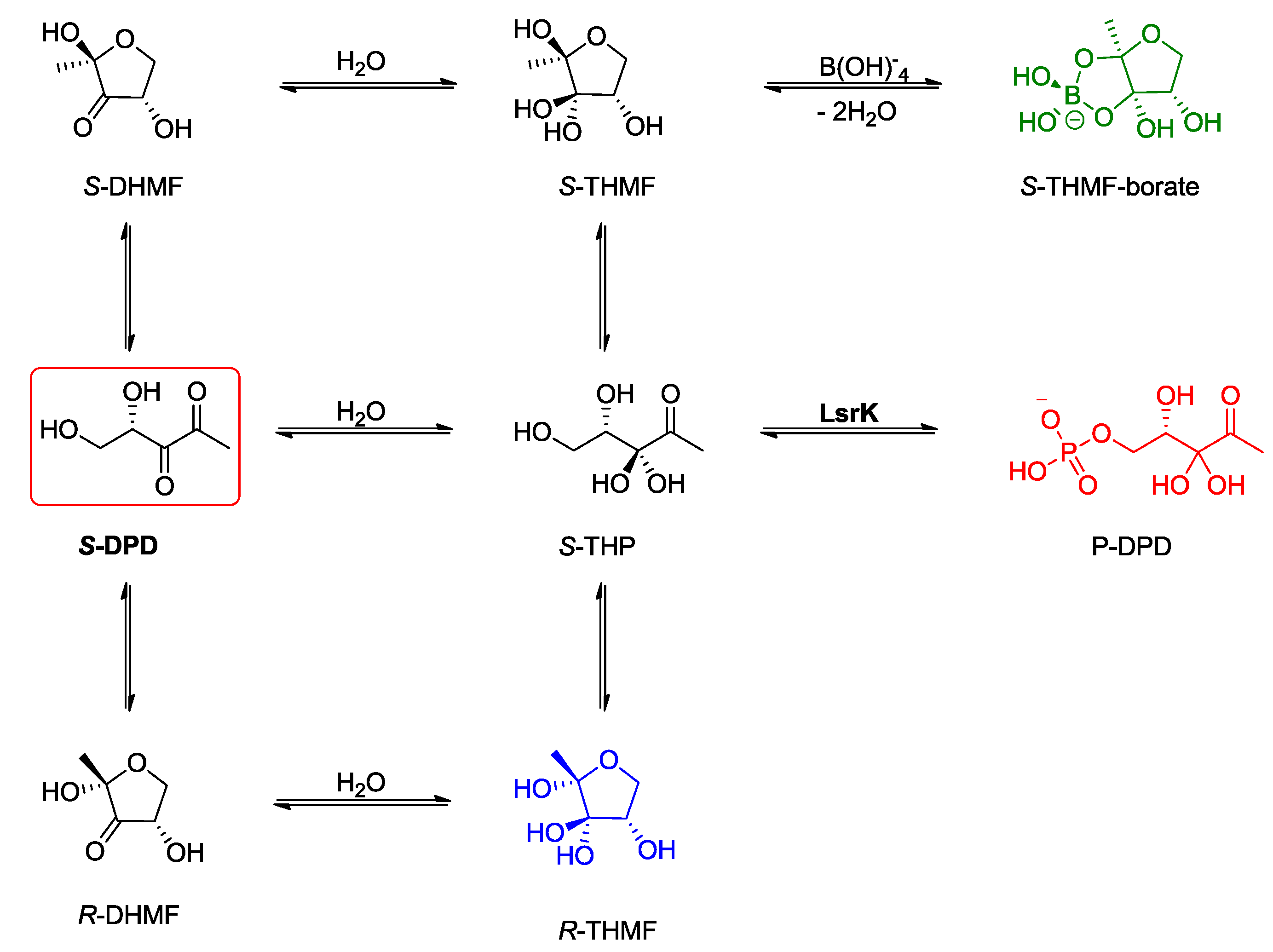
:1. Introduction
2. Results and Discussion
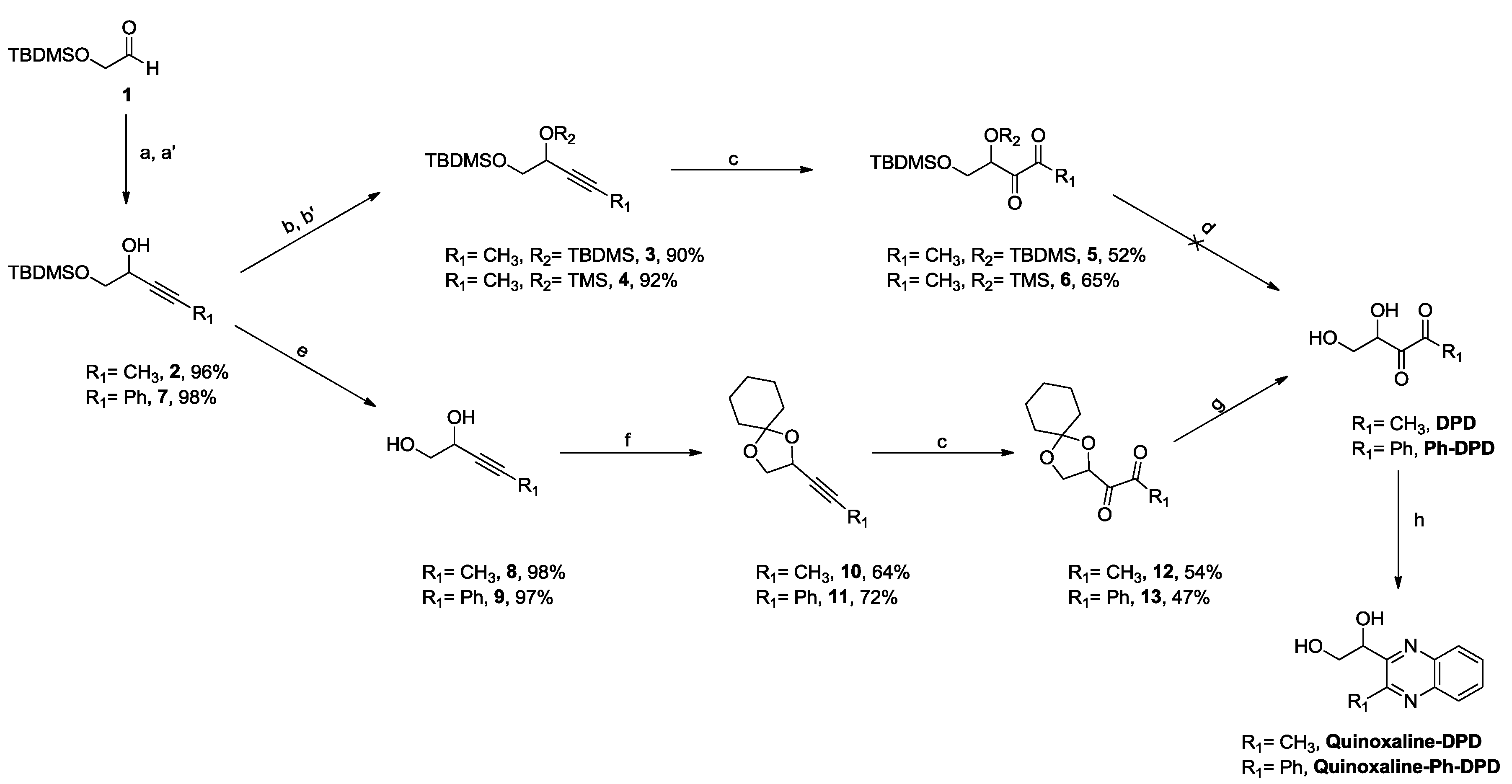
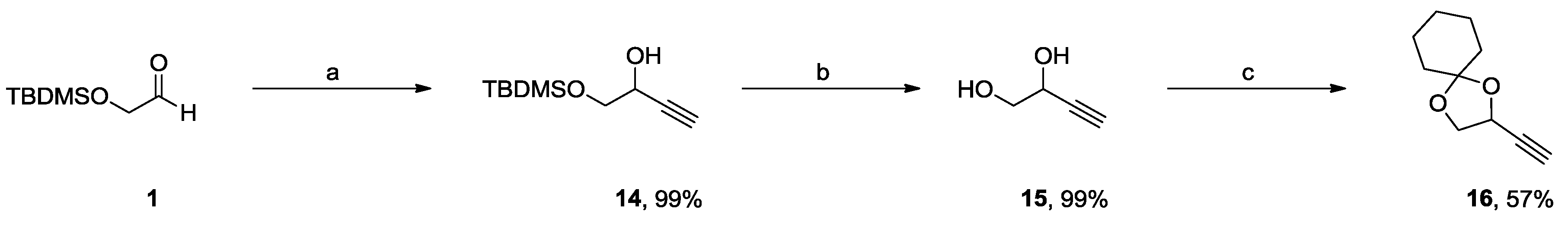
2.1. Synthesis of DPD and Ph-DPD
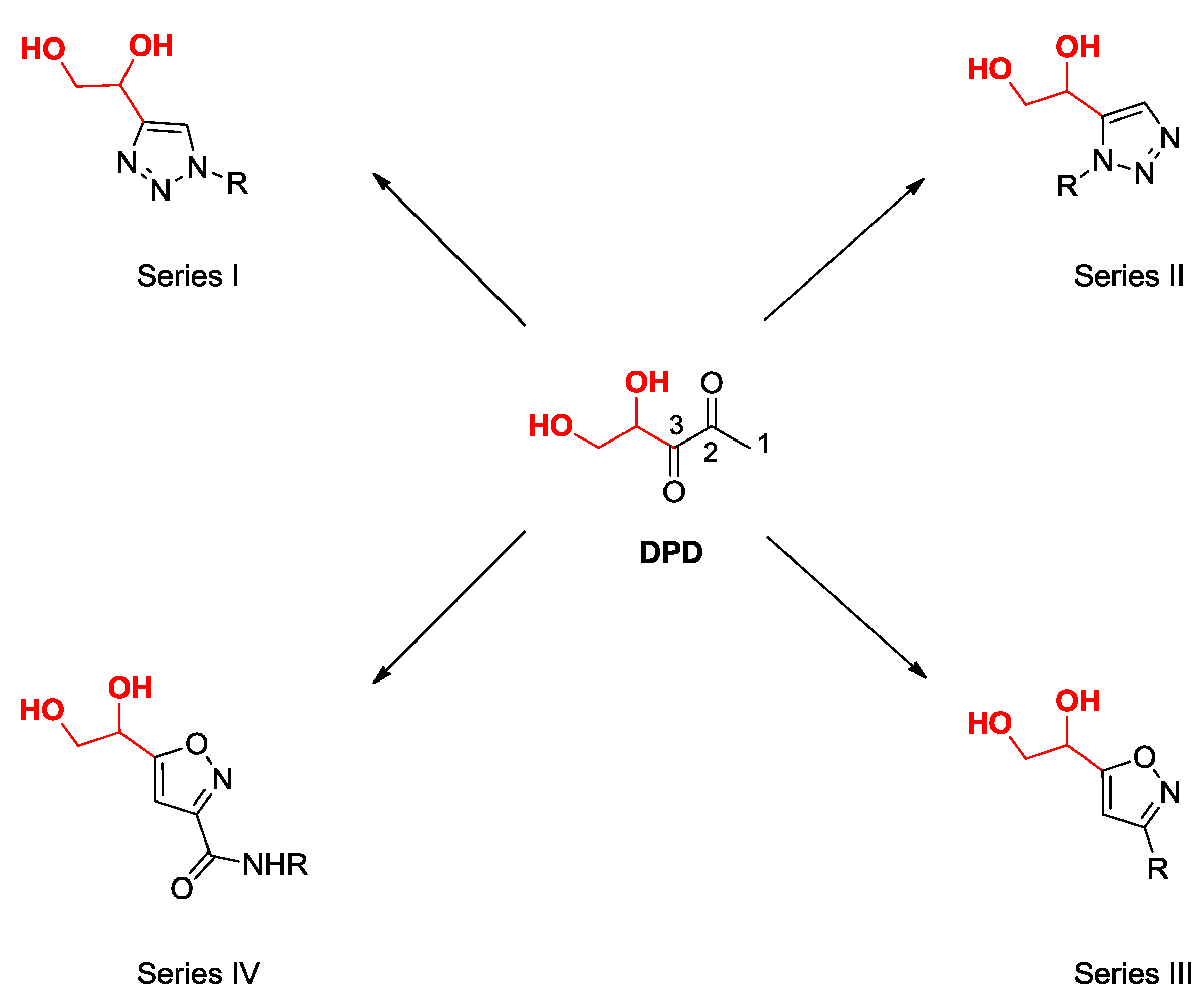
2.2. Synthesis of DPD-Related Compounds
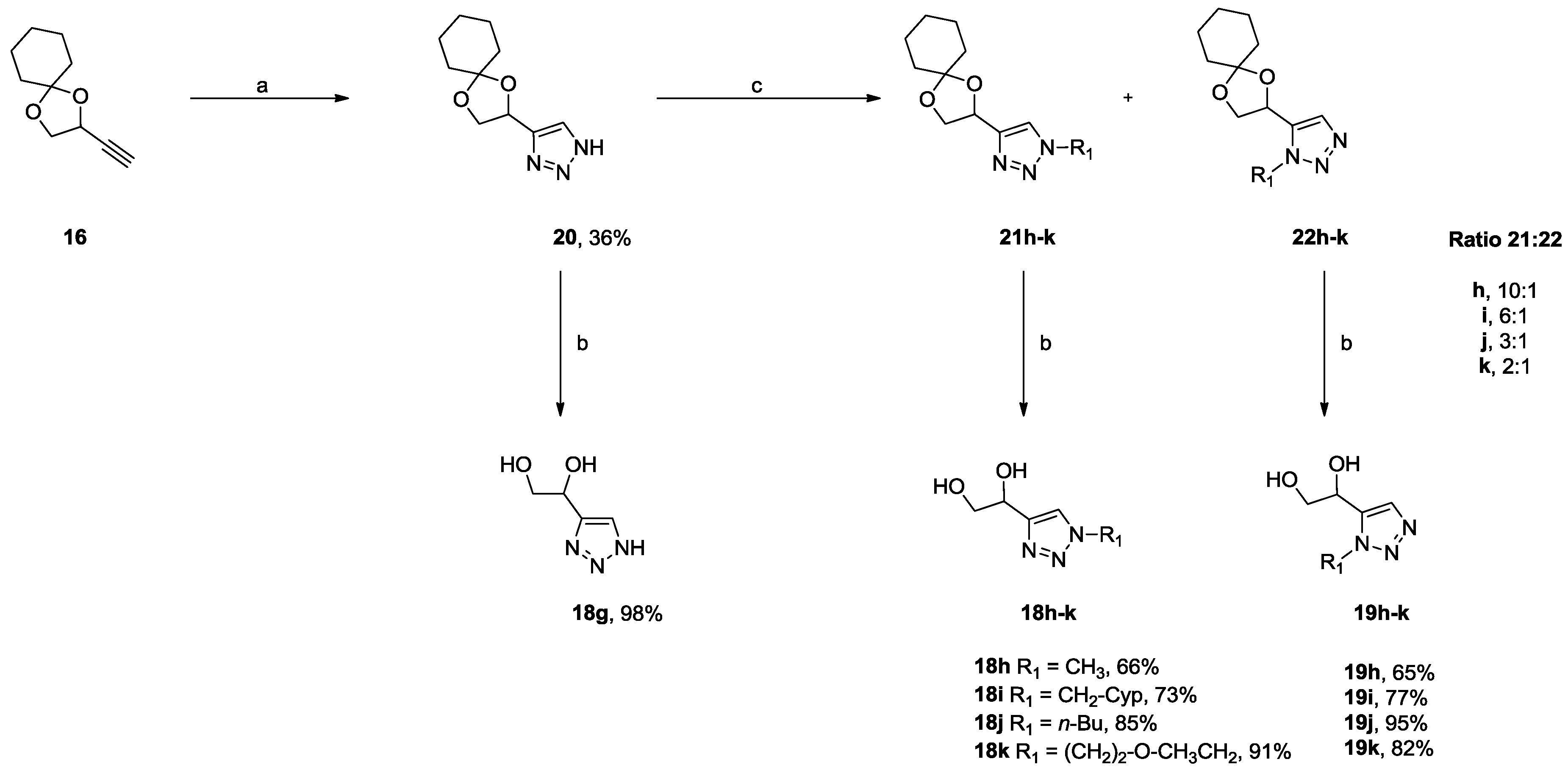
2.2.1. 1,4- and 1,5-Disubstituted 1,2,3-Triazoles DPD-Derivatives (Series I and II)
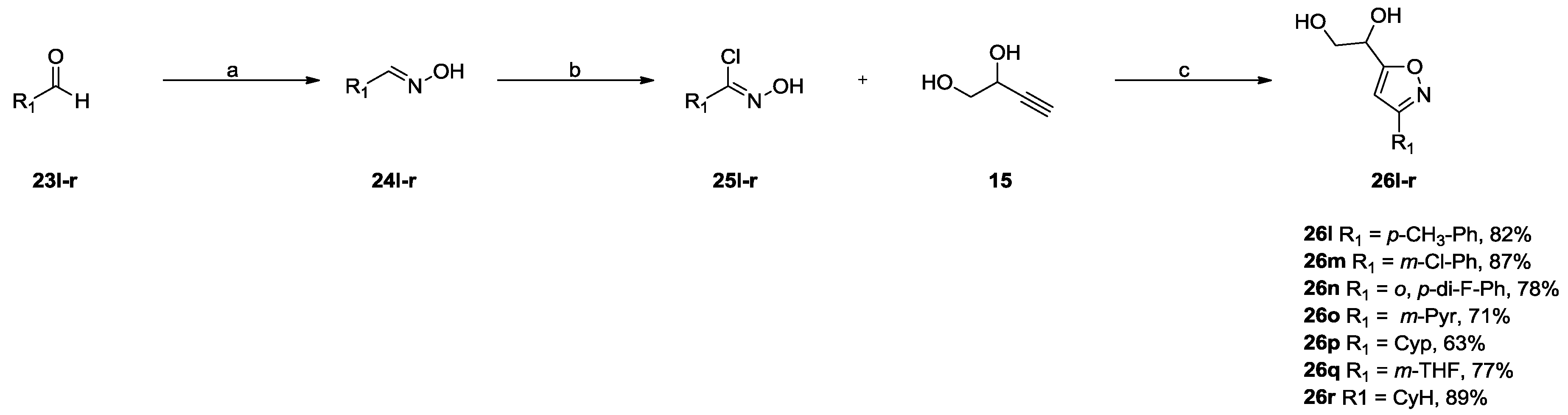
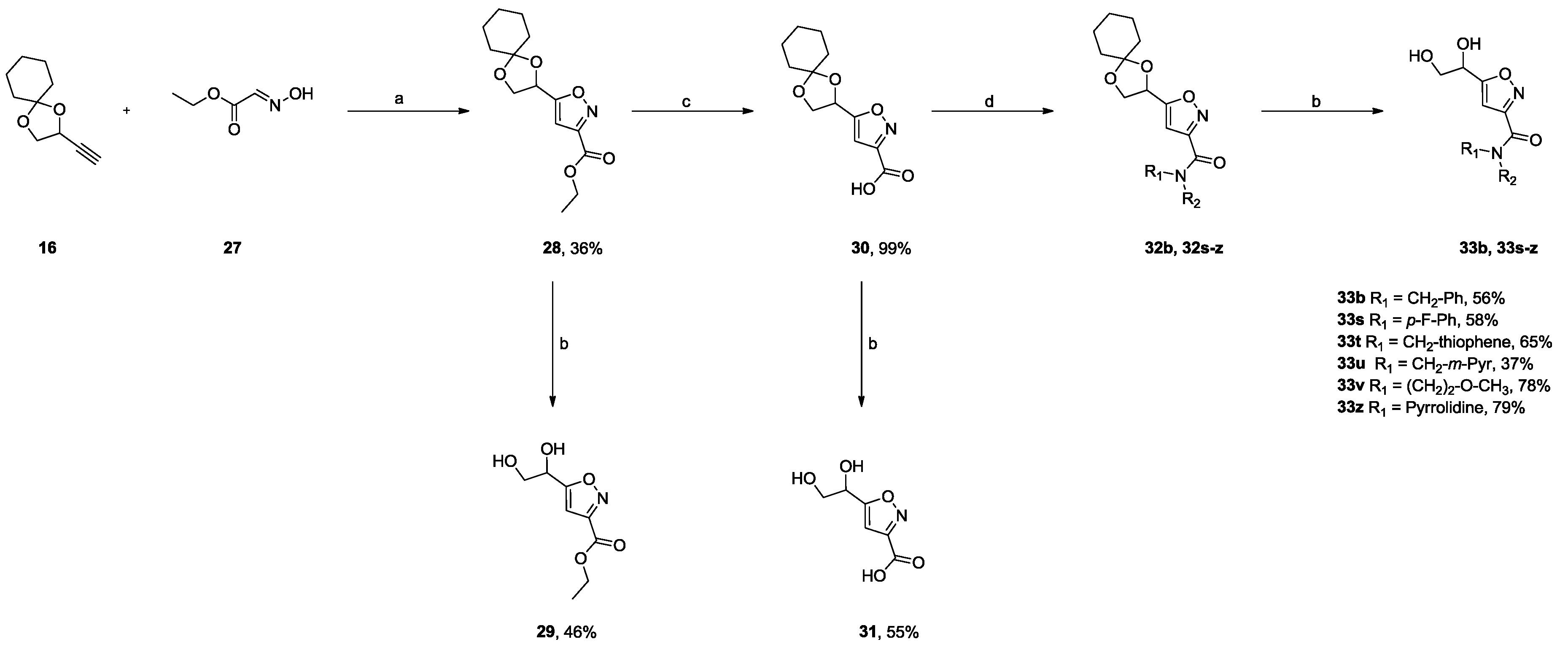
2.2.2. 3,5-Disubstituted Isoxazoles DPD-Derivatives (Series III and IV)
2.3. Biological Evaluation of Synthetized Compounds
3. Experimental
3.1. Chemistry
3.2. Synthesis of DPD and Ph-DPD
3.3. General Procedures for the Synthesis of 1,4- and 1,5-Disubstituted Triazoles DPD-Derivatives (Series I and II)
3.4. General Procedures for the Synthesis of 3,5-Disubstituted Isoxazoles DPD Derivatives (Series III and IV)
3.5. Biology
3.5.1. LsrK Overexpression and Purification
3.5.2. DPD Activity Evaluation
3.5.3. Screening of DPD-Related Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Surette, M.G.; Miller, M.B.; Bassler, B.L. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: A new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. USA 1999, 96, 1639–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Bassler, B.L.; Losick, R. Bacterially speaking. Cell 2006, 125, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.A.; Dickerson, T.J.; Janda, K.D. Interspecies and interkingdom communication mediated by bacterial quorum sensing. Chem. Soc. Rev. 2008, 37, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.-L.; Bassler, B.L. Bacterial Quorum-Sensing Network Architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Miller, M.B.; Vance, R.E.; Dziejman, M.; Bassler, B.L.; Mekalanos, J.J. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2002, 99, 3129–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, L.C.M.; Ferreira, R.B.R.; Buckner, M.M.C.; Finlay, B.B. Quorum sensing in bacterial virulence. Microbiology 2010, 156, 2271–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.A.A.M.; Petersen, F.C.; Scheie, A.A. AI-2 quorum sensing affects antibiotic susceptibility in Streptococcus anginosus. J. Antimicrob. Chemother. 2007, 60, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, D.G.; Parsek, M.R.; Pearson, J.P.; Iglewski, B.H.; Costerton, J.W.; Greenberg, E.P. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998, 280, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.H.; Palmer, R.J.; Blehert, D.S.; Campagna, S.R.; Semmelhack, M.F.; Egland, P.G.; Bassler, B.L.; Kolenbrander, P.E. Autoinducer 2: A concentration-dependent signal for mutualistic bacterial biofilm growth. Mol. Microbiol. 2006, 60, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Irie, Y.; Parsek, M.R. Quorum sensing and microbial biofilms. Curr. Top. Microbiol. Immunol. 2008, 322, 67–84. [Google Scholar] [PubMed]
- Smith, K.M.; Bu, Y.; Suga, H. Library screening for synthetic agonists and antagonists of a Pseudomonas aeruginosa autoinducer. Chem. Biol. 2003, 10, 563–571. [Google Scholar] [CrossRef]
- Suga, H.; Smith, K.M. Molecular mechanisms of bacterial quorum sensing as a new drug target. Curr. Opin. Chem. Biol. 2003, 7, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Geske, G.D.; Wezeman, R.J.; Siegel, A.P.; Blackwell, H.E. Small Molecule Inhibitors of Bacterial Quorum Sensing and Biofilm Formation. J. Am. Chem. Soc. 2005, 127, 12762–12763. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.B.; Givskov, M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int. J. Med. Microbiol. 2006, 296, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Geske, G.D.; O’Neill, J.C.; Miller, D.M.; Mattmann, M.E.; Blackwell, H.E. Modulation of Bacterial Quorum Sensing with Synthetic Ligands: Systematic Evaluation of N-Acylated Homoserine Lactones in Multiple Species and New Insights into Their Mechanisms of Action. J. Am. Chem. Soc. 2007, 129, 13613–13625. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Meijler, M.M.; Hom, L.G.; Kaufmann, G.F.; McKenzie, K.M.; Sun, C.; Moss, J.A.; Matsushita, M.; Janda, K.D. Synthesis and Biological Validation of a Ubiquitous Quorum-Sensing Molecule. Angew. Chem. Int. Ed. 2004, 43, 2106–2108. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Lowery, C.A.; McCague, K.C.; Janda, K.D. Uncharacterized 4,5-Dihydroxy-2,3-Pentanedione (DPD) Molecules Revealed ThrougH-NMR Spectroscopy: Implications for a Greater Signaling Diversity in Bacterial Species. Angew. Chem. Int. Ed. 2012, 51, 4204–4208. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schauder, S.; Potier, N.; Van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.T.; Xavier, K.B.; Campagna, S.R.; Taga, M.E.; Semmelhack, M.F.; Bassler, B.L.; Hughson, F.M. Salmonella typhimurium recognizes a chemically distinct form of the bacterial quorum-sensing signal AI-2. Mol. Cell 2004, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hixon, M.S.; Globisch, D.; Kaufmann, G.F.; Janda, K.D. Mechanistic Insights into the LsrK Kinase Required for Autoinducer-2 Quorum Sensing Activation. J. Am. Chem. Soc. 2013, 135, 7827–7830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.-H.; Eo, Y.; Grishaev, A.; Guo, M.; Smith, J.A.I.; Sintim, H.O.; Kim, E.-H.; Cheong, H.-K.; Bentley, W.E.; Ryu, K.-S. Crystal Structures of the LsrR Proteins Complexed with Phospho-AI-2 and Two Signal-Interrupting Analogues Reveal Distinct Mechanisms for Ligand Recognition. J. Am. Chem. Soc. 2013, 135, 15526–15535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penesyan, A.; Gillings, M.; Paulsen, I.T. Antibiotic discovery: Combatting bacterial resistance in cells and in biofilm communities. Mol. Basel Switz. 2015, 20, 5286–5298. [Google Scholar] [CrossRef] [PubMed]
- Kociolek, M. Quorum-Sensing Inhibitors and Biofilms. Anti-Infect. Agents Med. Chem. 2009, 8, 315–326. [Google Scholar] [CrossRef]
- Brackman, G.; Coenye, T. Quorum sensing inhibitors as anti-biofilm agents. Curr. Pharm. Des. 2015, 21, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Bassler, B.L. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 2005, 187, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Semmelhack, M.F.; Campagna, S.R.; Federle, M.J.; Bassler, B.L. An Expeditious Synthesis of DPD and Boron Binding Studies. Org. Lett. 2005, 7, 569–572. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, S.C.J.; Varszegi, C.; van Boxel, N.; Habel, L.W.; Metzger, K.; Daniels, R.; Marchal, K.; De Vos, D.; Vanderleyden, J. Chemical synthesis of (S)-4,5-dihydroxy-2,3-pentanedione, a bacterial signal molecule precursor, and validation of its activity in Salmonella typhimurium. J. Biol. Chem. 2005, 280, 19563–19568. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.A.; Park, J.; Kaufmann, G.F.; Janda, K.D. An Unexpected Switch in the Modulation of AI-2-Based Quorum Sensing Discovered through Synthetic 4,5-Dihydroxy-2,3-pentanedione Analogues. J. Am. Chem. Soc. 2008, 130, 9200–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadirvel, M.; Stimpson, W.T.; Moumene-Afifi, S.; Arsic, B.; Glynn, N.; Halliday, N.; Williams, P.; Gilbert, P.; McBain, A.J.; Freeman, S.; et al. Synthesis and bioluminescence-inducing properties of autoinducer (S)-4,5-dihydroxypentane-2,3-dione and its enantiomer. Bioorg. Med. Chem. Lett. 2010, 20, 2625–2628. [Google Scholar] [CrossRef] [PubMed]
- Ascenso, O.S.; Marques, J.C.; Santos, A.R.; Xavier, K.B.; Rita Ventura, M.; Maycock, C.D. An efficient synthesis of the precursor of AI-2, the signalling molecule for inter-species quorum sensing. Bioorg. Med. Chem. 2011, 19, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gamby, S.; Zheng, Y.; Sintim, H. Small Molecule Inhibitors of AI-2 Signaling in Bacteria: State-of-the-Art and Future Perspectives for Anti-Quorum Sensing Agents. Int. J. Mol. Sci. 2013, 14, 17694–17728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, M.; Soulère, L.; Queneau, Y.; Doutheau, A. A Baylis–Hillman/ozonolysis route towards (±) 4,5-dihydroxy-2,3-pentanedione (DPD) and analogues. Tetrahedron Lett. 2005, 46, 6495–6498. [Google Scholar] [CrossRef]
- Smith, J.A.I.; Wang, J.; Nguyen-Mau, S.-M.; Lee, V.; Sintim, H.O. Biological screening of a diverse set of AI-2 analogues in Vibrio harveyi suggests that receptors which are involved in synergistic agonism of AI-2 and analogues are promiscuous. Chem. Commun. 2009, 45, 7033–7035. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Tarantino, M.; Rossino, G.; Rui, M.; Juza, M.; Collina, S. Approaches for multi-gram scale isolation of enantiomers for drug discovery. Expert Opin. Drug Discov. 2017, 12, 1253–1269. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Delgado, O.; Florence, G.J.; Lyothier, I.; O’Brien, M.; Scott, J.P.; Sereinig, N. A second-generation total synthesis of (+)-discodermolide: The development of a practical route using solely substrate-based stereocontrol. J. Org. Chem. 2005, 70, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Peyton, L.R.; Gallagher, S.; Hashemzadeh, M. Triazole antifungals: A review. Drugs Today Barc. Spain 1998 2015, 51, 705–718. [Google Scholar] [CrossRef]
- Lal, K.; Yadav, P. Recent Advancements in 1,4-Disubstituted 1H-1,2,3-Triazoles as Potential Anticancer Agents. Anticancer Agents Med. Chem. 2018, 18, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-H.; Wang, Y. Recent Researches in Triazole Compounds as Medicinal Drugs. Curr. Med. Chem. 2012, 19, 239–280. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Alam, M.S.; Hamid, H. 1,2,3-Triazoles: Scaffold with medicinal significance. Inflamm. Cell Signal. 2014, 1. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, S.A.; Budagumpi, S.; Nagaraja, B.M. Triazole: A Promising Antitubercular Agent. Chem. Biol. Drug Des. 2015, 86, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Shahar Yar, M.; Sharma, P.C. Recent advances and future perspectives of triazole analogs as promising antiviral agents. Mini Rev. Med. Chem. 2011, 11, 84–96. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.P.; Badillo, J.J.; Arevalo, G.E.; Silva-García, A.; Franz, A.K. Catalytic Stereoselective Synthesis of Diverse Oxindoles and Spirooxindoles from Isatins. ACS Comb. Sci. 2012, 14, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte, C.; Mayer, P.; Straub, B.F. Isolation of a copper(I) triazolide: A “click” intermediate. Angew. Chem. Int. Ed. 2007, 46, 2101–2103. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Wang, X.; Xu, J.; Zhao, J.; Zhang, Q.; Hu, Y. Carboxylic Acid-Promoted Copper(I)-Catalyzed Azide−Alkyne Cycloaddition. J. Org. Chem. 2010, 75, 7002–7005. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Cheng, G.; Su, D.; Xu, J.; Wang, X.; Hu, Y. Copper(I) Acetate: A Structurally Simple but Highly Efficient Dinuclear Catalyst for Copper-Catalyzed Azide-Alkyne Cycloaddition. Adv. Synth. Catal. 2010, 352, 1587–1592. [Google Scholar] [CrossRef]
- Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. Acid–Base Jointly Promoted Copper(I)-Catalyzed Azide–Alkyne Cycloaddition. J. Org. Chem. 2011, 76, 6832–6836. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-catalyzed azide-alkyne cycloaddition: Scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, L.; De Sarlo, F.; Machetti, F. 1,4-Diazabicyclo[2.2.2]octane (DABCO) as an Efficient Reagent for the Synthesis of Isoxazole Derivatives from Primary Nitro Compounds and Dipolarophiles: The Role of the Base. Eur. J. Org. Chem. 2006, 2006, 4852–4860. [Google Scholar] [CrossRef]
- Chand, P.; Kotian, P.L.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T.L.; Babu, Y.S.; Bantia, S.; Elliott, A.J.; Montgomery, J.A. Systematic Structure-Based Design and Stereoselective Synthesis of Novel Multisubstituted Cyclopentane Derivatives with Potent Antiinfluenza Activity. J. Med. Chem. 2001, 44, 4379–4392. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Atack, J.R.; Braun, M.P.; Cato, B.P.; Chambers, M.S.; O’Connor, D.; Cook, S.M.; Hobbs, S.C.; Maxey, R.; Szekeres, H.J.; et al. Pharmacokinetics and metabolism studies on (3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy) pyrazolo[1,5-d][1,2,4]triazine, a functionally selective GABAA α5 inverse agonist for cognitive dysfunction. Bioorg. Med. Chem. Lett. 2006, 16, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.L.; Liauw, A.Y.; Ellis, C.D.; Pruitt, J.R.; Carini, D.J.; Bostrom, L.L.; Huang, P.P.; Harrison, K.; Knabb, R.M.; Thoolen, M.J.; et al. Design and Synthesis of Isoxazoline Derivatives as Factor Xa Inhibitors. 1. J. Med. Chem. 1999, 42, 2752–2759. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, Y.A.M.; Inagaki, F.; Takahashi, R.; Mukai, C. A new procedure for the preparation of 2-vinylindoles and their [4+2] cycloaddition reaction. Tetrahedron 2011, 67, 5133–5141. [Google Scholar] [CrossRef]
- Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E.F. An Extremely Facile Synthesis of Furans, Pyrroles, and Thiophenes by the Dehydrative Cyclization of Propargyl Alcohols. Org. Lett. 2009, 11, 4624–4627. [Google Scholar] [CrossRef] [PubMed]
- Gamby, S.; Roy, V.; Guo, M.; Smith, J.A.I.; Wang, J.; Stewart, J.E.; Wang, X.; Bentley, W.E.; Sintim, H.O. Altering the communication networks of multispecies microbial systems using a diverse toolbox of AI-2 analogues. ACS Chem. Biol. 2012, 7, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Morriello, G.; Devita, R.; Mills, S.; Moyes, C.; Lin, P. Anti-Hypercholesterolemic Compounds 2010. U.S. Patent 20070078098A1, 5 April 2007. [Google Scholar]
- Mames, A.; Stecko, S.; Mikołajczyk, P.; Soluch, M.; Furman, B.; Chmielewski, M. Direct, Catalytic Synthesis of Carbapenams via Cycloaddition/Rearrangement Cascade Reaction: Unexpected Acetylenes’ Structure Effect. J. Org. Chem. 2010, 75, 7580–7587. [Google Scholar] [CrossRef] [PubMed]
- Jen, T.; Mendelsohn, B.A.; Ciufolini, M.A. Oxidation of α-Oxo-Oximes to Nitrile Oxides with Hypervalent Iodine Reagents. J. Org. Chem. 2011, 76, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Miller, S.T.; Lu, W.; Kim, J.H.; Rabinowitz, J.; Pelczer, I.; Semmelhack, M.F.; Bassler, B.L. Phosphorylation and Processing of the Quorum-Sensing Molecule Autoinducer-2 in Enteric Bacteria. ACS Chem. Biol. 2007, 2, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchikama, K.; Zhu, J.; Lowery, C.A.; Kaufmann, G.F.; Janda, K.D. C4-Alkoxy-HPD: A Potent Class of Synthetic Modulators Surpassing Nature in AI-2 Quorum Sensing. J. Am. Chem. Soc. 2012, 134, 13562–13564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, V.; Meyer, M.T.; Smith, J.A.I.; Gamby, S.; Sintim, H.O.; Ghodssi, R.; Bentley, W.E. AI-2 analogs and antibiotics: A synergistic approach to reduce bacterial biofilms. Appl. Microbiol. Biotechnol. 2013, 97, 2627–2638. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are all available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Oxidant and eq | Time | Yield (%) |
|---|---|---|---|---|
| 1 | Acetone | KMnO4/NaHCO3/MgSO4 3.8/0.6/2.0 | Overnight | No reaction |
| 2 | Acetone | KMnO4/NaHCO3/MgSO4 3.9/0.6/4.2 | Overnight | Traces |
| 3 | CCl4/ACN (1:1) | NaIO4/RuO2·H2O 2.2 eq/2.5% mol | 3 h | Traces |
| 4 | CCl4/ACN (1:1) | NaIO4/RuO2·H2O 4.4 eq/2.5% mol | 3 h | 23 |
| 5 | CHCl3/ACN/H2O (1:1:1) | NaIO4/RuO2·H2O 4.4 eq/2.5% mol | 3 h | 52 |

| Entry | R1 | Azide, eq | Solvent | Catalyst | Product | Yield (%) a | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | (CH2)2-Ph | 17a, 1.1 | THF | CuI (10% mol) DIPEA (15% mol) | 18a | 58 | [44] |
| 2 | (CH2)2-Ph | 17a, 1.05 | DCM | CuI (2% mol) DIPEA (4% mol) AcOH (cat) | 18a | 72 | [48] |
| 3 | (CH2)2-Ph | 17a, 1.0 | t-BuOH/H2O (1:1) | CuSO4·5H2O (5% mol) Na Ascorbate (0.5 eq) | 18a | 89 | [49] |
| 4 | (CH2)2-Ph | 17a, 1.0 | 1,4-dioxane | (Cp*RuCl(PPh3)2) (2% mol) | 19a | 87 | [50] |
| 5 | (CH2)-Ph | 17b, 1.0 | t-BuOH/H2O (1:1) | CuSO4·5H2O (5% mol) Na Ascorbate (0.5 eq) | 18b | 60 | [49] |
| 6 | (CH2)2-o-F-Ph | 17c, 1.0 | t-BuOH/H2O (1:1) | CuSO4·5H2O (5% mol) Na Ascorbate (0.5 eq) | 18c | 62 | [49] |
| 7 | (CH2)2-m-Pyr | 17d, 1.0 | t-BuOH/H2O (1:1) | CuSO4·5H2O (5% mol) Na Ascorbate (0.5 eq) | 18d | 88 | [49] |
| 8 | (CH2)5-CN | 17e, 1.0 | t-BuOH/H2O (1:1) | CuSO4·5H2O (5% mol) Na Ascorbate (0.5 eq) | 18e | 72 | [49] |
| 9 | (CH2)2-CyH | 17f, 1.0 | t-BuOH/H2O (1:1) | CuSO4·5H2O (5% mol) Na Ascorbate (0.5 eq) | 18f | 73 | [49] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stotani, S.; Gatta, V.; Medda, F.; Padmanaban, M.; Karawajczyk, A.; Tammela, P.; Giordanetto, F.; Tzalis, D.; Collina, S. A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing. Molecules 2018, 23, 2545. https://doi.org/10.3390/molecules23102545
Stotani S, Gatta V, Medda F, Padmanaban M, Karawajczyk A, Tammela P, Giordanetto F, Tzalis D, Collina S. A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing. Molecules. 2018; 23(10):2545. https://doi.org/10.3390/molecules23102545
Chicago/Turabian StyleStotani, Silvia, Viviana Gatta, Federico Medda, Mohan Padmanaban, Anna Karawajczyk, Päivi Tammela, Fabrizio Giordanetto, Dimitrios Tzalis, and Simona Collina. 2018. "A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing" Molecules 23, no. 10: 2545. https://doi.org/10.3390/molecules23102545