Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus

Abstract
:1. Introduction
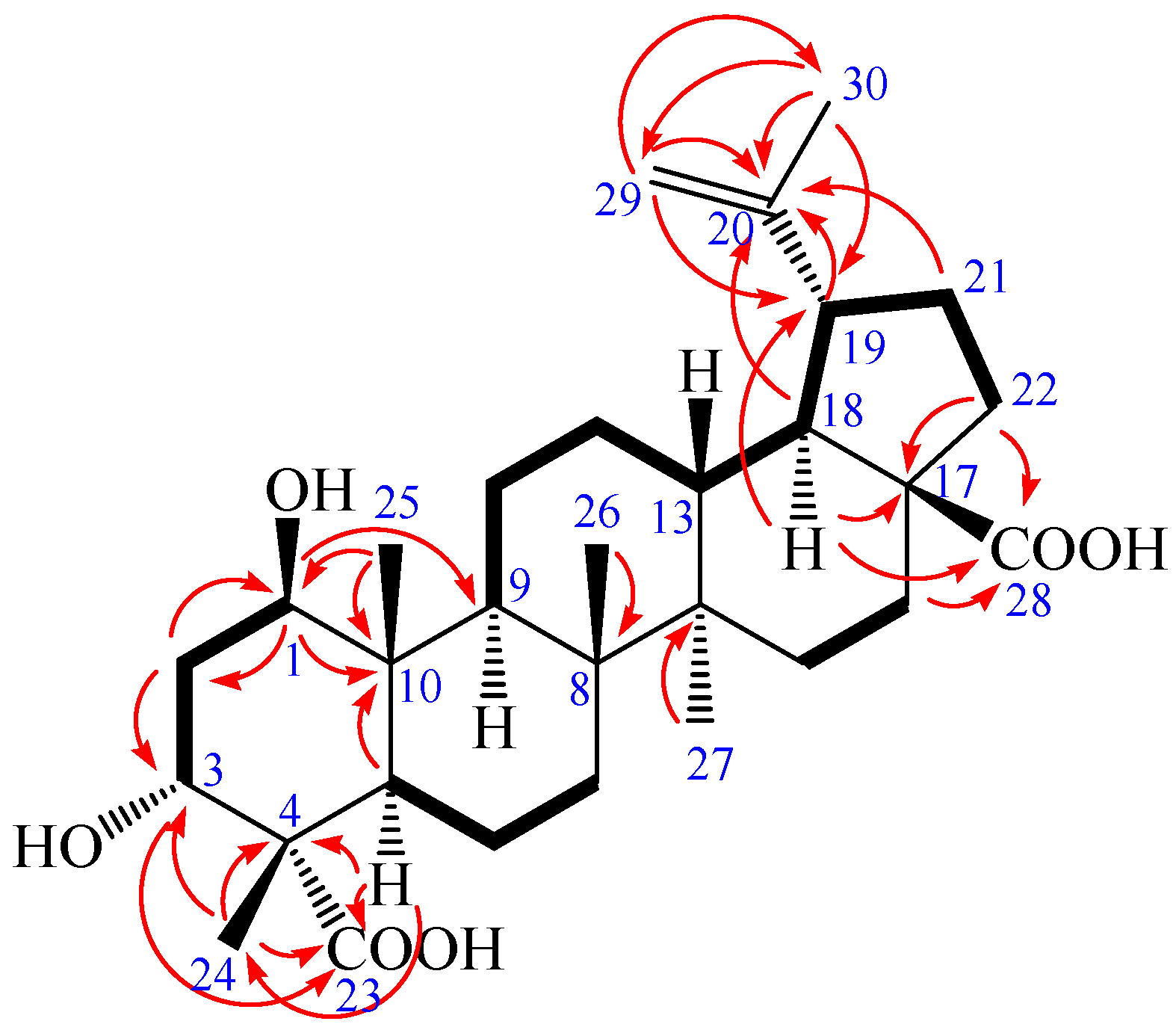
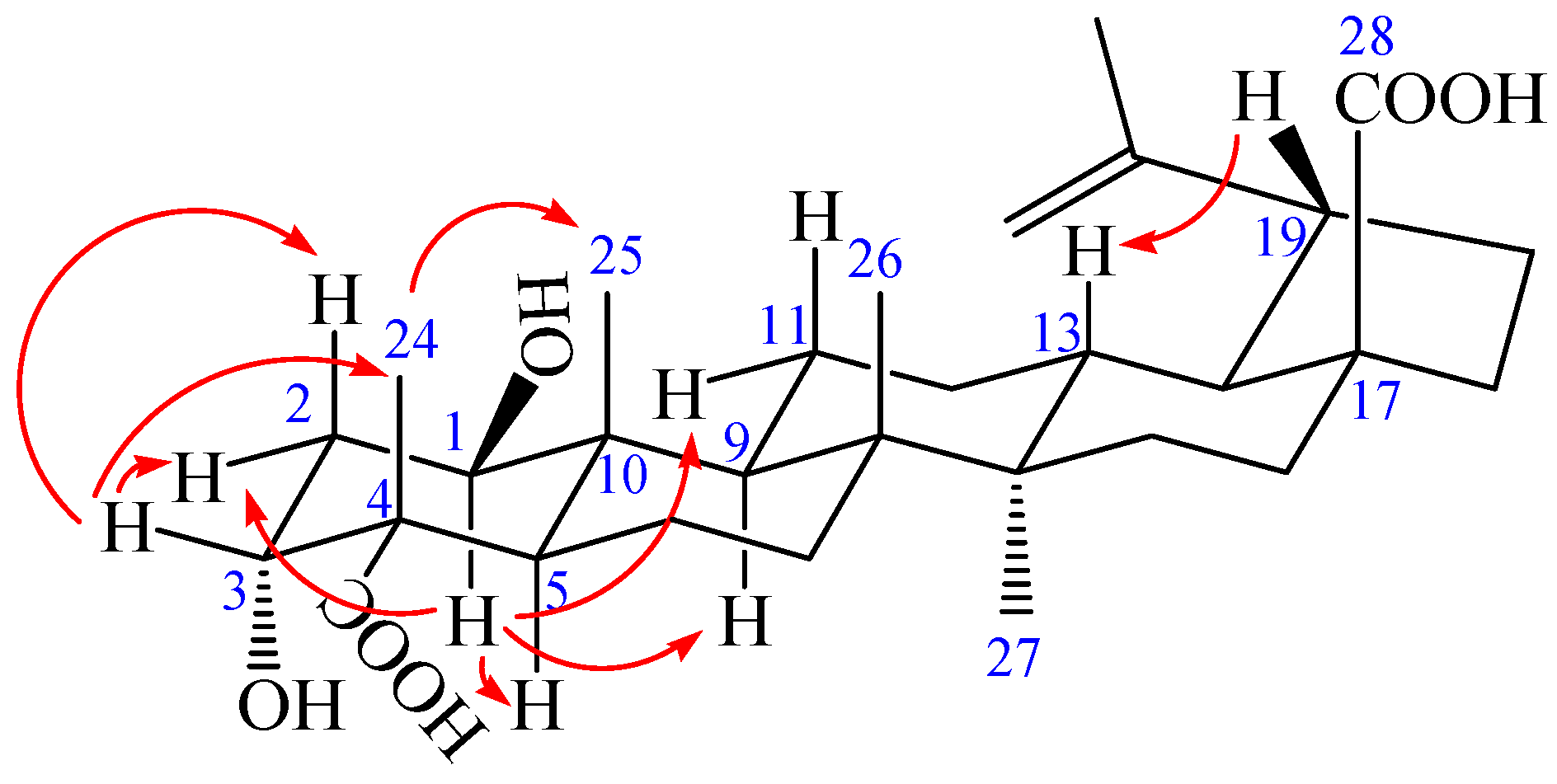
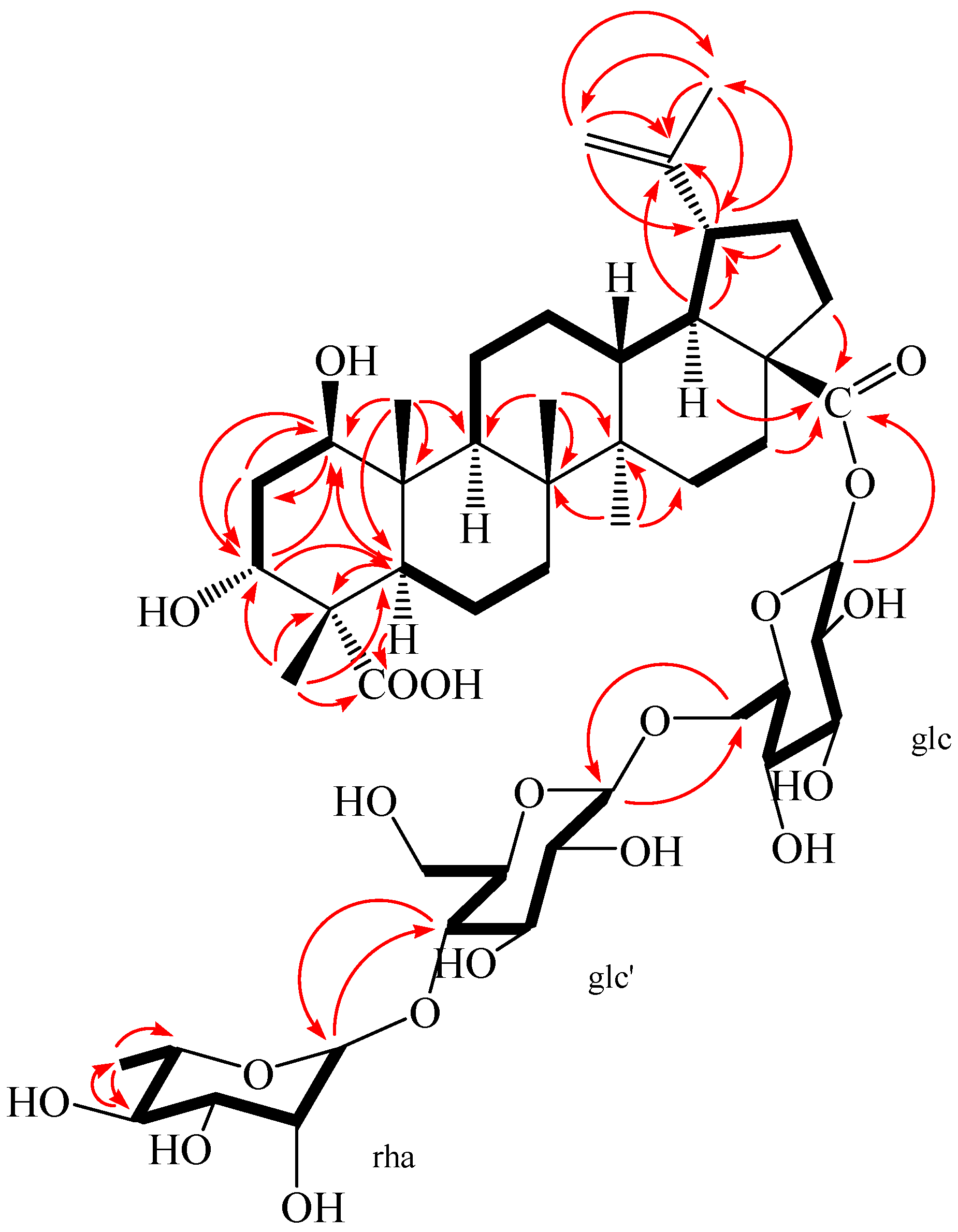
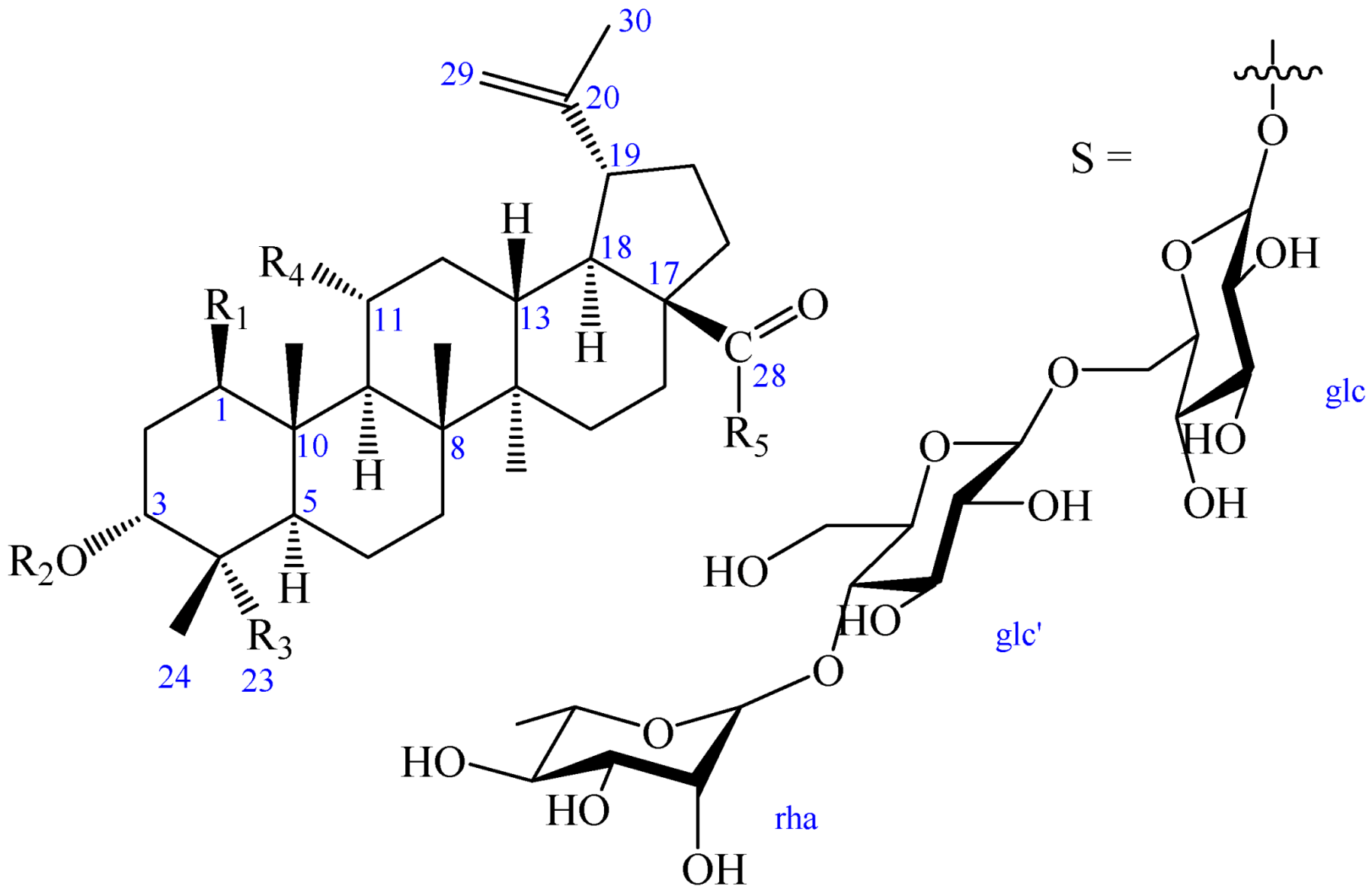
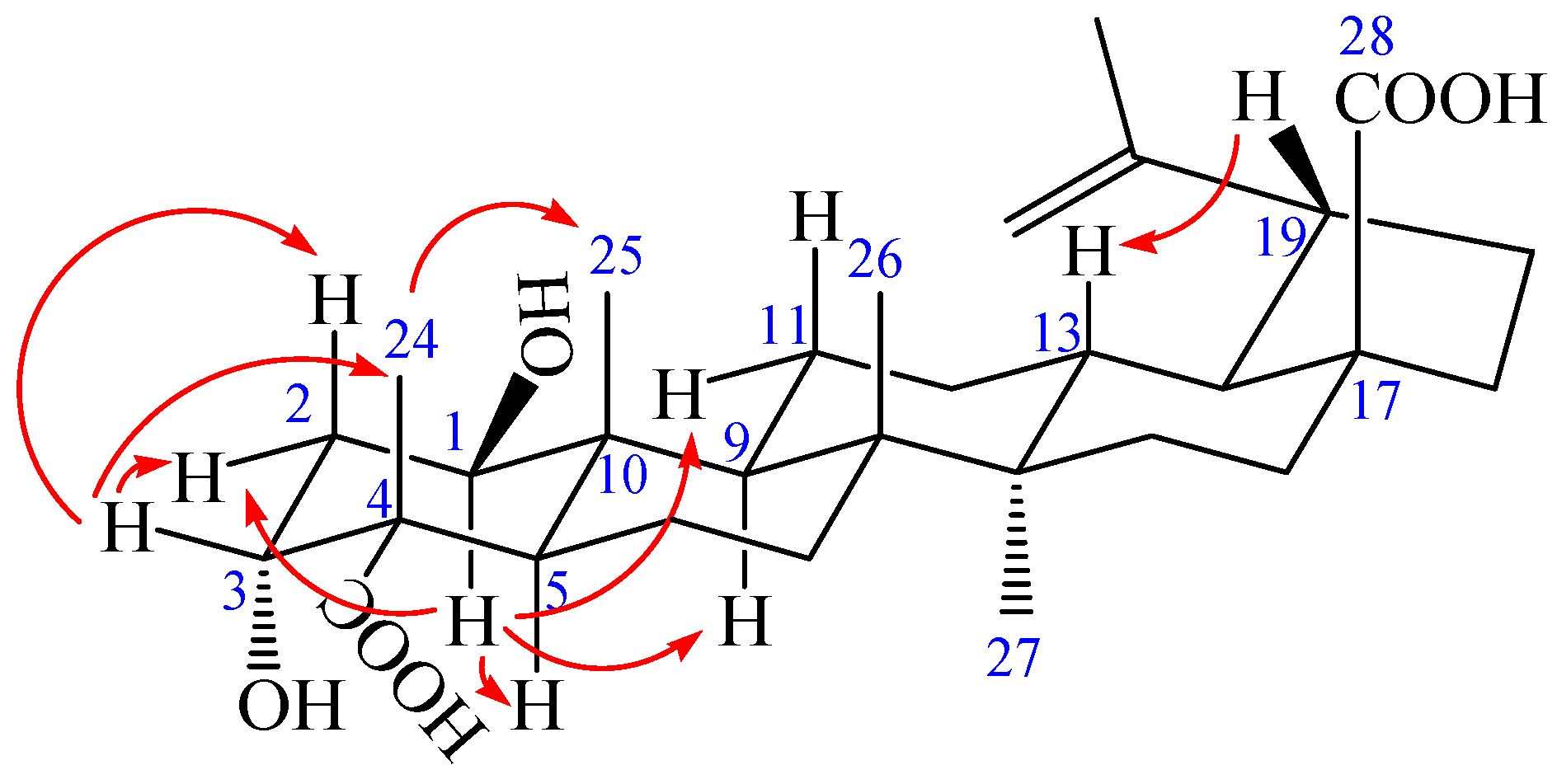
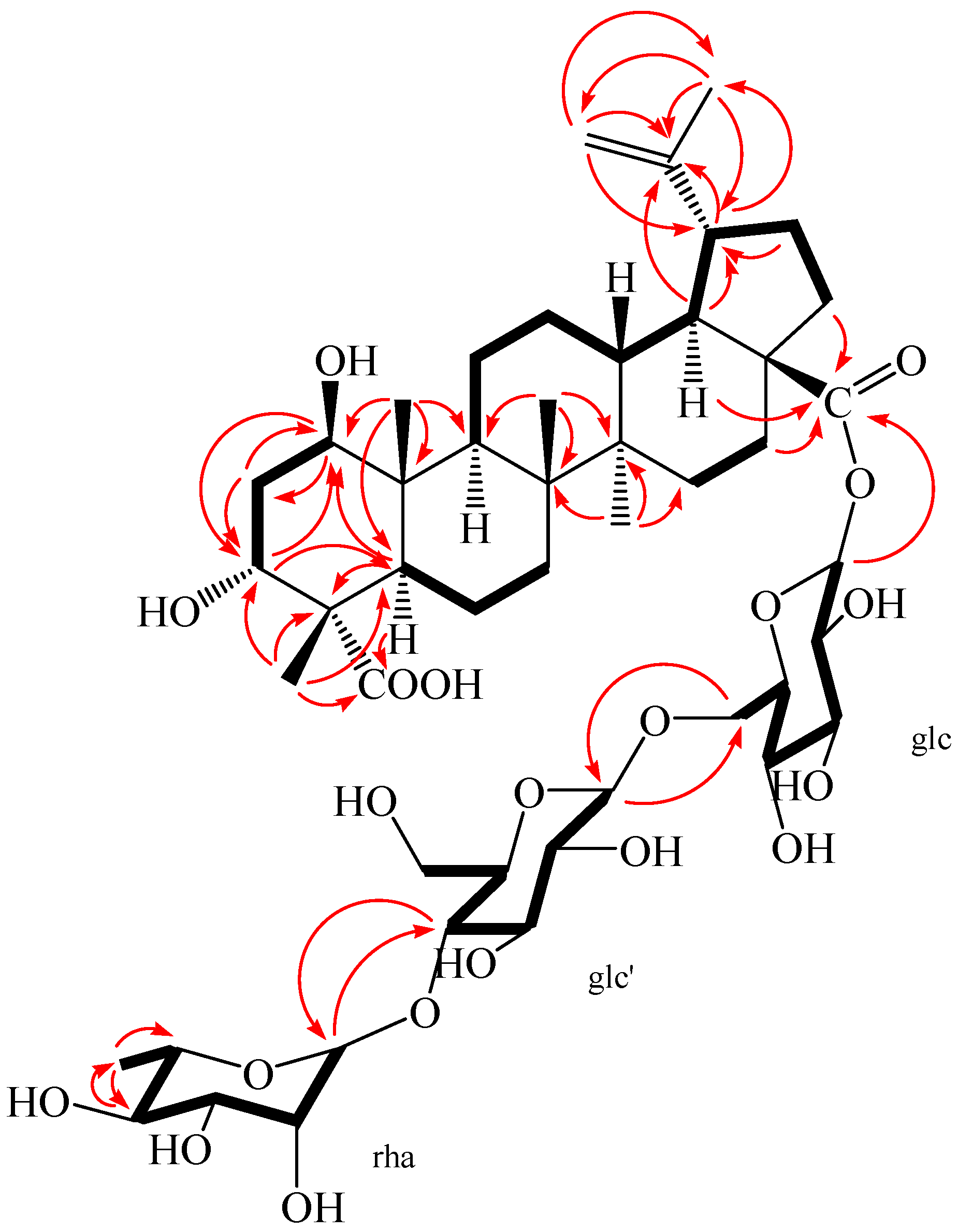
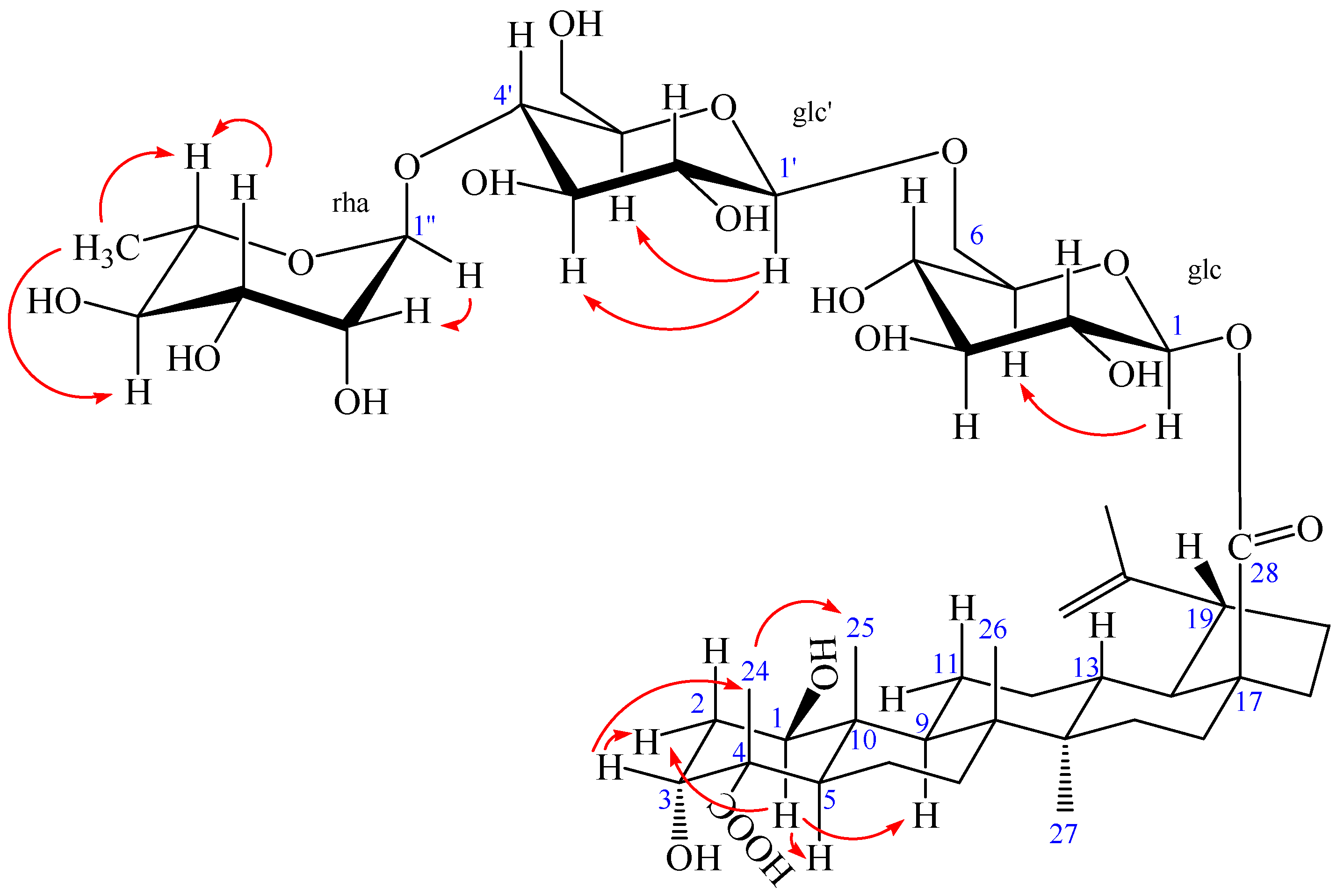
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Collection and Identification of Biological Materials
3.3. Extraction and Isolation
3.4. Alkaline Hydrolysis of 2
3.5. MTT Assay for Cell Viability
3.6. Nitrite Assay
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia of the People’s Republic of China; Medical Science and Technology Press: Beijing, China, 2015; Volume 1, p. 79. [Google Scholar]
- Ni, N.; Liu, X.Q. Advances in studies on plants of Acanthopanax Miq. in Araliaceae. Chin. Tradit. Herb. Drugs 2006, 37, 1895–1900. [Google Scholar]
- Zhang, B.X.; Li, N.; Zhang, Z.P.; Liu, H.B.; Zhou, R.R.; Zhong, B.Y.; Zou, M.X.; Dai, X.H.; Xiao, M.F.; Liu, X.Q.; et al. Protective effect of Acanthopanax gracilistylus-extracted Acankoreanogenin A on mice with fulminant hepatitis. Int. Immunopharmacol. 2011, 11, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Yook, C.S.; Liu, X.Q.; Chang, S.Y.; Park, S.Y.; Nohara, T. Lupane-triterpene glycosides from the leaves of Acanthopanax gracilistylus. Chem. Pharm. Bull. 2002, 50, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Chang, S.Y.; Park, S.Y.; Nohara, T.; Yook, C.S. A new lupane-triterpene glycoside from the leaves of Acanthopanax gracilistylus. Arch. Pharm. Res. 2002, 25, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Chang, S.Y.; Yook, C.S. Lupane-triterpenoids from the leaves of Acanthopanax gracilistylus. J. Lanzhou Univ. Nat. Sci. 2006, 42, 86–91. [Google Scholar]
- Zou, Q.P.; Liu, X.Q.; Lee, H.K.; Oh, O.J. Lupane-triterpenoids from the methanol extracts of leaves of Acanthopanax gracilistylus W. W. Smith. J. Lanzhou Univ. Nat. Sci. 2011, 47, 120–126. [Google Scholar]
- Tang, X.Y.; Ma, Y.C.; Li, P.J. Separation and identification of the anti-inflammatory diterpene from the root cortices of Acanthopanax gracilistylus W. W. Smith. Chin. J. Chin. Mater. Med. 1995, 20, 231–232. [Google Scholar]
- Wu, Z.Y.; Zhang, Y.B.; Zhu, K.K.; Luo, C.; Zhang, J.X.; Cheng, C.R.; Feng, R.H.; Yang, W.Z.; Zeng, F.; Wang, Y.; et al. Anti-inflammatory diterpenoids from the root bark of Acanthopanax gracilistylus. J. Nat. Prod. 2014, 77, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Zhang, C.Y.; Yin, W.J.; Liu, Z.X.; Yook, C.S. Analysis of volatile oil components of Acanthopanax gracilistylus. Chin. Tradit. Herb. Drugs 2001, 32, 1074–1075. [Google Scholar]
- Liu, X.Q.; Park, S.Y.; Yook, C.S. Studies on the constituents of the stem barks of Acanthopanax gracilistylus W. W. Smith. Nat. Prod. Sci. 2002, 8, 23–25. [Google Scholar]
- Liu, X.Q.; Yook, C.S.; Chang, S.Y. Chemical constituents of Acanthopanax gracilistylus. Chin. Tradit. Herb. Drugs 2004, 35, 250–252. [Google Scholar]
- Zhang, J.Y.; Pu, S.B.; Qian, S.H.; Liu, D. New cerebrosides from Acanthopanax gracilistylus. Chin. J. Nat. Med. 2011, 9, 105–107. [Google Scholar]
- Liu, X.Q.; Chang, S.Y.; Ro, S.H.; Yook, C.S. Constituents of Acanthopanax gracilistylus W. W. Smith. Nat. Med. 2002, 56, 215–216. [Google Scholar]
- An, S.Y.; Qian, S.H.; Jiang, J.Q.; Wang, K.C. Chemical constituents in leaves of Acanthopanax gracilistylus. Chin. Tradit. Herb. Drugs 2009, 40, 1528–1534. [Google Scholar]
- Xian, L.N.; Qian, S.H.; Li, Z.L. Studies on the chemical constituents from the stems of Acanthopanax gracilistylus. J. Chin. Med. Mater. 2010, 33, 538–542. [Google Scholar]
- Zhang, J.Y.; Pu, S.B.; Qian, S.H.; Liu, D.; Wang, K.C. Studies on the chemical constituents in fruits of Acanthopanax gracilistylus. J. Chin. Med. Mater. 2011, 34, 226–229. [Google Scholar]
- Song, X.H.; Xu, G.J.; Jin, R.L. Studies on chemical constituents of the root bark of Acanthopanax gracilistylus. J. Chin. Pharm. Univ. 1987, 18, 203–204. [Google Scholar]
- Dai, L.; Liu, X.Q.; Xie, X.; Liu, H.Y. Characterization of stereostructure by X-ray and technology of extracting in combination hydrolysis in situ of acankoreanogenin from leaves of Acanthopanax gracilistylus W. W. Smith. J. Cent. South Univ. 2014, 21, 3063–3070. [Google Scholar] [CrossRef]
- Shan, B.E.; Zeki, K.; Sugiura, T.; Yoshida, Y.; Yamashita, U. Chinese medicinal herb, Acanthopanax gracilistylus, extract induces cell cycle arrest of human tumor cells in vitro. Cancer Sci. 2000, 91, 383–389. [Google Scholar] [CrossRef]
- Shan, B.E.; Si, C.Y.; Zhang, J.Z. The Isolation of Anti-tumor Component of Acanthopanax gracilistylus. Teratog. Carcinog. Mutagen. 2004, 4, 203–205. [Google Scholar]
- Li, X.J.; Dai, L.; Li, Z.; Zhang, X.D.; Liu, X.Q.; Zou, Q.P.; Xie, X. Anti-inflammatory activities of lupane-triterpenoids in vitro and their phytochemical fingerprinting from leaves of Acanthopanax gracilistylus. Nat. Prod. Sci. 2015, 21, 104–110. [Google Scholar]
- Zou, Q.P.; Liu, X.Q.; Huang, J.J.; Yook, C.S.; Whang, W.K.; Lee, H.K.; Kwon, O.K. Inhibitory effects of lupane-type triterpenoid saponins from the leaves of Acanthopanax gracilistylus on lipopolysaccharide-induced TNF-α, IL-1β and high-mobility group box 1 release in macrophages. Mol. Med. Rep. 2017, 16, 9149–9156. [Google Scholar] [PubMed]
- Liu, X.Q.; Zou, Q.P.; Huang, J.J.; Yook, C.S.; Whang, W.K.; Lee, H.K.; Kwon, O.K. Inhibitory effects of 3α-hydroxy-lup-20(29)-en-23, 28-dioic acid on lipopolysaccharide-induced TNF-α, IL-1β, and the high mobility group box 1 release in macrophages. Biosci. Biotechnol. Biochem. 2017, 81, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Yook, C.S.; Kim, I.H.; Hahn, D.R.; Nohara, T.; Chang, S.Y. A lupane-triterpene glycoside from leaves of two Acanthopanax. Phytochemistry 1998, 49, 839–843. [Google Scholar] [CrossRef]
- Chang, S.Y.; Yook, C.S.; Nohara, T. Two new lupane-triterpene glycosides from leaves of Acanthopanax koreanum. Chem. Pharm. Bull. 1998, 46, 163–165. [Google Scholar] [CrossRef]
- Nhiem, N.X.; Tung, N.H.; Van Kiem, P.; Van Minh, C.; Ding, Y.; Hyun, J.H.; Kang, H.K.; Kim, Y.H. Lupane triterpene glycosides from leave of Acanthopanax koreanum and their cytotoxic activity. Chem. Pharm. Bull. 2009, 57, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Nhiem, N.X.; Van Kiem, P.; Van Minh, C.; Tai, B.H.; Yen, P.H.; Tung, N.H.; Tung, N.H.; Hyun, J.H.; Kang, H.K.; Kim, Y.H. Lupane-type triterpene glycosides from the leaves of Acanthopanax koreanum and their in vitro cytotoxicity. Planta Med. 2010, 76, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Ahmed, W.; Malik, A. A lupene-type triterpene from Mimusops elengi. Phytochemistry 1995, 39, 255–257. [Google Scholar] [CrossRef]
- Lischewski, M.; Ty, P.D.; Schmidt, J.; Preiss, A.; Phiet, H.V.; Adam, G. Natural products from Vietnamese plants. Part 8. 3α,11α-Dihydroxylup-20(29)-ene-23,28-dioic acid from Schefflera octophylla. Phytochemistry 1984, 23, 1695–1697. [Google Scholar] [CrossRef]
- Chang, S.Y.; Yook, C.S.; Nohara, T. Lupane-triterpene glycosides from leaves of Acanthopanax koreanum. Phytochemistry 1999, 50, 1369–1374. [Google Scholar] [CrossRef]
- Titheradge, M.A. The enzymatic measurement of nitrate and nitrite. Methods Mol. Biol. 1998, 100, 83–91. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
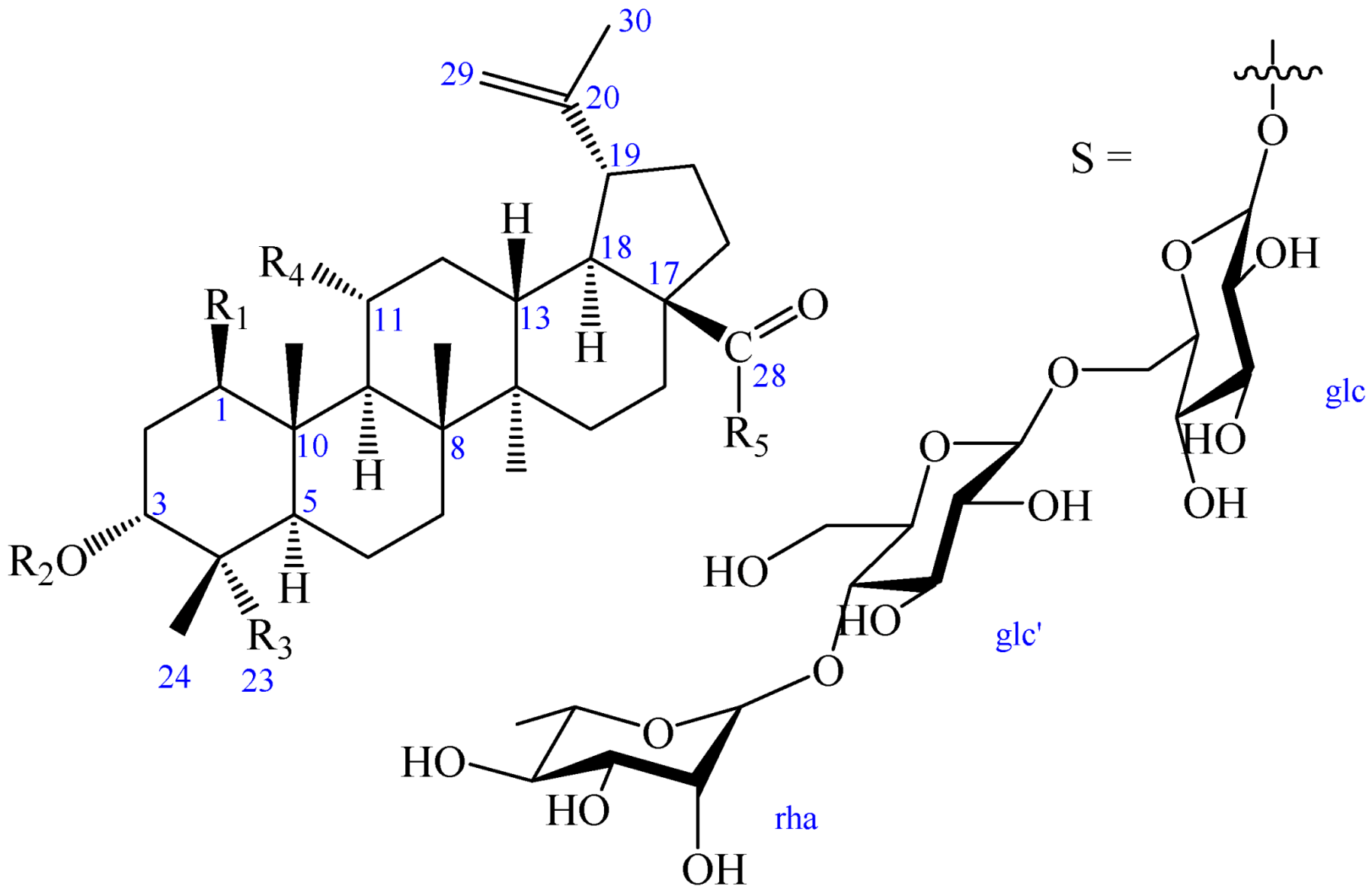
| R1 | R2 | R3 | R4 | R5 | |
| 1 | OH | OH | COOH | H | OH |
| 2 | OH | OH | COOH | H | S |
| 2a | OH | OH | COOH | H | OH |
| 3 | H | OH | COOH | OH | OH |
| 4 | H | -β-d-glc | CH3 | OH | S |
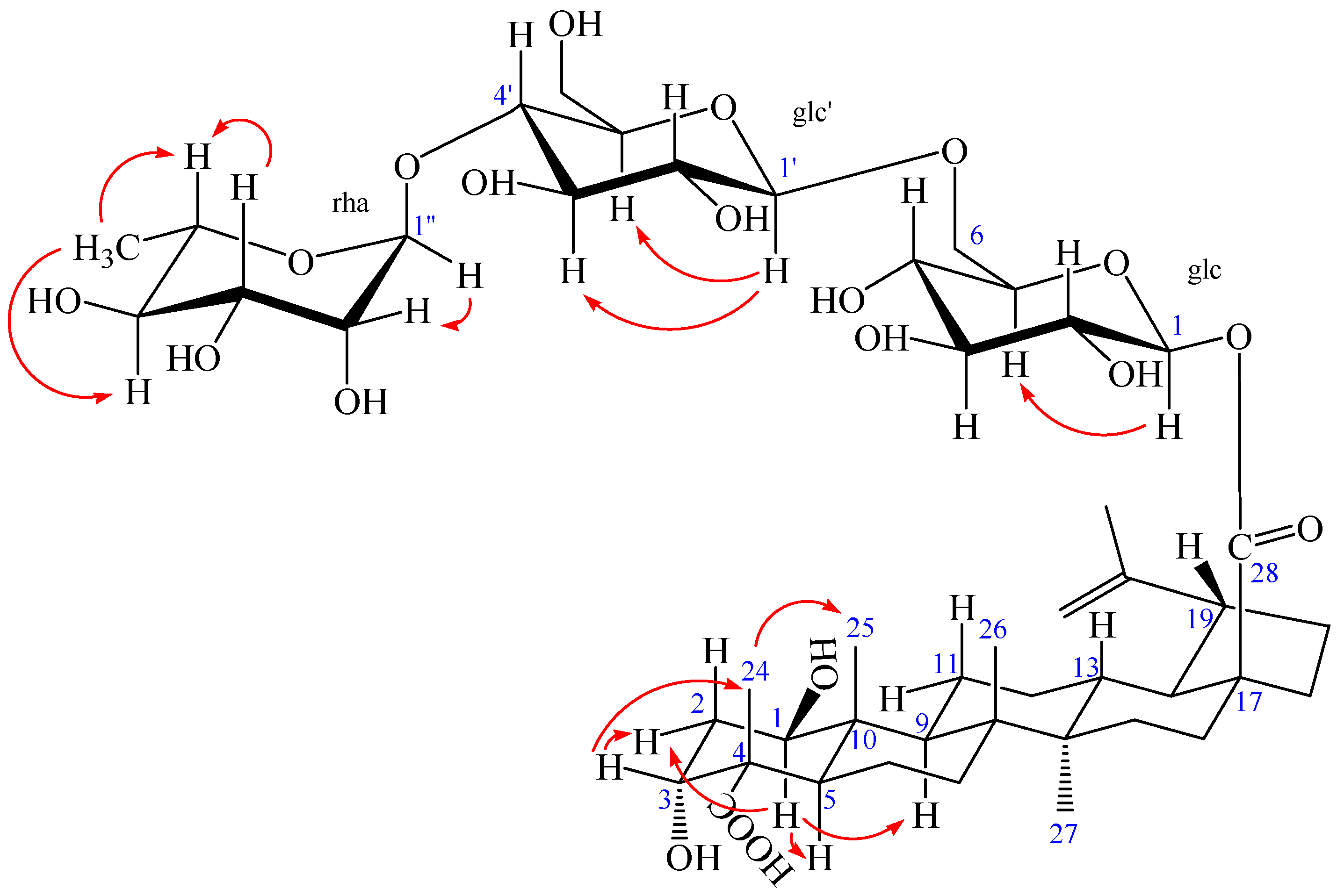

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 δC a,c | δH a,d [mult. (J in Hz)] | 2 δC b,c | δH b,d [mult. (J in Hz)] |
|---|---|---|---|---|
| Aglycone | ||||
| 1 | 75.46 | 3.84 (1H, dd, 8.16, 3.96) | 76.33 | 3.80 (1H, m) |
| 2 | 37.36 | 1.82 (1H, m); 1.87 (1H, m) | 36.79 | 1.72 (1H, m); 1.80 (1H, m) |
| 3 | 73.99 | 3.82 (1H, t, 2.40) | 74.14 | 3.67 (1H, m) |
| 4 | 52.27 | - | 52.24 | - |
| 5 | 45.25 | 1.95 (1H, m) | 46.11 | 1.87 (1H, m) |
| 6 | 22.09 | 1.35 (1H, m); 1.57 (1H, m) | 22.17 | 1.26 (1H, m); 1.58 (1H, m) |
| 7 | 35.19 | 1.31 (1H, m); 1.56 (1H, m) | 35.09 | 1.30 (1H, m); 1.55 (1H, m) |
| 8 | 42.76 | - | 42.96 | - |
| 9 | 52.98 | 1.72 (1H, dd, 10.08, 2.68) | 53.06 | 1.72 (1H, m) |
| 10 | 44.33 | - | 44.47 | - |
| 11 | 24.68 | 1.32 (1H, m); 2.43 (1H, brd, 9.80) | 24.82 | 1.36 (1H, m); 2.28 (1H, m) |
| 12 | 26.87 | 1.12 (1H, dd, 10.52, 3.76); 1.68 (1H, m) | 26.94 | 1.12 (1H, m); 1.68 (1H, m) |
| 13 | 39.14 | 2.34 (1H, td, 10.32, 3.0) | 39.13 | 2.27 (1H, m) |
| 14 | 43.72 | - | 43.82 | - |
| 15 | 30.75 | 1.20 (1H, m); 1.46 (1H, m) | 30.86 | 1.15 (1H, m); 1.54 (1H, m) |
| 16 | 33.15 | 1.48 (1H, m); 2.25 (1H, dt, 9.84, 2.8) | 32.95 | 1.44 (1H, m); 2.33 (1H, m) |
| 17 | 56.98 | - | 57.93 | - |
| 18 | 50.30 | 1.64 (1H, m) | 50.60 | 1.65 (1H, m) |
| 19 | 48.24 | 3.05 (1H, td, 8.76, 4.0) | 48.36 | 3.00 (1H, m) |
| 20 | 151.89 | - | 151.77 | - |
| 21 | 31.61 | 1.34 (1H, m); 1.90 (1H, m) | 31.55 | 1.37 (1H, m); 1.94 (1H, m) |
| 22 | 37.84 | 1.48 (1H, m); 1.92 (1H, m) | 37.68 | 1.48 (1H, m); 1.94 (1H, m) |
| 23 | 178.16 | - | 182.6 | - |
| 24 | 17.92 | 1.17 (3H, s) | 18.08 | 1.09 (3H, s) |
| 25 | 13.24 | 0.97 (3H, s) | 13.17 | 0.95 (3H, s) |
| 26 | 17.31 | 0.99 (3H, s) | 17.14 | 0.98 (3H, s) |
| 27 | 15.40 | 1.07 (3H, s) | 15.10 | 1.03 (3H, s) |
| 28 | 177.84 | - | 176.40 | - |
| 29 | 110.26 | 4.58 (1H, m); 4.72 (1H, d, 1.68) | 110.41 | 4.58 (1H, brs); 4.72 (1H, brs) |
| 30 | 19.72 | 1.71 (3H, s) | 19.49 | 1.70 (3H, s) |
| C-28 O-glc | ||||
| 1 | 95.26 | 5.45 (1H, d, 6.56) | ||
| 2 | 74.00 | 3.33 (1H, m) | ||
| 3 | 78.28 | 3.42 (1H, m) | ||
| 4 | 70.95 | 3.43 (1H, m) | ||
| 5 | 78.06 | 3.54 (1H, m) | ||
| 6 | 69.55 | 3.81 (1H, m); 4.11 (1H, dd, 9.48, 1.36) | ||
| glc′(1→6)glc | ||||
| 1′ | 104.56 | 4.37 (1H, d, 6.28) | ||
| 2′ | 75.32 | 3.23 (1H, m) | ||
| 3′ | 76.71 | 3.45 (1H, m) | ||
| 4′ | 79.51 | 3.53 (1H, m) | ||
| 5′ | 76.89 | 3.30 (1H, m) | ||
| 6′ | 61.90 | 3.65 (1H, m); 3.80 (1H, m) | ||
| rha(1→4)glc′ | ||||
| 1″ | 102.92 | 4.84 (1H, overlapped) | ||
| 2″ | 72.44 | 3.81 (1H, m) | ||
| 3″ | 72.16 | 3.62 (1H, m) | ||
| 4″ | 73.75 | 3.38 (1H, m) | ||
| 5″ | 70.64 | 3.95 (1H, m) | ||
| 6″ | 17.84 | 1.25 (3H, d, 4.96) |
| Compounds | Inhibition | Cell Viability |
|---|---|---|
| 1 | 38.6% in 80 μM | 86.44% |
| 3 | 40.4% in 80 μM | 83.90% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.-J.; Zou, Q.-P.; Wang, X.; Kim, K.-W.; Lu, M.-F.; Ko, S.-K.; Yook, C.-S.; Kim, Y.-C.; Liu, X.-Q. Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus. Molecules 2018, 23, 87. https://doi.org/10.3390/molecules23010087
Li X-J, Zou Q-P, Wang X, Kim K-W, Lu M-F, Ko S-K, Yook C-S, Kim Y-C, Liu X-Q. Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus. Molecules. 2018; 23(1):87. https://doi.org/10.3390/molecules23010087
Chicago/Turabian StyleLi, Xiao-Jun, Qin-Peng Zou, Xiang Wang, Kwan-Woo Kim, Mao-Fang Lu, Sung-Kwon Ko, Chang-Soo Yook, Youn-Chul Kim, and Xiang-Qian Liu. 2018. "Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus" Molecules 23, no. 1: 87. https://doi.org/10.3390/molecules23010087
APA StyleLi, X.-J., Zou, Q.-P., Wang, X., Kim, K.-W., Lu, M.-F., Ko, S.-K., Yook, C.-S., Kim, Y.-C., & Liu, X.-Q. (2018). Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus. Molecules, 23(1), 87. https://doi.org/10.3390/molecules23010087