Molecular Basis for Converting (2S)-Methylsuccinyl-CoA Dehydrogenase into an Oxidase

 ,
,  and
and
Abstract
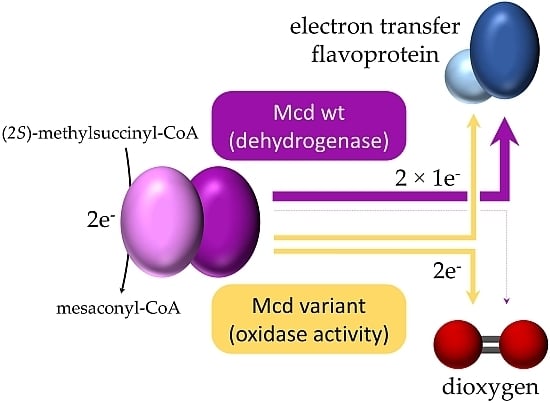
:
1. Introduction
2. Results
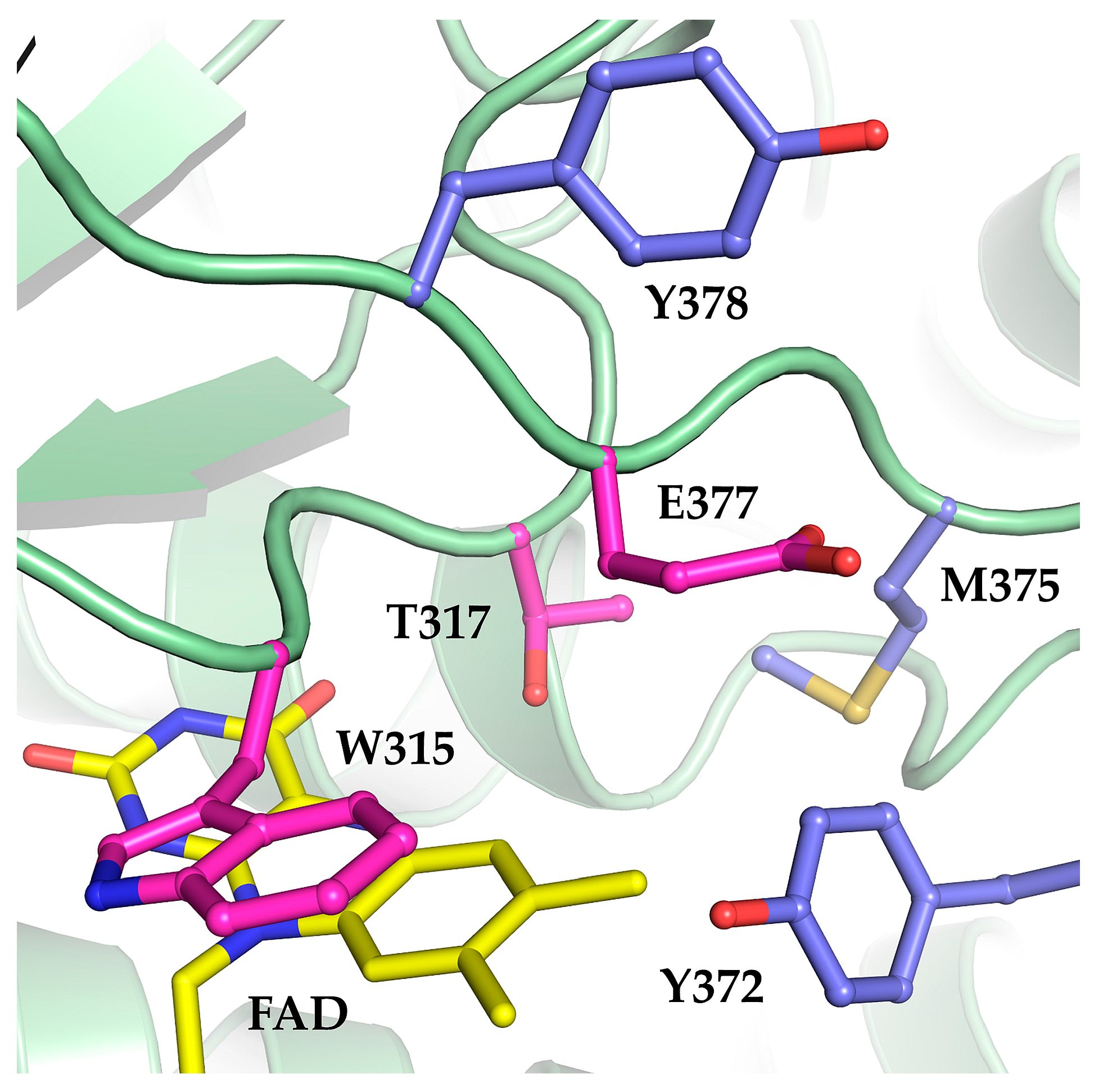
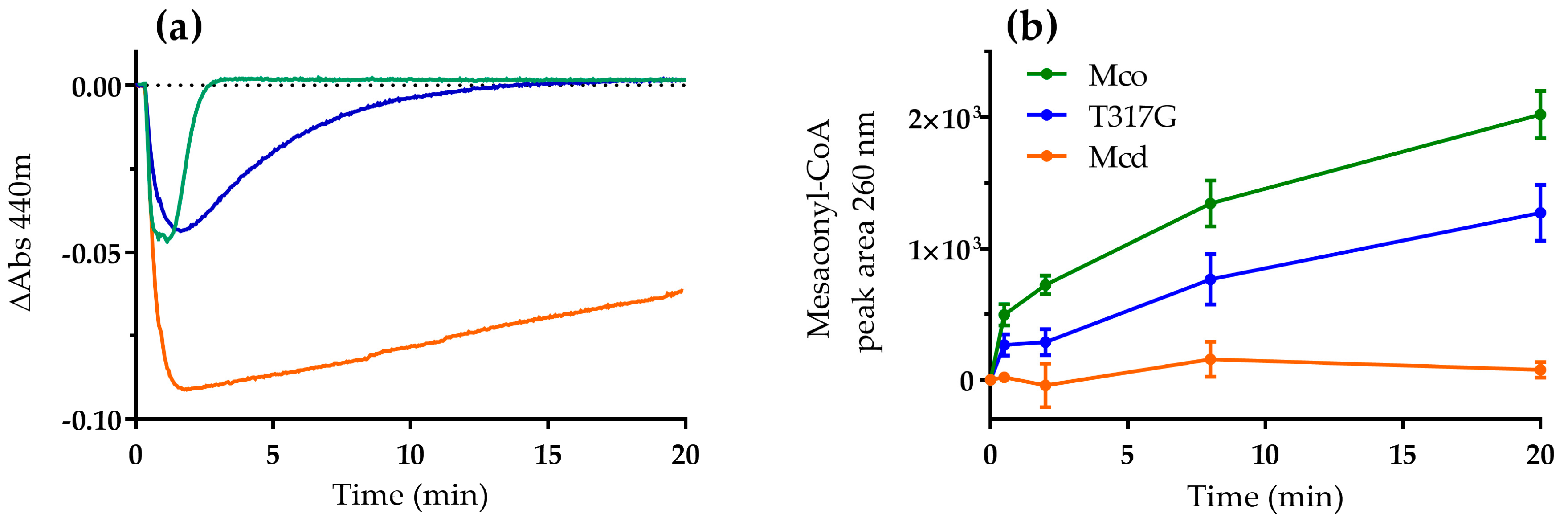
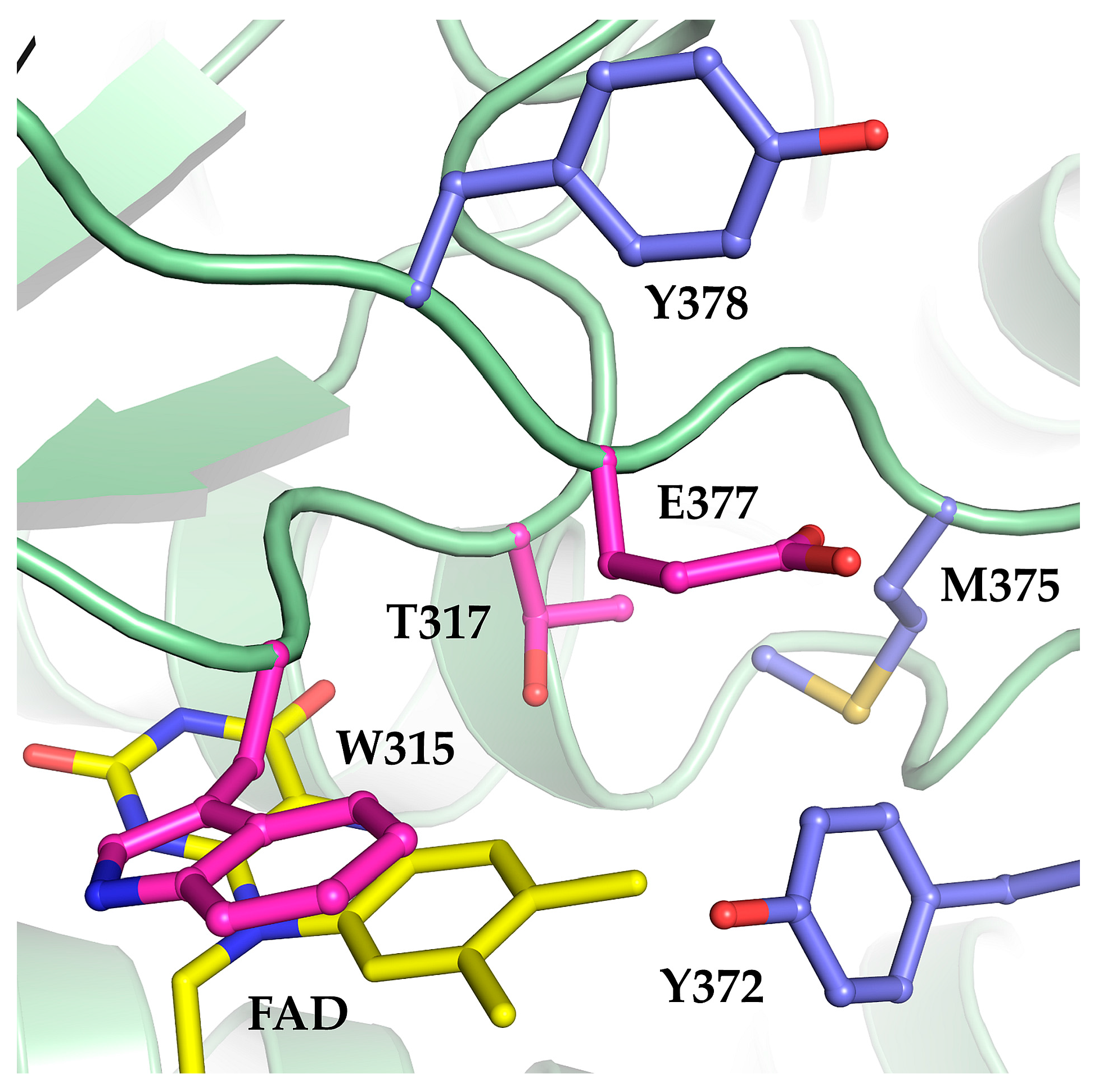
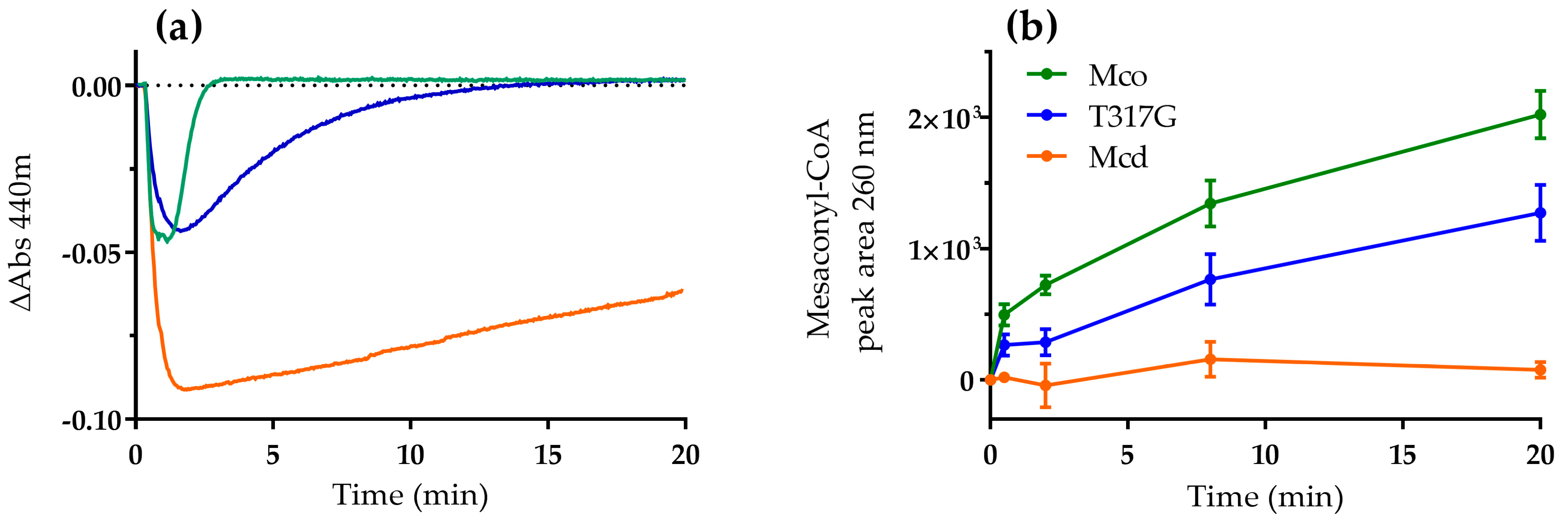
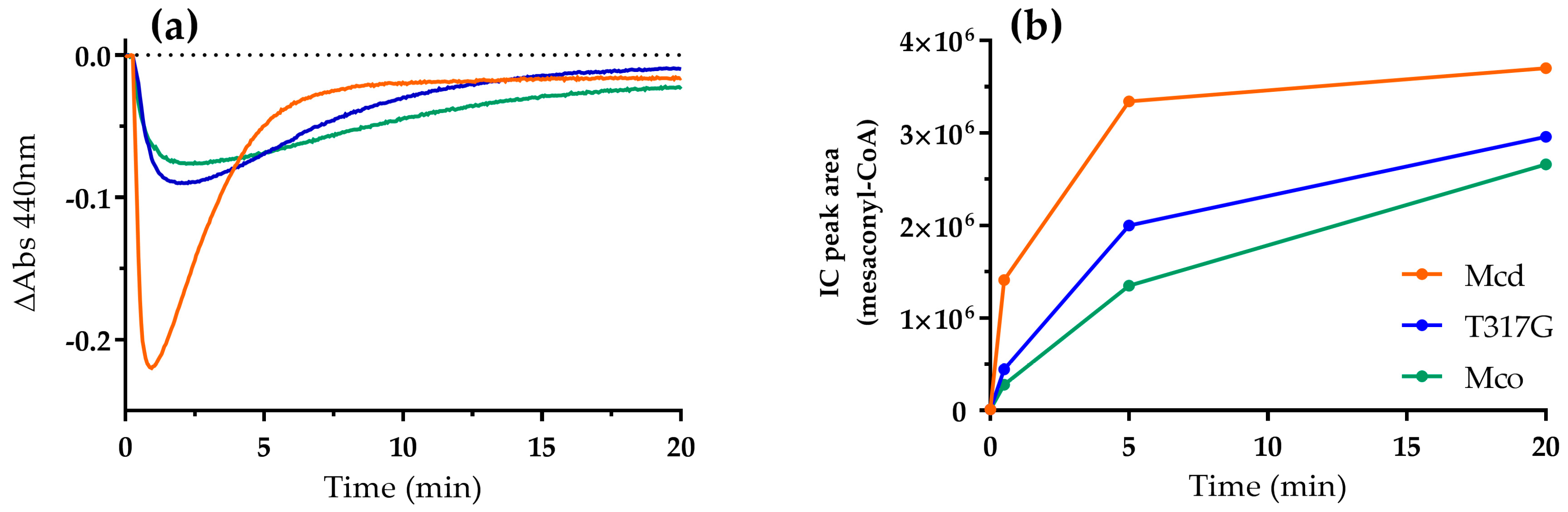
2.1. The Oxidase Activity of Engineered Mco Cannot Be Improved by Increasing FAD’s Solvent Accessibility
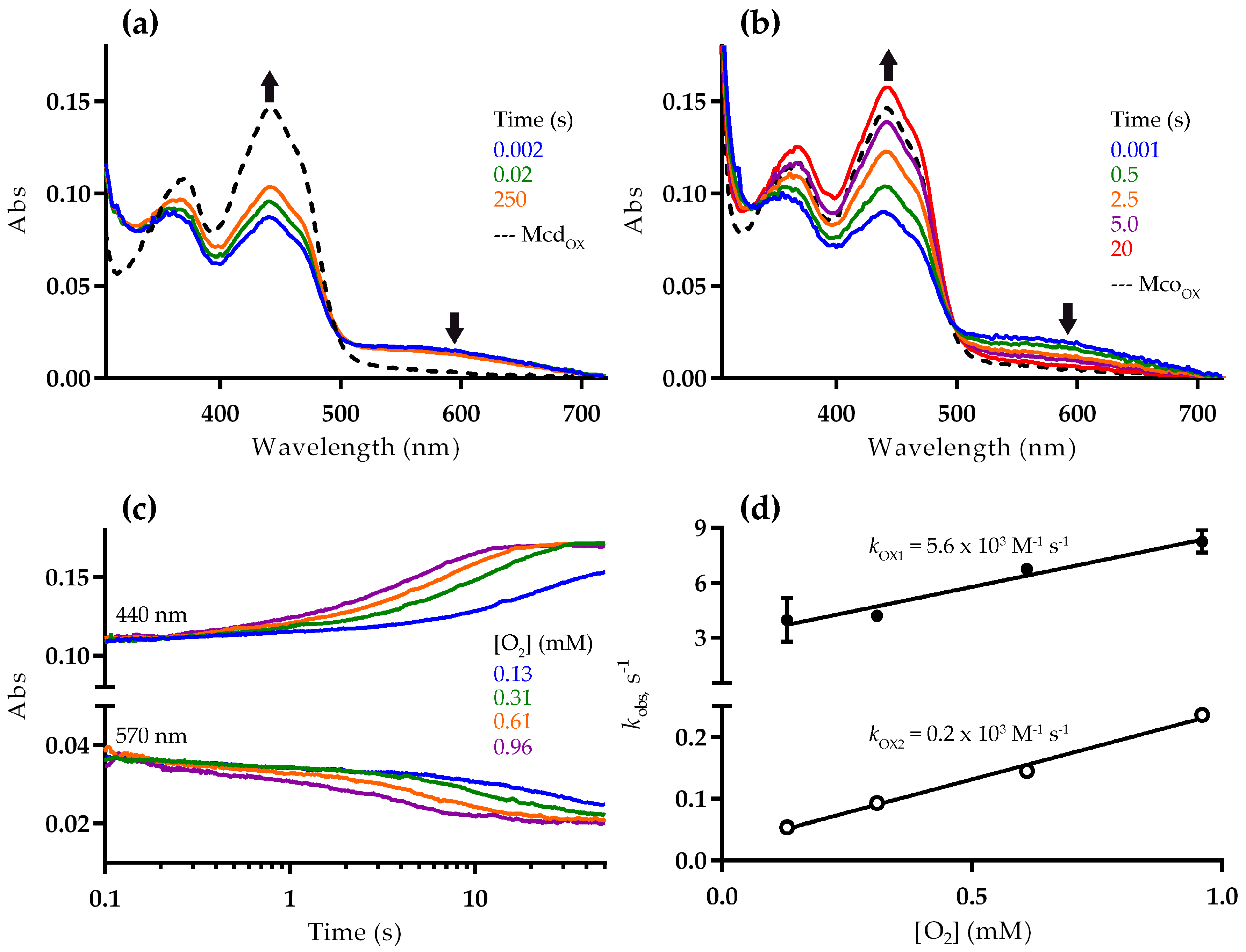
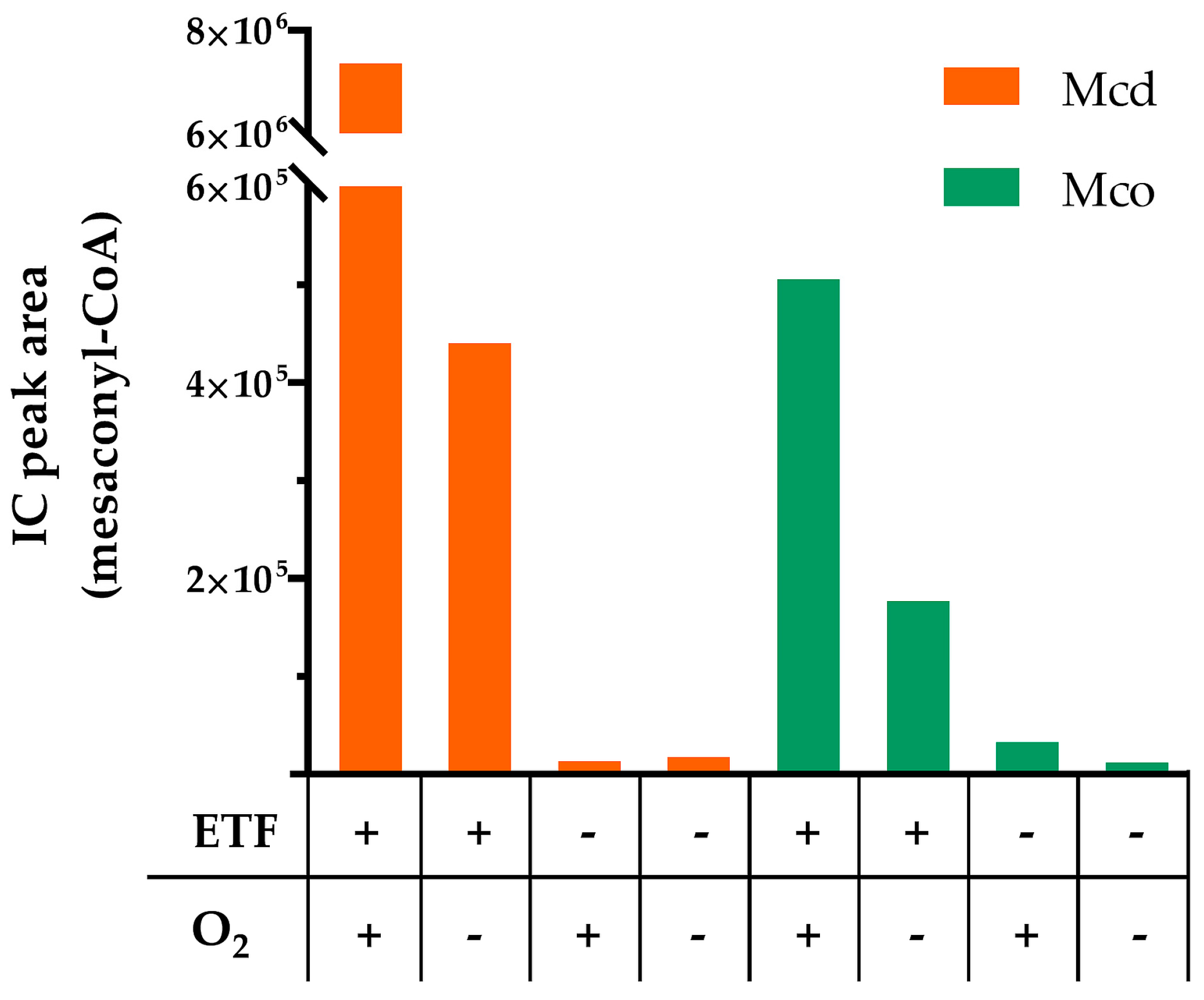
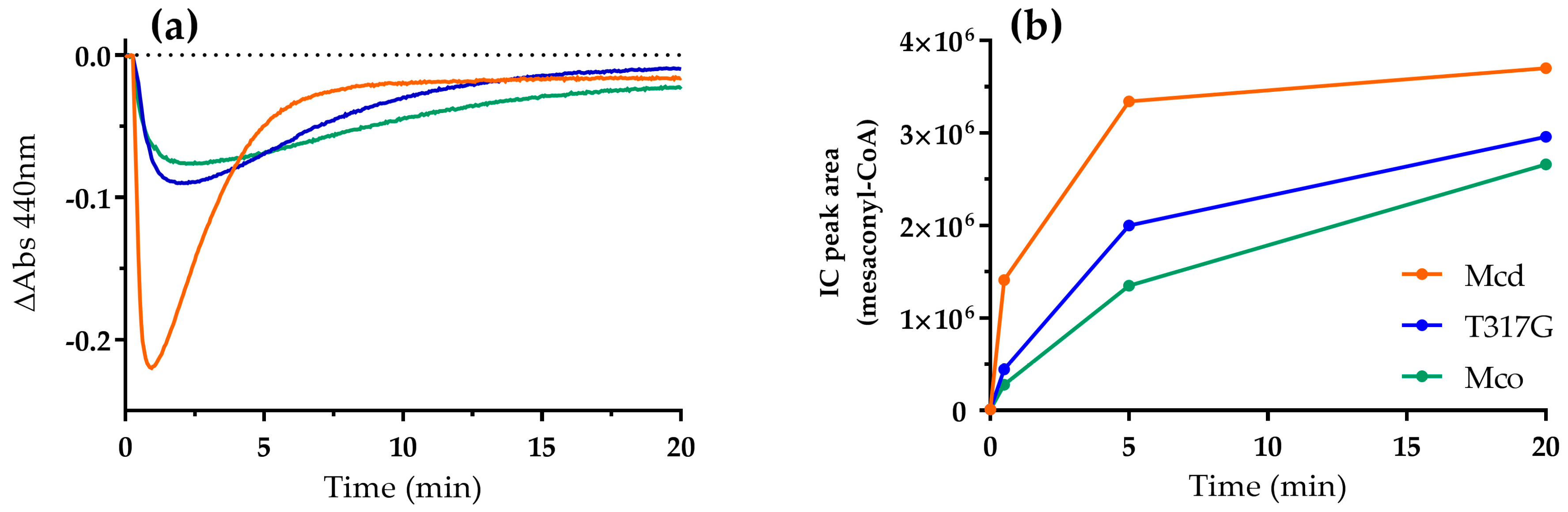
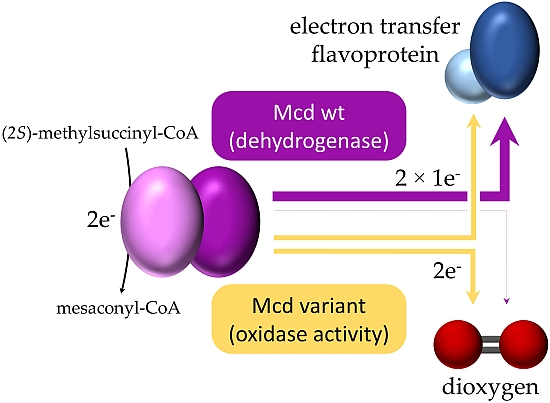
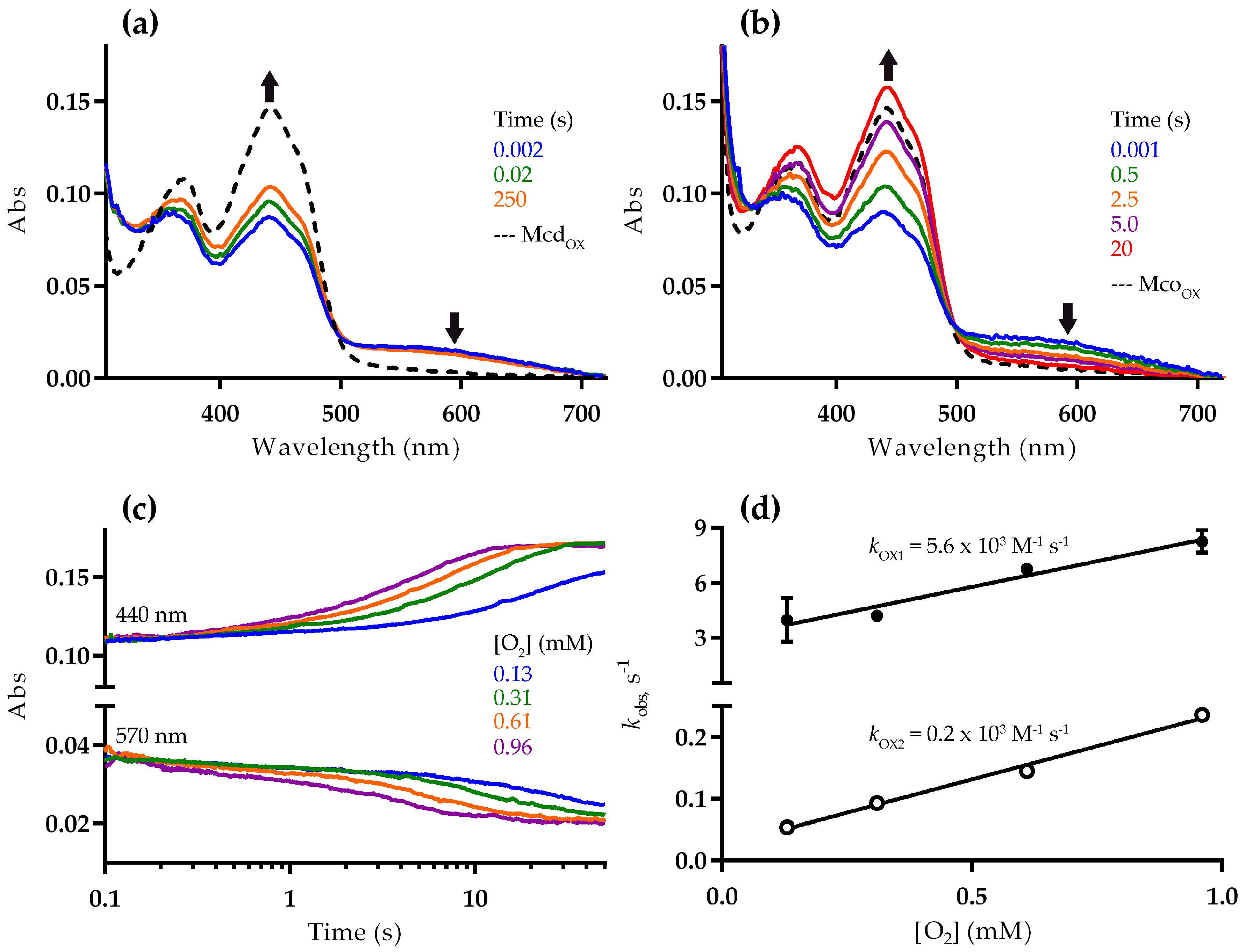
2.2. Mco Shows an Improved Oxidative Half-Reaction with Dioxygen
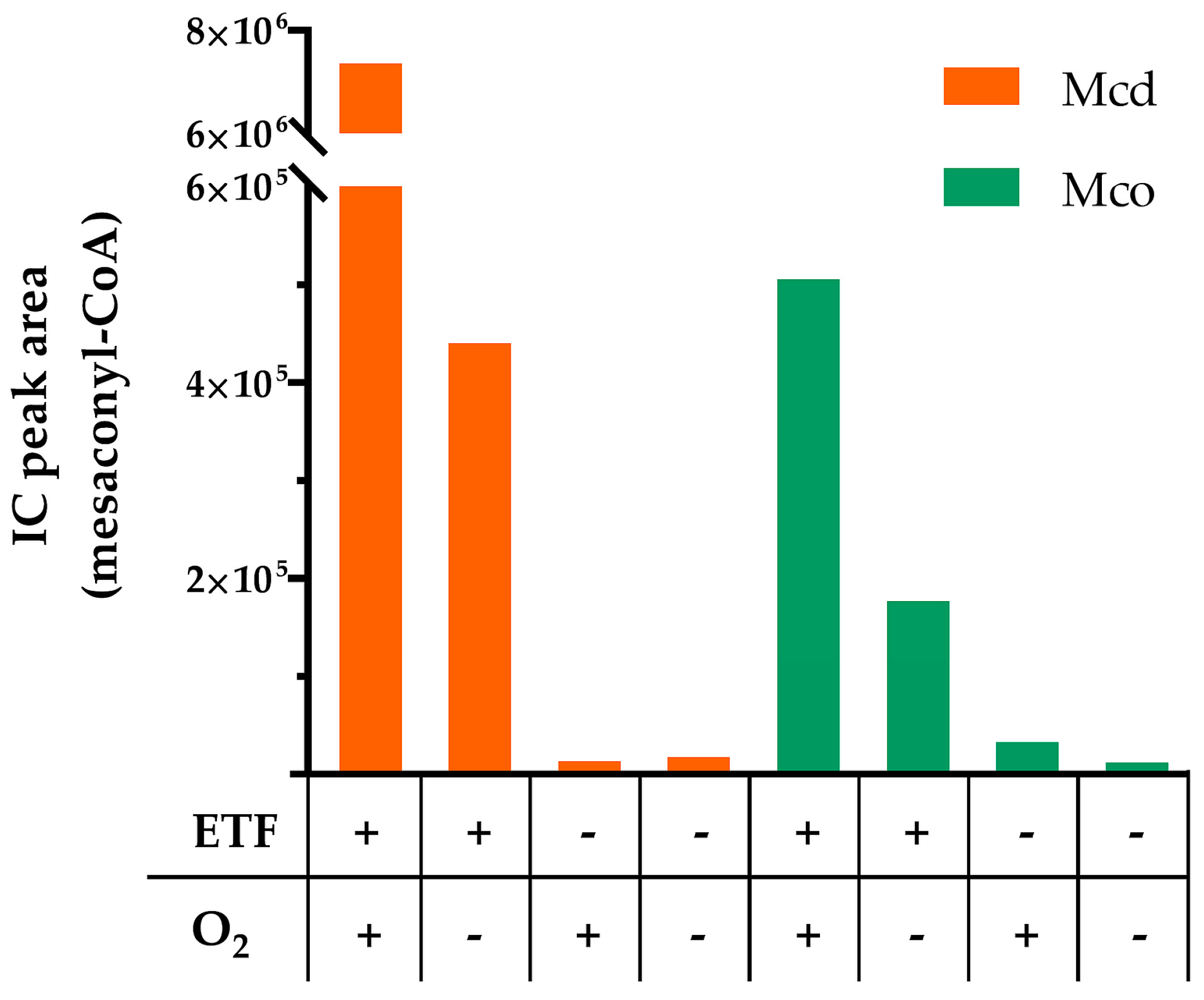
2.3. Mco Still Interacts with ETF, but at a Decreased Rate
3. Discussion
4. Materials and Methods
4.1. Cloning
4.2. Heterologous Protein Production and Purification
4.2.1. Mcd Variants
4.2.2. ETF
4.3. Chemical Synthesis of Methylsuccinyl-CoA
4.4. Enzyme Assays
4.4.1. LC and LC-MS-Based Assays
4.4.2. Spectrophotometric Assays
4.4.3. Spectrophotometric Assay to Determine FAD Released after Mco Reduction
4.4.4. Spectrophotometric Stopped-Flow Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frerman, F.E. Acyl-CoA dehydrogenases, electron transfer flavoprotein and electron transfer flavoprotein dehydrogenase. Biochem. Soc. Trans. 1988, 16, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L.; Beinert, H. On the mechanism of dehydrogenation of fatty acyl derivatives of coenzyme A. II. The electron-transferring flavoprotein. J. Biol. Chem. 1956, 218, 717–731. [Google Scholar] [PubMed]
- Ruzicka, F.J.; Beinert, H. A new iron-sulfur flavoprotein of the respiratory chain. A component of the fatty acid beta oxidation pathway. J. Biol. Chem. 1977, 252, 8440–8445. [Google Scholar] [PubMed]
- Thorpe, C.; Kim, J.J. Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 1995, 9, 718–725. [Google Scholar] [PubMed]
- Ikeda, Y.; Hine, D.G.; Okamura-Ikeda, K.; Tanaka, K. Mechanism of action of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases. Direct evidence for carbanion formation as an intermediate step using enzyme-catalyzed C-2 proton/deuteron exchange in the absence of C-3 exchange. J. Biol. Chem. 1985, 260, 1326–1337. [Google Scholar] [PubMed]
- Ghisla, S.; Massey, V.; Lhoste, J.M.; Mayhew, S.G. Fluorescence and optical characteristics of reduced flavines and flavoproteins. Biochemistry 1974, 13, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Toogood, H.S.; Leys, D.; Scrutton, N.S. Dynamics driving function: New insights from electron transferring flavoproteins and partner complexes. FEBS J. 2007, 274, 5481–5504. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Thorpe, C. Reactivity of medium-chain acyl-CoA dehydrogenase toward molecular oxygen. Biochemistry 1991, 30, 7895–7901. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [Google Scholar] [PubMed]
- Kim, J.J.; Miura, R. Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur. J. Biochem. 2004, 271, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Mattevi, A. To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci. 2006, 31, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Schwander, T.; Schada von Borzyskowski, L.; Burgener, S.; Cortina, N.S.; Erb, T.J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 2016, 354, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Fuchs, G.; Alber, B.E. (2S)-Methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-CoA pathway for acetyl-CoA assimilation. Mol. Microbiol. 2009, 73, 992–1008. [Google Scholar] [CrossRef] [PubMed]
- DuPlessis, E.R.; Pellett, J.; Stankovich, M.T.; Thorpe, C. Oxidase activity of the acyl-CoA dehydrogenases. Biochemistry 1998, 37, 10469–10477. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Retey, J.; Fuchs, G.; Alber, B.E. Ethylmalonyl-CoA mutase from Rhodobacter sphaeroides defines a new subclade of coenzyme B12-dependent acyl-CoA mutases. J. Biol. Chem. 2008, 283, 32283–32293. [Google Scholar] [CrossRef] [PubMed]
- Ghisla, S.; Thorpe, C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem. 2004, 271, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.G.; Lau, S.M.; Powell, P.J.; Thorpe, C. Reductive half-reaction in medium-chain acyl-CoA dehydrogenase: modulation of internal equilibrium by carboxymethylation of a specific methionine residue. Biochemistry 1992, 31, 8523–8529. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.C.; Massey, V. Green butyryl-coenzyme A dehydrogenase. An enzyme-acyl-coenzyme A complex. Biochem. J. 1971, 125, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.V.; Gomes, C.M. Mechanism of superoxide and hydrogen peroxide generation by human electron-transfer flavoprotein and pathological variants. Free Radic. Biol. Med. 2012, 53, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Wang, M.; Paschke, R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. USA 1993, 90, 7523–7527. [Google Scholar] [CrossRef] [PubMed]
- Toogood, H.S.; van Thiel, A.; Scrutton, N.S.; Leys, D. Stabilization of non-productive conformations underpins rapid electron transfer to electron-transferring flavoprotein. J. Biol. Chem. 2005, 280, 30361–30366. [Google Scholar] [CrossRef] [PubMed]
- Gadda, G. Oxygen activation in flavoprotein oxidases: The importance of being positive. Biochemistry 2012, 51, 2662–2669. [Google Scholar] [CrossRef] [PubMed]
- Gygli, G.; Lucas, M.F.; Guallar, V.; van Berkel, W.J.H. The ins and outs of vanillyl alcohol oxidase: Identification of ligand migration paths. PLoS Comput. Biol. 2017, 13, e1005787. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Riley, C.; Chenprakhon, P.; Thotsaporn, K.; Winter, R.T.; Alfieri, A.; Forneris, F.; van Berkel, W.J.; Chaiyen, P.; Fraaije, M.W.; et al. Multiple pathways guide oxygen diffusion into flavoenzyme active sites. Proc. Natl. Acad. Sci. USA 2009, 106, 10603–10608. [Google Scholar] [CrossRef] [PubMed]
- Leferink, N.G.; Fraaije, M.W.; Joosten, H.J.; Schaap, P.J.; Mattevi, A.; van Berkel, W.J. Identification of a gatekeeper residue that prevents dehydrogenases from acting as oxidases. J. Biol. Chem. 2009, 284, 4392–4397. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.; Pedersen, L.; Arent, S.; Henriksen, A. Controlling electron transfer in Acyl-CoA oxidases and dehydrogenases: A structural view. J. Biol. Chem. 2006, 281, 31012–31020. [Google Scholar] [CrossRef] [PubMed]
- Kass, I.J.; Sampson, N.S. Evaluation of the role of His447 in the reaction catalyzed by cholesterol oxidase. Biochemistry 1998, 37, 17990–18000. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.P.; Klinman, J.P. Catalysis of electron transfer during activation of O2 by the flavoprotein glucose oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Bruckner, R.C.; Jorns, M.S. Identification of the oxygen activation site in monomeric sarcosine oxidase: Role of Lys265 in catalysis. Biochemistry 2008, 47, 9124–9135. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; De Bellis, L.; Ciurli, A.; Kondo, M.; Hayashi, M.; Nishimura, M. A novel acyl-CoA oxidase that can oxidize short-chain acyl-CoA in plant peroxisomes. J. Biol. Chem. 1999, 274, 12715–12721. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Peter, D.; Vögeli, B.; Cortina, N.; Erb, T. A Chemo-Enzymatic Road Map to the Synthesis of CoA Esters. Molecules 2016, 21, 517. [Google Scholar] [CrossRef] [PubMed]
- Macheroux, P. UV-Visible Spectroscopy as a Tool to Study Flavoproteins. In Flavoprotein Protocols, 1 ed.; Chapman, S.K., Reid, G.A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 1999; Volume 131, pp. 1–7. [Google Scholar]
- Van Beek, H.L.; Romero, E.; Fraaije, M.W. Engineering Cyclohexanone Monooxygenase for the Production of Methyl Propanoate. ACS Chem. Biol. 2017, 12, 291–299. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Backbone | Relevant Features | Source |
|---|---|---|---|
| pTE22 | pET16b | mcd from R. sphaeroides, N-terminal His10-tag, T7 promoter, amp resistance marker | [13] |
| pTE392 | pCDFDuet-1 | etfA and etfB from R. sphaeroides, etfA N-terminal His6-tag, T7 promoter, streptomycin resistance marker | This work |
| pTE801 | pET16b | Mcd from R. sphaeroides (T317G), N-terminal His10-tag,T7 promoter, amp resistance marker | [12] |
| pTE813 | pET16b | mcd from R. sphaeroides (W315F, T317G, E377N), “Mco”; N-terminal His10-tag,T7 promoter, amp resistance marker | [12] |
| Template | Mutation | Fw Primer 5′ to 3′ | Rv Primer 5′ to 3′ | Plasmid |
|---|---|---|---|---|
| pTE813 | Y372I | CGAGATCGAGGTGCTGGGCAT CCGCGGCATGAAGAACTATG | CATAGTTCTTCATGCCGCGGA TGCCCAGCACCTCGATCTCG | pTE1218 |
| pTE813 | M375S | CTGGGCTACCGCGGCTCGAAG AACTATGAGATC | GATCTCATAGTTCTTCGAGCC GCGGTAGCCCAG | pTE1219 |
| pTE813 | M378G | GCATGAAGAACGGCGAGATCG GCTTC | GAAGCCGATCTCGCCGTTCTT CATGC | pTE1220 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgener, S.; Schwander, T.; Romero, E.; Fraaije, M.W.; Erb, T.J. Molecular Basis for Converting (2S)-Methylsuccinyl-CoA Dehydrogenase into an Oxidase. Molecules 2018, 23, 68. https://doi.org/10.3390/molecules23010068
Burgener S, Schwander T, Romero E, Fraaije MW, Erb TJ. Molecular Basis for Converting (2S)-Methylsuccinyl-CoA Dehydrogenase into an Oxidase. Molecules. 2018; 23(1):68. https://doi.org/10.3390/molecules23010068
Chicago/Turabian StyleBurgener, Simon, Thomas Schwander, Elvira Romero, Marco W. Fraaije, and Tobias J. Erb. 2018. "Molecular Basis for Converting (2S)-Methylsuccinyl-CoA Dehydrogenase into an Oxidase" Molecules 23, no. 1: 68. https://doi.org/10.3390/molecules23010068