Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea



Abstract
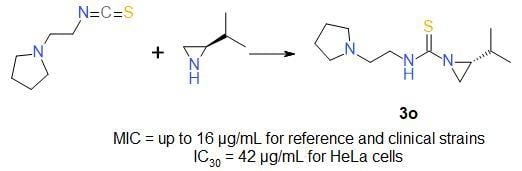
:
1. Introduction
2. Results and Discussion
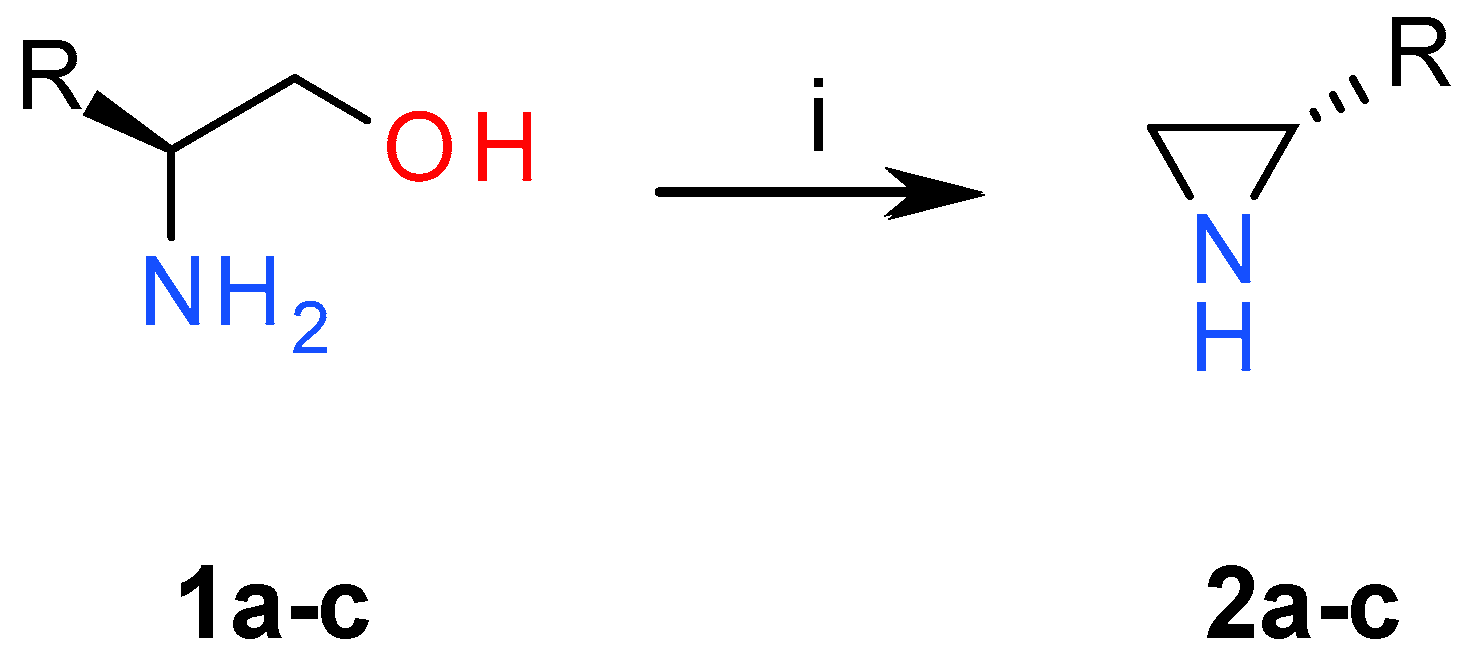
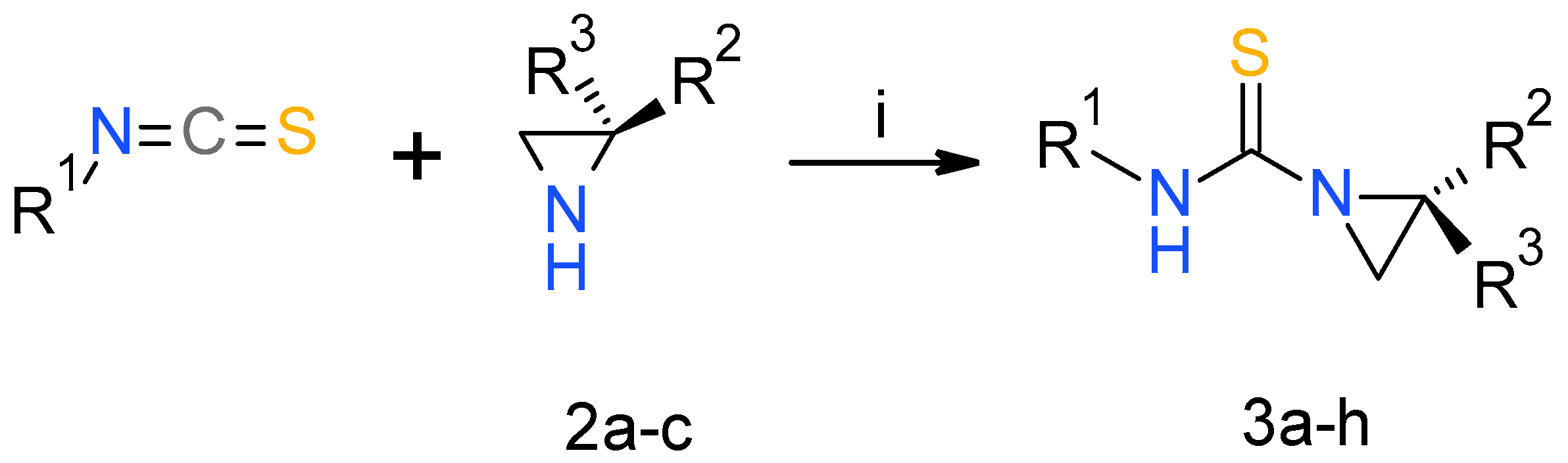
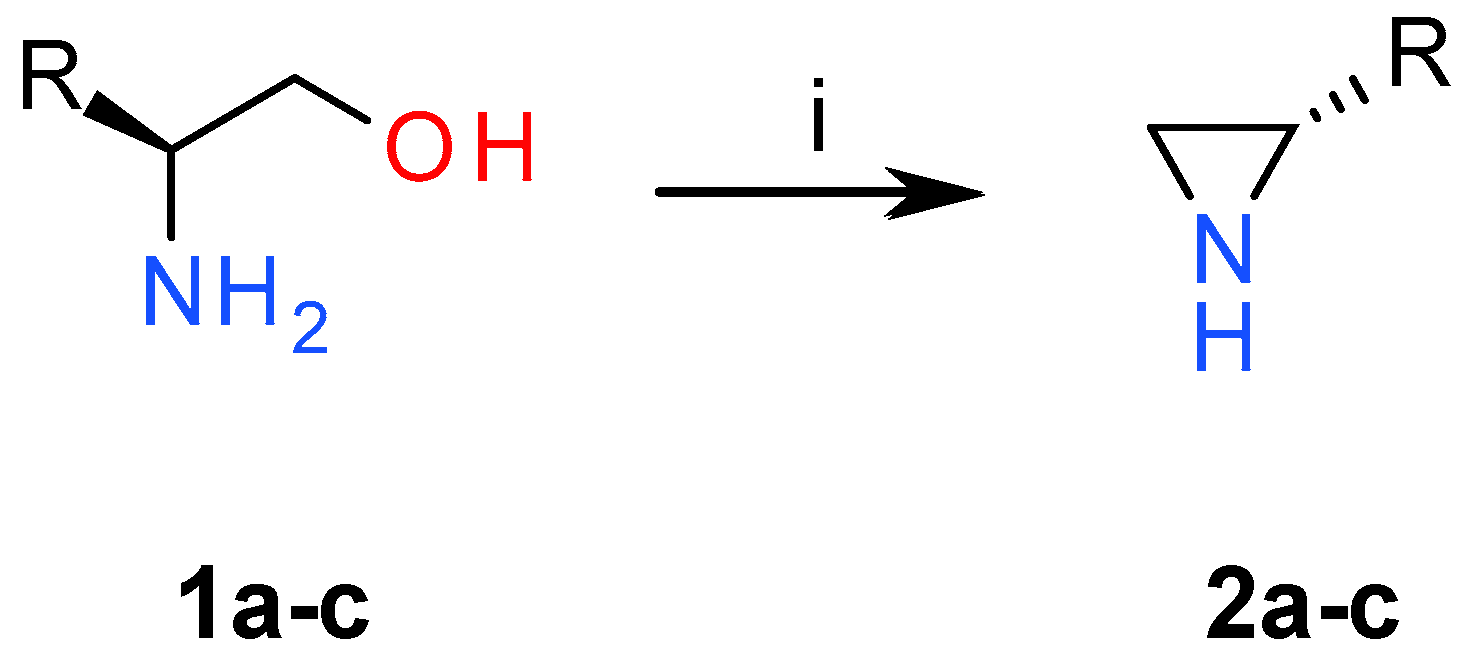
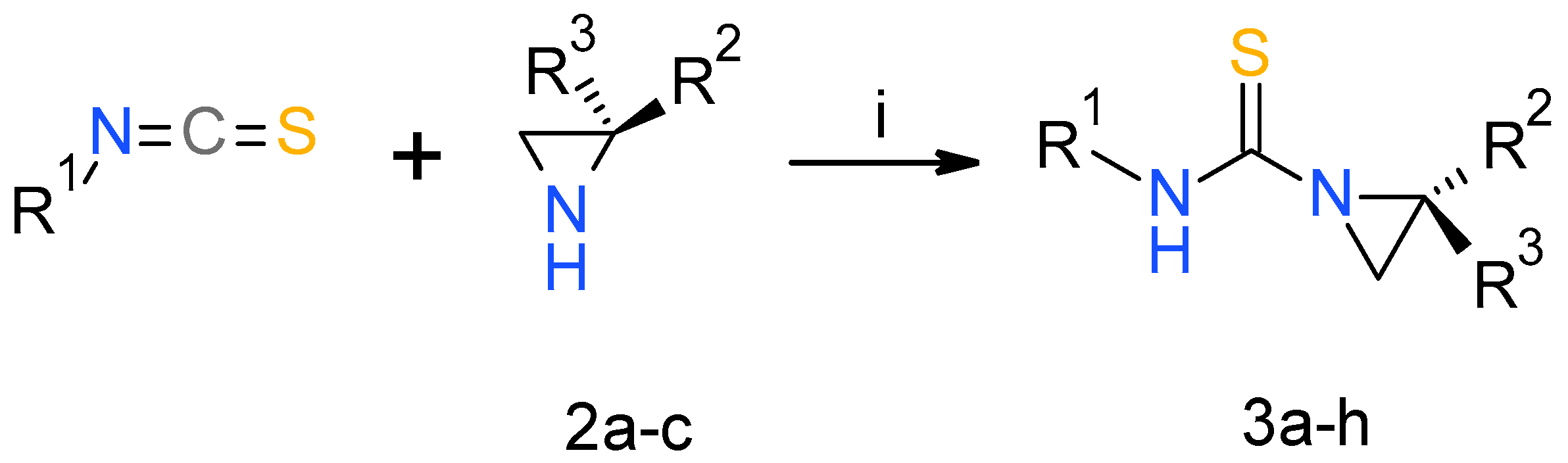
2.1. Chemistry
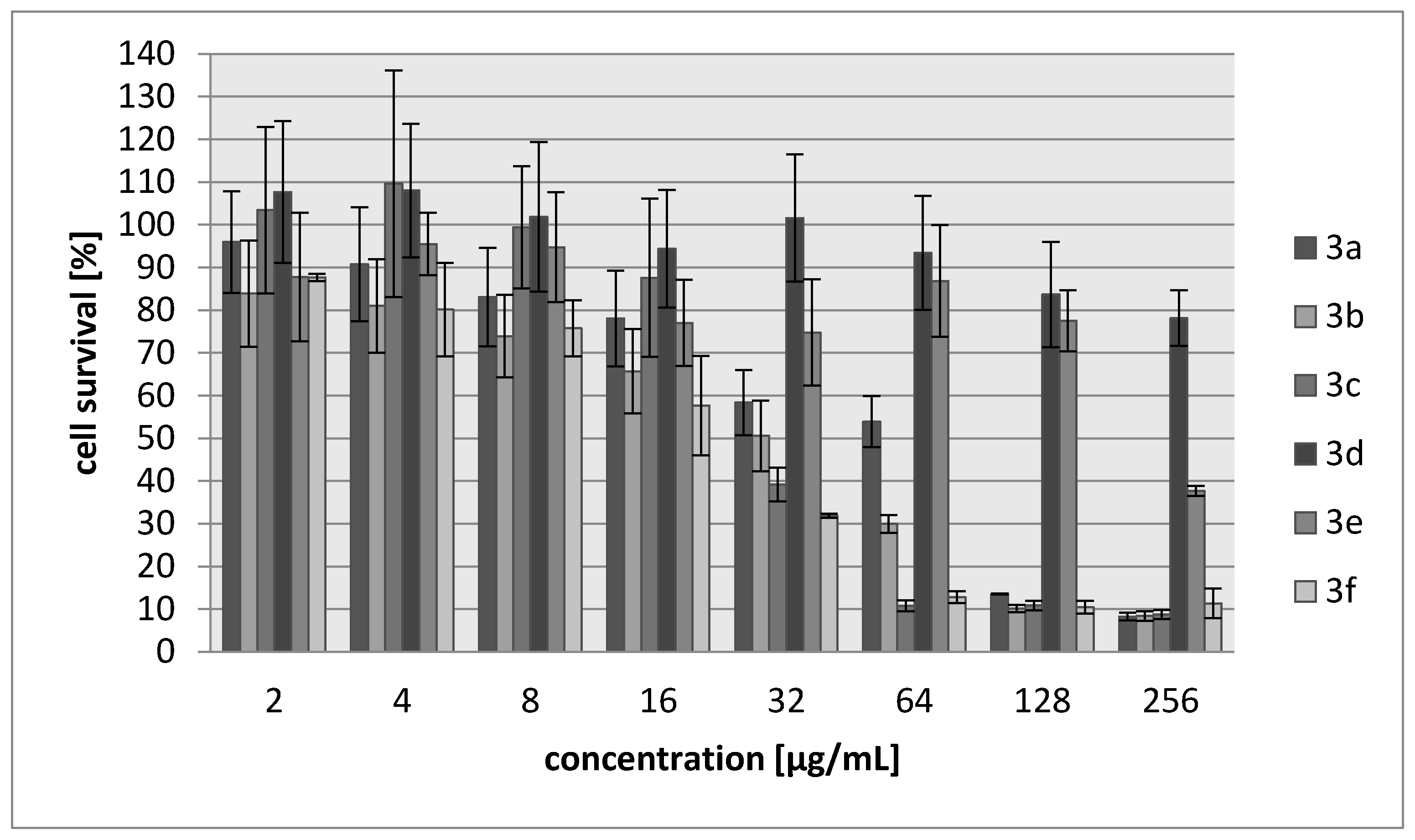
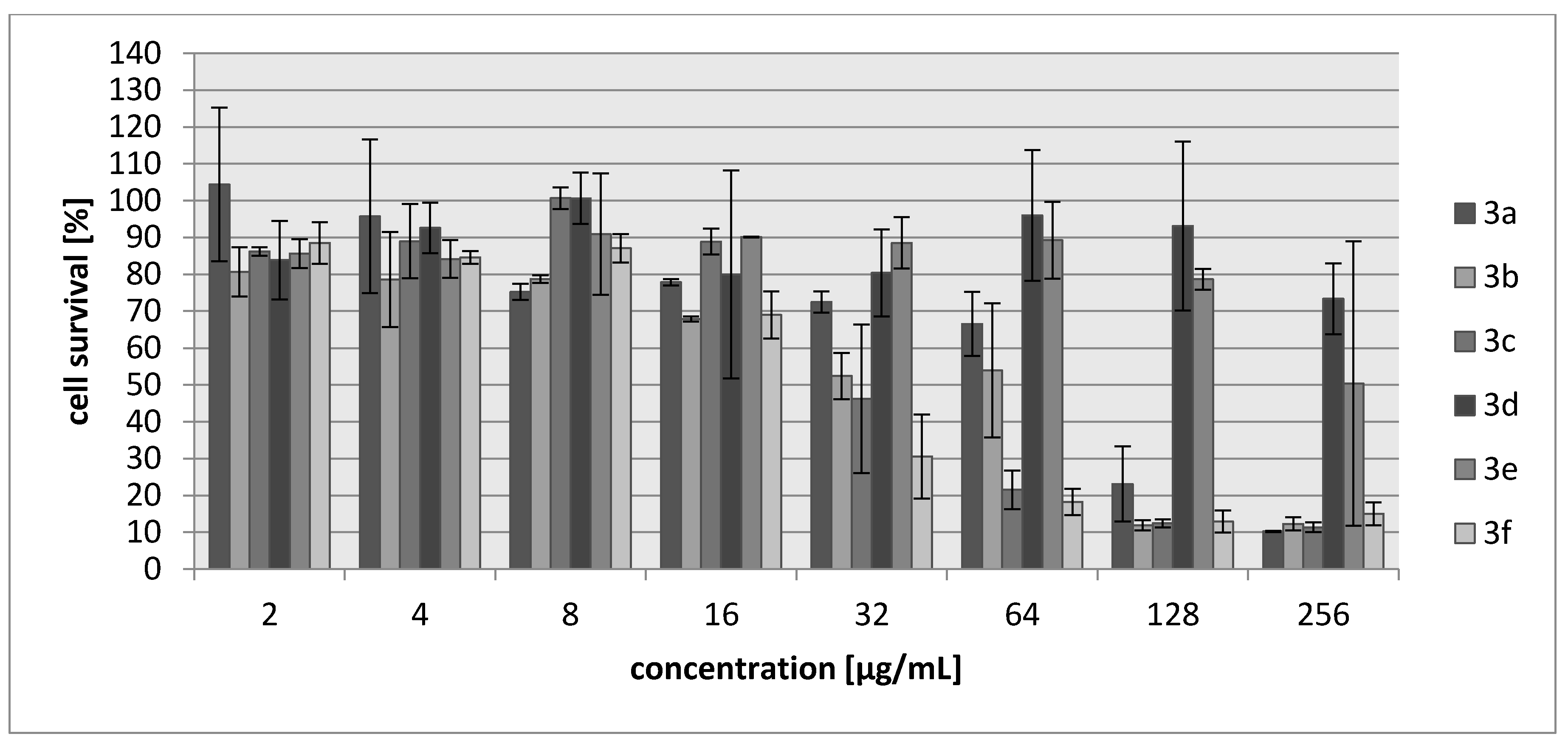
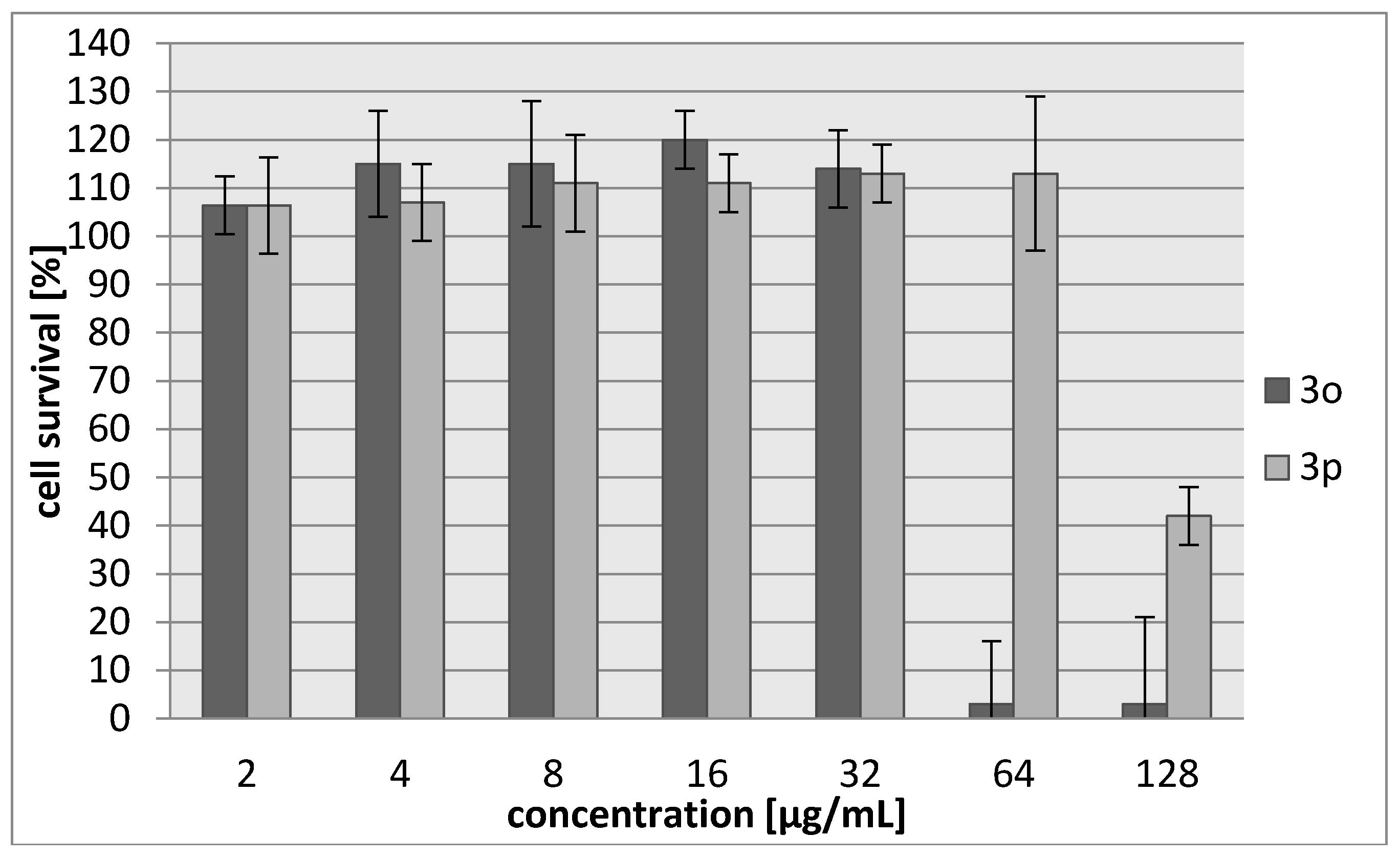
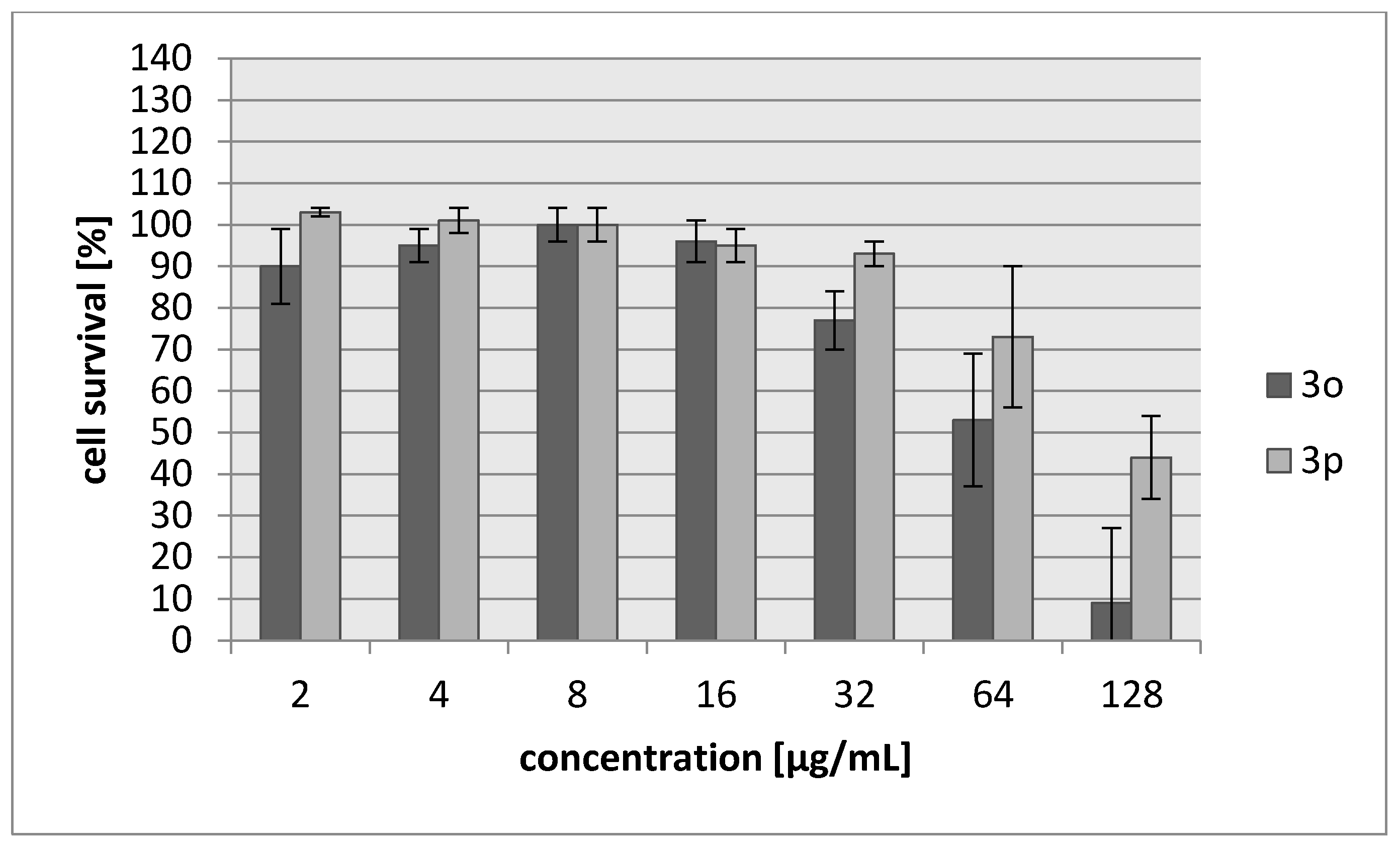
2.2. Biology
3. Experimental Section
3.1. Chemistry
3.2. General Procedure for Synthesis of Urea and Thiourea Derivatives 3
3.3. Biology
3.3.1. Antibacterial Assay
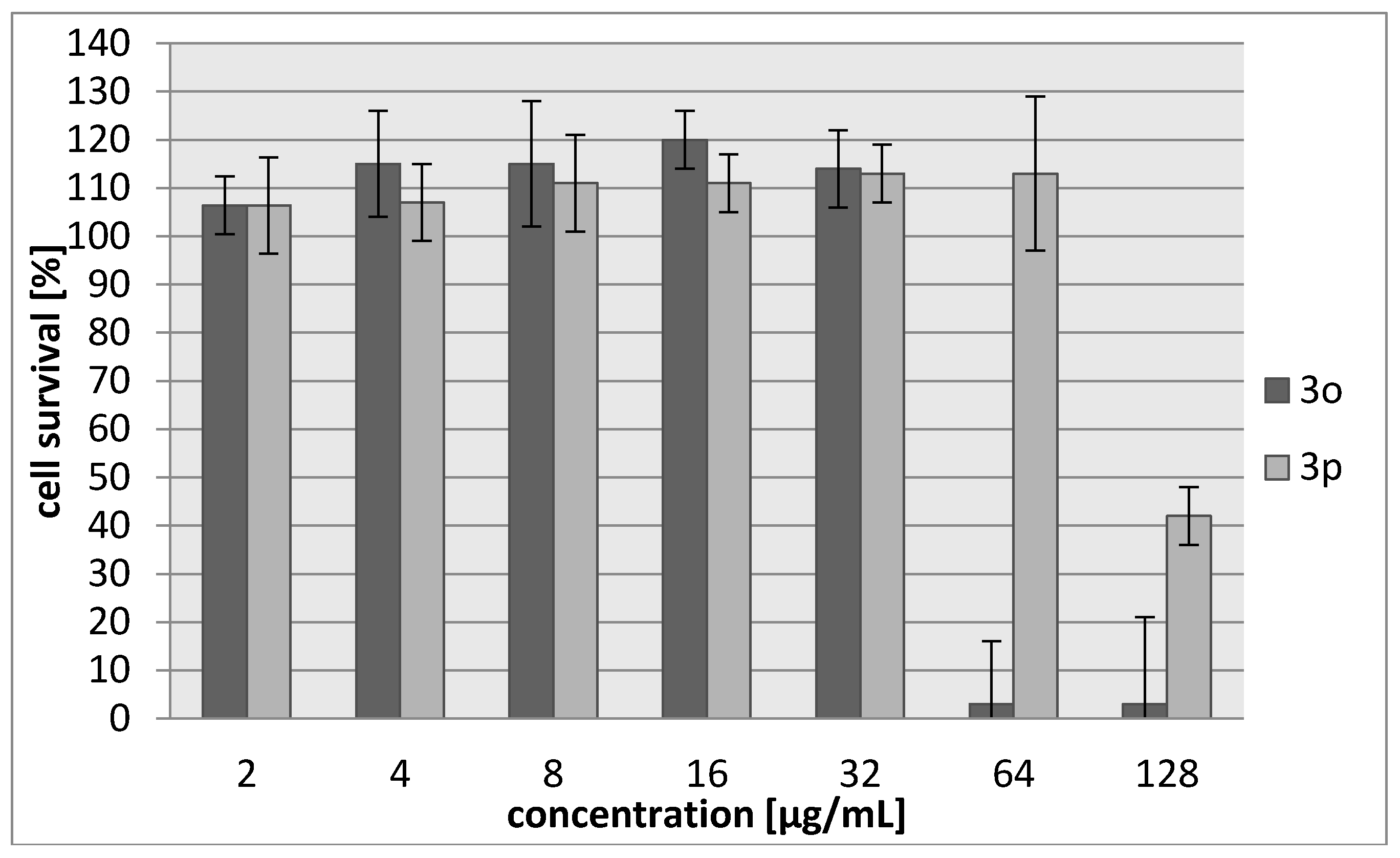
3.3.2. Cytotoxicity Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yudin, A.K. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006; p. 494. [Google Scholar]
- Morieux, P.; Stables, J.P.; Kohn, H. Synthesis and anticonvulsant activities of N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamide derivatives. Bioorg. Med. Chem. 2006, 16, 8968–8975. [Google Scholar] [CrossRef] [PubMed]
- Cimarelli, C.; Fratoni, D.; Palmieri, G. A Convenient Synthesis of New Diamine, Amino Alcohol and Aminophosphines Chiral Auxiliaries Based on Limonene Oxide. Tetrahedron Asymmetry 2009, 20, 2234–2239. [Google Scholar] [CrossRef]
- Leśniak, S.; Pieczonka, A.M.; Jarzyński, S.; Justyna, K.; Rachwalski, M. ChemInform Abstract: Synthesis and Evaluation of the Catalytic Properties of Semicarbazides Derived from N-Triphenylmethyl-aziridine-2-carbohydrazides. Tetrahedron Asymmetry 2013, 24, 1341–1344. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Leśniak, S.; Rachwalski, M. Direct asymmetric aldol condensation catalyzed by aziridine semicarbazide zinc(II) complexes. Tetrahedron Lett. 2014, 55, 2373–2375. [Google Scholar] [CrossRef]
- Jarzyński, S.; Leśniak, S.; Pieczonka, A.M.; Rachwalski, M. N-Trityl-aziridinyl alcohols as highly efficient chiral catalysts in asymmetric additions of organozinc species to aldehydes. Tetrahedron Asymmetry 2015, 26, 35–40. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Leśniak, S.; Jarzyński, S.; Rachwalski, M. Aziridinylethers as highly enantioselective ligands for the asymmetric addition of organozinc species to carbonyl compounds. Tetrahedron Asymmetry 2015, 26, 148–151. [Google Scholar] [CrossRef]
- Lesniak, S.; Rachwalski, M.; Pieczonka, A.M. Optically Pure Aziridinyl Ligands as Useful Catalysts in the Stereocontrolled Synthesis. Curr. Org. Chem. 2014, 18, 3045–3065. [Google Scholar] [CrossRef]
- Cheung, L.L.W.; Zhi, H.; Decker, S.M.; Yudin, A.K. Skeletal Fusion of Small Heterocycles with Amphoteric Molecules. Angew. Chem. Int. Ed. 2011, 50, 11798–11802. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.M.D.; Levitsky, D.O.; Dembitsky, V.M. Aziridine alkaloids as potential therapeutic agents. Eur. J. Med. Chem. 2009, 44, 3373–3387. [Google Scholar] [CrossRef] [PubMed]
- Wakaki, S.; Marumo, H.; Tomioka, K.; Shimizu, G.; Kato, E.; Kamada, H.; Kudo, S.; Fujimoto, Y. Isolation of new fractions of antitumor mitomycins. Antibiot. Chemother. 1958, 8, 228–235. [Google Scholar]
- Herr, R.R.; Bergy, M.E.; Eble, T.E.; Jahnke, H.K. Porfiromycin, a new antibiotic. Antimicrob. Agents Ann. 1960, 8, 23–31. [Google Scholar]
- Zang, H.; Gates, K.S. DNA Binding and Alkylation by the “Left Half” of Azinomycin B. Biochemistry 2000, 39, 14968–14975. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, K.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; McClure, T.; Dorr, R.T. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem. Pharm. 2000, 60, 749–758. [Google Scholar] [CrossRef]
- Dvorakova, K.; Waltmire, C.N.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; Dorr, R.T. Induction of mitochondrial changes in myeloma cells by imexon. Blood 2001, 97, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Sheveleva, E.V.; Landowski, T.H.; Samulitis, B.K.; Bartholomeusz, G.; Powis, G.; Dorr, R.T. Imexon Induces an Oxidative Endoplasmic Reticulum Stress Response in Pancreatic Cancer Cells. Mol. Cancer Res. 2012, 10, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Moulder, S.; Dhillon, N.; Ng, C.; Hong, D.; Wheler, J.; Naing, A.; Tse, S.; La Paglia, A.; Dorr, R.; Hersh, E.; et al. A phase I trial of imexon, a pro-oxidant, in combination with docetaxel for the treatment of patients with advanced breast, non-small cell lung and prostate cancer. Investig. New Drugs 2010, 28, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Miller, T.P.; Friedberg, J.W.; Peterson, D.R.; Baran, A.M.; Herr, M.; Spier, C.M.; Cui, H.; Roe, D.J.; Persky, D.O.; et al. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2014, 124, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.; Haag, R.; Bosies, E.; Bicker, U.; Kampe, W. Use of Imexone as an Immunosuppressive Agent. Patent EP 352652 A2, 31 January 1990. [Google Scholar]
- Kinoshita, S.; Uzu, K.; Nakano, M.; Shimizu, A.; Takahashi, T. Mitomycin derivatives. 1. Preparation of mitosane and mitosene compounds and their biological activities. J. Med. Chem. 1971, 14, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Stapley, E.O.; Hendlin, D.; Jackson, M.; Miller, A.K.; Hernandez, S.; Martinez, M.; Martinez, M. Azirinomycin. I. Microbial production and biological characteristics. J. Antibiot. 1971, 24, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Tristram, E.W.; Wolf, F.J. Azirinomycin. II. Isolation and chemical characterization as 3-methyl-2(2H) azirinecarboxylic acid. J. Antibiot. 1971, 24, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Argoudelis, A.D.; Reusser, F.; Whaley, H.A. The Antibiotic U-47,929 and Its Preparation. U.S. Patent 542,226, 24 February 1976. [Google Scholar]
- Harada, K.I.; Tomita, K.; Fujii, K.; Masuda, K.; Mikami, Y.; Yazawa, K.; Komaki, H. Isolation and Structural Characterization of Siderophores, Madurastatins, Produced by a Pathogenic Actinomadura madurae. J. Antibiot. 2004, 57, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Iinuma, H.; Kinoshita, N.; Ikeda, T.; Sawa, T.; Hamada, M.; Takeuchi, T. Azicemicins A and B, a New Antimicrobial Agent Produced by Amycolatopsis. J. Antibiot. 1995, 48, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.R.; Colson, K.L.; Klohr, S.E.; Zein, N.; Langley, D.R.; Lee, M.S.; Matson, J.A.; Doyle, T.W. Isolation, Structure Determination, and Proposed Mechanism of Action for Artifacts of Maduropeptin Chromophore. J. Am. Chem. Soc. 1994, 116, 9351–9352. [Google Scholar] [CrossRef]
- Budzisz, E.; Bobka, R.; Hauss, A.; Roedel, J.N.; Wirth, S.; Lorenz, I.-P.; Rozalska, B.; Więckowska-Szakiel, M.; Krajewska, U.; Rozalski, M. Synthesis, structural characterization, antimicrobial and cytotoxic effects of aziridine, 2-aminoethylaziridine and azirine complexes of copper(II) and palladium(II). Dalton Trans. 2012, 41, 5925–5933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swapnaja, K.J.M.; Yennam, S.; Chavali, M.; Poornachandra, Y.; Kumar, C.G.; Muthusamy, K.; Jayaraman, V.B.; Arumugam, P.; Balasubramanian, S.; Sriram, K.K. Design, synthesis and biological evaluation of diaziridinyl quinone isoxazole hybrids. Eur. J. Med. Chem. 2016, 117, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Moonen, K.; Laureyn, I.; Stevens, C.V. Synthetic methods for azaheterocyclic phosphonates and their biological activity. Chem. Rev. 2004, 104, 6177–6215. [Google Scholar] [CrossRef] [PubMed]
- Dogan, Ö.; Babiz, H.; Gözen, A.G.; Budak, S. Synthesis of 2-aziridinyl phosphonates by modified Gabriel–Cromwell reaction and their antibacterial activities. Eur. J. Med. Chem. 2011, 46, 2485–2489. [Google Scholar] [CrossRef] [PubMed]
- Chebanov, V.A.; Zbruyev, A.I.; Desenko, S.M.; Orlov, V.D.; Yaremenko, F.G. Three-Membered Azaheterocycles Based on α, β-Unsaturated Ketones. Curr. Org. Chem. 2008, 12, 792–812. [Google Scholar] [CrossRef]
- Singh, G.S.; D’hooghe, M.; De Kimpe, N. Synthesis and Reactivity of C-Heteroatom-Substituted Aziridines. Chem. Rev. 2007, 107, 2080–2135. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, G.; Meiresonne, T.; De Kimpe, N.; Mangelinckx, S. Synthesis and Reactivity of 2-(Carboxymethyl)aziridine Derivatives. Chem. Rev. 2014, 114, 7954–8015. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, N.; Xu, J. An improved and mild Wenker synthesis of aziridines. Synthesis 2010, 20, 3423–3428. [Google Scholar]
- Fishbein, P.L.; Kohn, H. Synthesis and antineoplastic activity of 1a-formyl and 1a-thioformyl derivatives of mitomycin C and 2-methylaziridine. J. Med. Chem. 1987, 30, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Kwan, B.W.; Chowdhury, N.; Wood, T.K. Combatting bacterial infections by killing persister cells with mitomycin C. Environ. Microbiol. 2015, 17, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Tokushige, M.; Umezawa, H. Specific inhibition of aspartase by S-2,3-dicarboxyaziridine. Biochem. Int. 1988, 16, 449–452. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 3a–3s are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | R3 | Yield (%) |
|---|---|---|---|---|
| 3a | Bu | iPr | H | 92 |
| 3b | Me | iPr | H | 90 |
| 3c | cHex | iPr | H | 89 |
| 3d | Me | Me | H | 89 |
| 3e | Me | Me | Me | 94 |
| 3f | cHex | Me | H | 97 |
| 3g | cHex | Me | Me | 96 |
| 3h | Allyl | iPr | H | 76 |
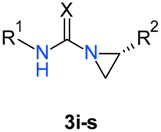
| Compound | R1 | R2 | X | Yield (%) |
|---|---|---|---|---|
| 3i | CH2Ph | iPr | S | 90 |
| 3j | CH2Ph | Me | S | 89 |
| 3k | CHPh2 | iPr | S | 88 |
| 3l | CHPh2 | Me | S | 85 |
| 3m | 2-(4-morpholino)ethyl | iPr | S | 92 |
| 3n | 2-(4-morpholino)ethyl | Me | S | 93 |
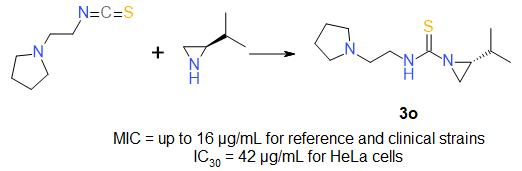
| 3o | 2-piperidinoethyl | iPr | S | 95 |
| (R)-3o | 2-piperidinoethyl | iPr | S | 90 |
| 3p | 2-piperidinoethyl | Me | S | 91 |
| 3r | Bu | iPr | O | 96 |
| 3s | cHex | iPr | O | 98 |
| Tested Strain | E. coli NCTC 8196 NCTC 8196 | S. aureus ATCC 6538 ATCC 6538 | S. aureus ATCC 29213 ATCC 29213 | S. epidermidis ATCC 12228 ATCC 12228 | ||||
|---|---|---|---|---|---|---|---|---|
| Compound | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC |
| 3a | >512 | nd | 32 | 128 | 32 | 128 | 32 | 64 |
| 3b | 32 | 32 | 32 | 64 | 32 | 64 | 16 | 16 |
| 3c | >512 | nd | 32 | 64 | 32 | 64 | 32 | 64 |
| 3d | 128 | 128 | 256 | 512 | 256 | 512 | 256 | 512 |
| 3e | >512 | nd | 128 | 128 | 128 | 128 | 128 | 128 |
| 3f | 256 | 512 | 16 | 128 | 16 | 128 | 16 | 16 |
| NTF | 8 | 8 | 16 | 32 | 16 | 32 | 8 | 8 |
| AMP | 4 | 4 | 1 | 1 | 2 | 4 | 1 | 1 |
| STR | 1 | 2 | 1 | 1 | 1 | 2 | >512 | >512 |
| Compound | 3a | 3b | 3c | 3f | OX | AMP | NTF | STR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tested Strain | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC |
| naso-pharynx isolates | ||||||||||||||||
| S. aureus | ||||||||||||||||
| C4 | 32 | 64 | 32 | 64 | 32 | 64 | 16 | 16 | 0.25 | 0.25 | 512 | 512 | 16 | 16 | 8 | 8 |
| C7 | 32 | 64 | 32 | 32 | 32 | 64 | 16 | 16 | 0.25 | 0.25 | 64 | 128 | 16 | 16 | 8 | 16 |
| C8 | 32 | 64 | 32 | 32 | 32 | 64 | 16 | 16 | 0.25 | 0.25 | 512 | >512 | 16 | 16 | 8 | 16 |
| C19 | 32 | 64 | 32 | 32 | 32 | 64 | 16 | 16 | 0.5 | 0.5 | 64 | 128 | 32 | 32 | 8 | 8 |
| ulcers/furuncles isolates | ||||||||||||||||
| D12 | 32 | 64 | 16 | 32 | 32 | 128 | 8 | 16 | 0.5 | 0.5 | 256 | 256 | 32 | 32 | 8 | 16 |
| F1 | 32 | 64 | 32 | 128 | 64 | 128 | 16 | 64 | 0.25 | 0.25 | 128 | 128 | 32 | 32 | 8 | 8 |
| F7 | 32 | 64 | 32 | 128 | 32 | 64 | 16 | 64 | 0.25 | 0.25 | 4 | 4 | 16 | 16 | 8 | 16 |
| F12 | 32 | 64 | 16 | 32 | 32 | 64 | 8 | 16 | 0.25 | 0.25 | 64 | 128 | 16 | 32 | 8 | 32 |
| bone isolates | ||||||||||||||||
| D14 | 32 | 64 | 32 | 32 | 32 | 64 | 16 | 16 | 0.5 | 0.5 | 256 | 512 | 32 | 32 | 8 | 16 |
| D15 (MRSA) | 32 | 64 | 32 | 32 | 32 | 64 | 16 | 16 | >512 | >512 | >512 | >512 | 32 | 32 | 256 | >512 |
| D17 (MRSA) | 32 | 64 | 16 | 16 | 32 | 64 | 8 | 32 | >512 | >512 | >512 | >512 | 32 | 32 | 256 | >512 |
| D20 | 32 | 64 | 32 | 32 | 64 | 128 | 16 | 64 | 0.5 | 0.5 | 128 | 256 | 16 | 32 | 8 | 8 |
| Compound | IC30 (µg/mL)/(µM) L929 Cells | HeLa Cells |
|---|---|---|
| 3a | 68/340 | 83/388 |
| 3b | 27/171 | 28/177 |
| 3c | 34/147 | 69/304 |
| 3d | >250/>1922 | >250/1922 |
| 3e | 214/1485 | 250/1735 |
| 3f | 20/102 | 22/111 |
| Cisplatin | 7.24/24 | 6.53/22 |
| Tested Strain | E. coli NCTC 8196 NCTC 8196 | S. aureus ATCC 6538 ATCC 6538 | S. aureus ATCC 29213 ATCC 29213 | S. epidermidis ATCC 12228 ATCC 12228 |
|---|---|---|---|---|
| Compound | MIC (µg/mL) | |||
| 3j | 256 | 128 | 128 | 128 |
| 3k | 256 | na* | Na | 256 |
| 3l | 256 | 256 | 256 | 256 |
| 3m | na | 128 | 256 | 128 |
| 3o | 64 | 8 | 8 | 4 |
| 3p | 256 | 64 | 64 | 64 |
| NTF | 8 | 16 | 16 | 8 |
| AMP | 4 | 1 | 2 | 1 |
| STR | 1 | 1 | 1 | >512 |
| Tested Strain | E. coli NCTC 8196 NCTC 8196 | S. aureus ATCC 6538 ATCC 6538 | S. aureus ATCC 29213 ATCC 29213 | S. epidermidis ATCC 12228 ATCC 12228 |
|---|---|---|---|---|
| Compound | MBC (µg/mL) | |||
| 3o | 128 | 16 | 16 | 8 |
| 3p | 256 | 128 | 128 | 128 |
| Compound | 3o | 3p | OX | AMP | NTF | STR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tested Strain | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC |
| naso-pharynx isolates | ||||||||||||
| S. aureus | ||||||||||||
| C4 | 32 | 64 | 256 | 256 | 0.25 | 0.25 | 512 | 512 | 16 | 16 | 8 | 8 |
| C7 | 32 | 64 | 256 | 256 | 0.25 | 0.25 | 64 | 128 | 16 | 16 | 8 | 16 |
| C8 | 32 | 64 | 256 | 256 | 0.25 | 0.25 | 512 | >512 | 16 | 16 | 8 | 16 |
| C19 | 32 | 64 | 256 | 256 | 0.5 | 0.25 | 64 | 128 | 32 | 32 | 8 | 8 |
| ulcers/furuncles isolates | ||||||||||||
| D12 | 32 | 64 | 256 | 256 | 0.5 | 0.5 | 256 | 256 | 32 | 32 | 8 | 16 |
| F1 | 32 | 64 | 256 | 256 | 0.25 | 0.25 | 128 | 128 | 32 | 32 | 8 | 8 |
| F7 | 32 | 64 | 256 | 256 | 0.25 | 0.25 | 4 | 4 | 16 | 16 | 8 | 16 |
| F12 | 32 | 64 | 256 | 256 | 0.25 | 0.25 | 64 | 128 | 16 | 32 | 8 | 32 |
| bone isolates | ||||||||||||
| D14 | 32 | 64 | 256 | 256 | 0.5 | 0.5 | 256 | 512 | 32 | 32 | 8 | 16 |
| D15 (MRSA) | 32 | 64 | 256 | 512 | >512 | >512 | >512 | >512 | 32 | 32 | 256 | >512 |
| D17 (MRSA) | 32 | 64 | 256 | 512 | >512 | >512 | >512 | >512 | 32 | 32 | 256 | >512 |
| D20 | 32 | 64 | 256 | 512 | 0.5 | 0.5 | 128 | 256 | 16 | 32 | 8 | 8 |
| Compound | IC30 (µg/mL)/(µM) L929 Cells | HeLa Cells |
|---|---|---|
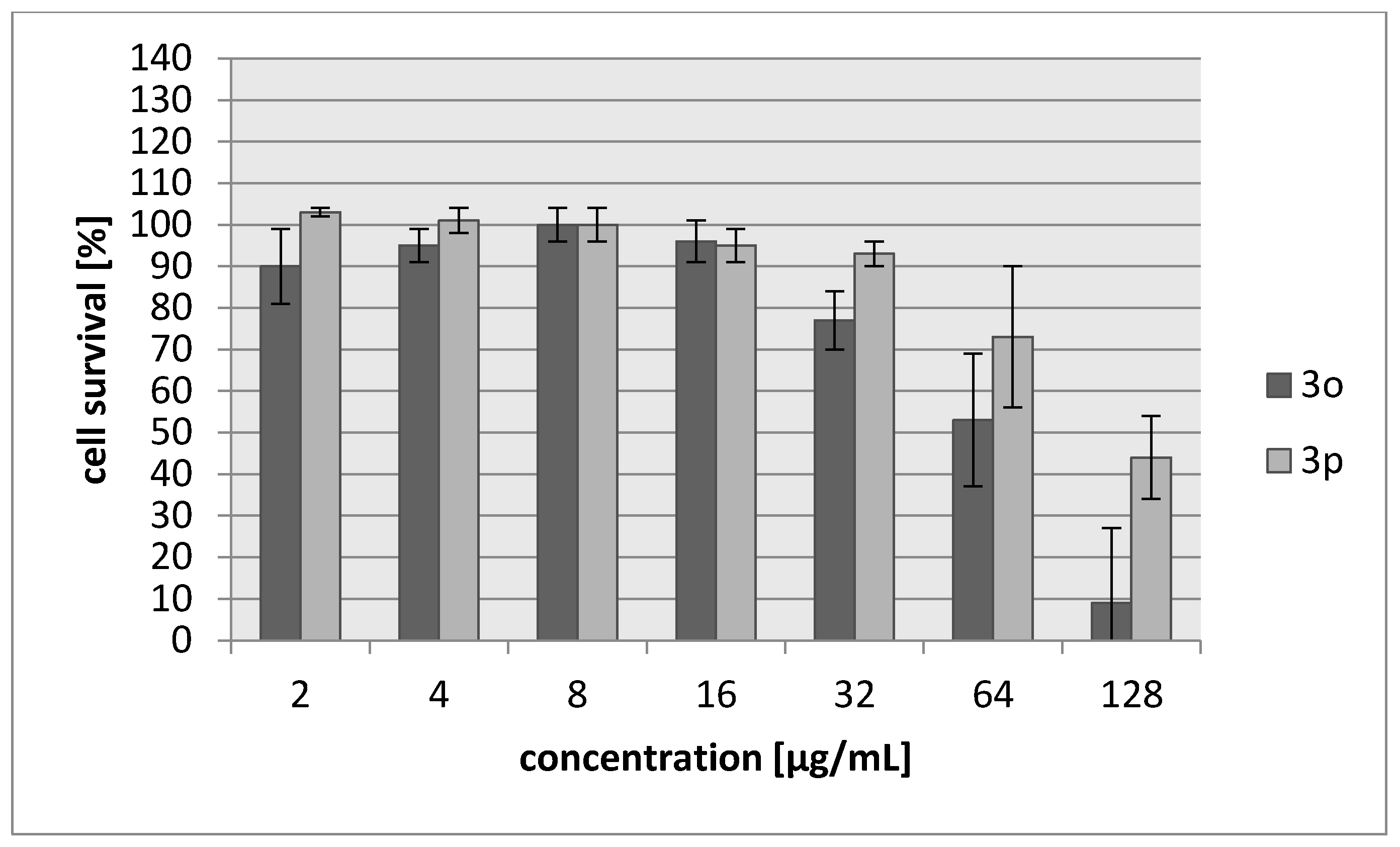
| 3o | 45/187 | 42/174 |
| 3p | 103/483 | 71/333 |
| Cisplatin | 7.24/24 | 6.53/22 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, A.; Pieczonka, A.M.; Rachwalski, M.; Leśniak, S.; Stączek, P. Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea. Molecules 2018, 23, 45. https://doi.org/10.3390/molecules23010045
Kowalczyk A, Pieczonka AM, Rachwalski M, Leśniak S, Stączek P. Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea. Molecules. 2018; 23(1):45. https://doi.org/10.3390/molecules23010045
Chicago/Turabian StyleKowalczyk, Aleksandra, Adam M. Pieczonka, Michał Rachwalski, Stanisław Leśniak, and Paweł Stączek. 2018. "Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea" Molecules 23, no. 1: 45. https://doi.org/10.3390/molecules23010045