Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
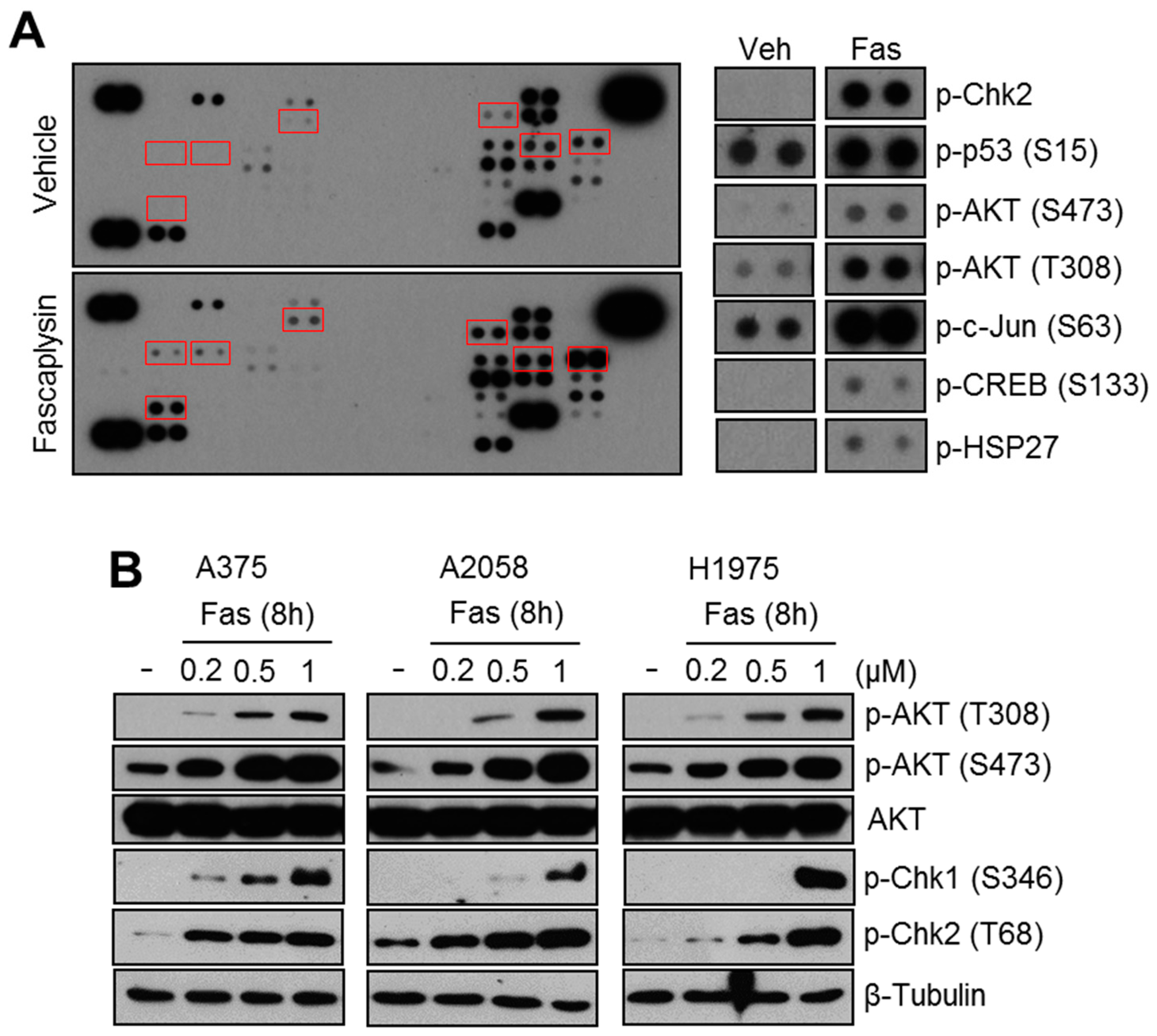
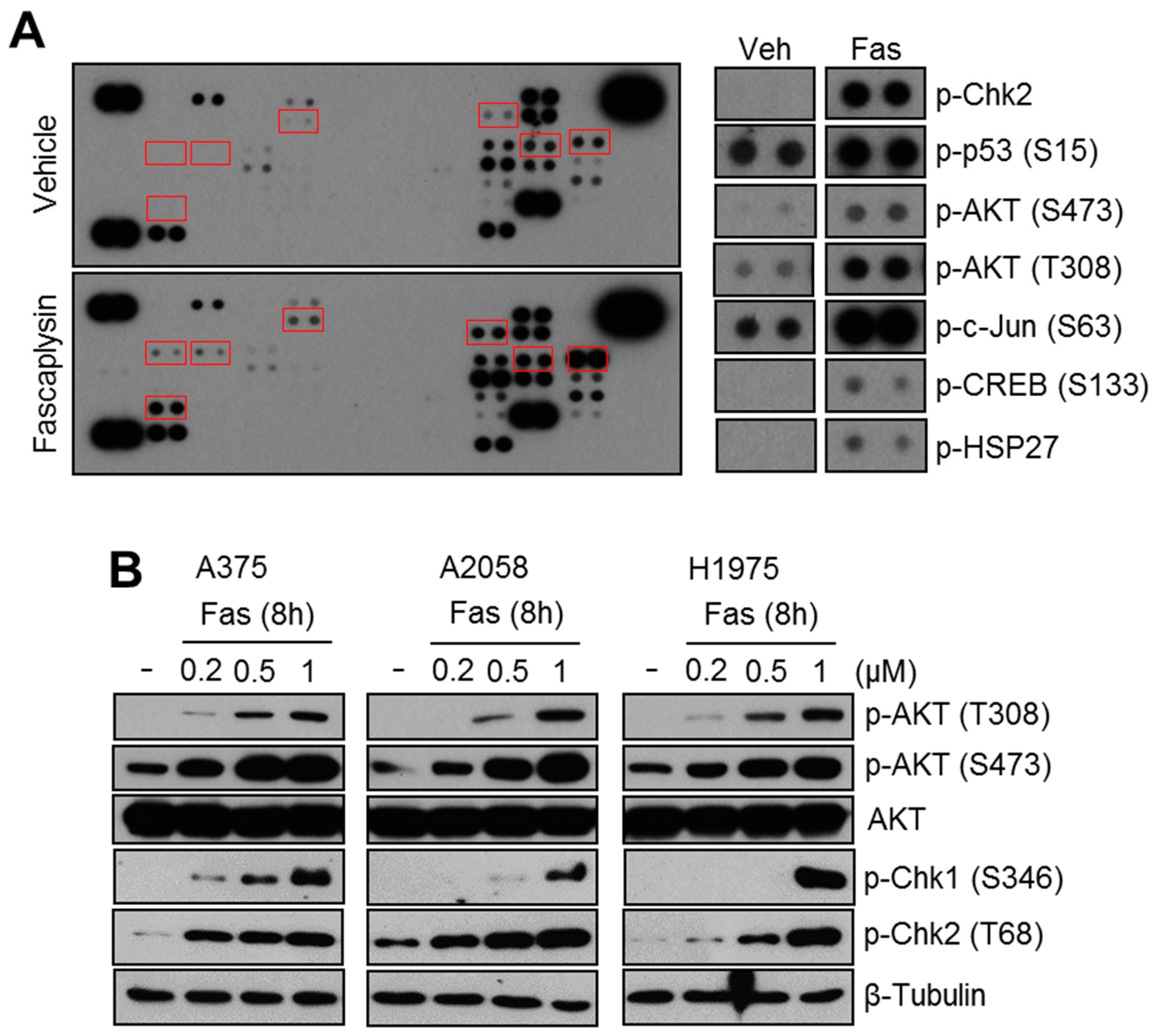
2.1. Fascaplysin Increases AKT Phosphorylation and DNA Damage Signaling
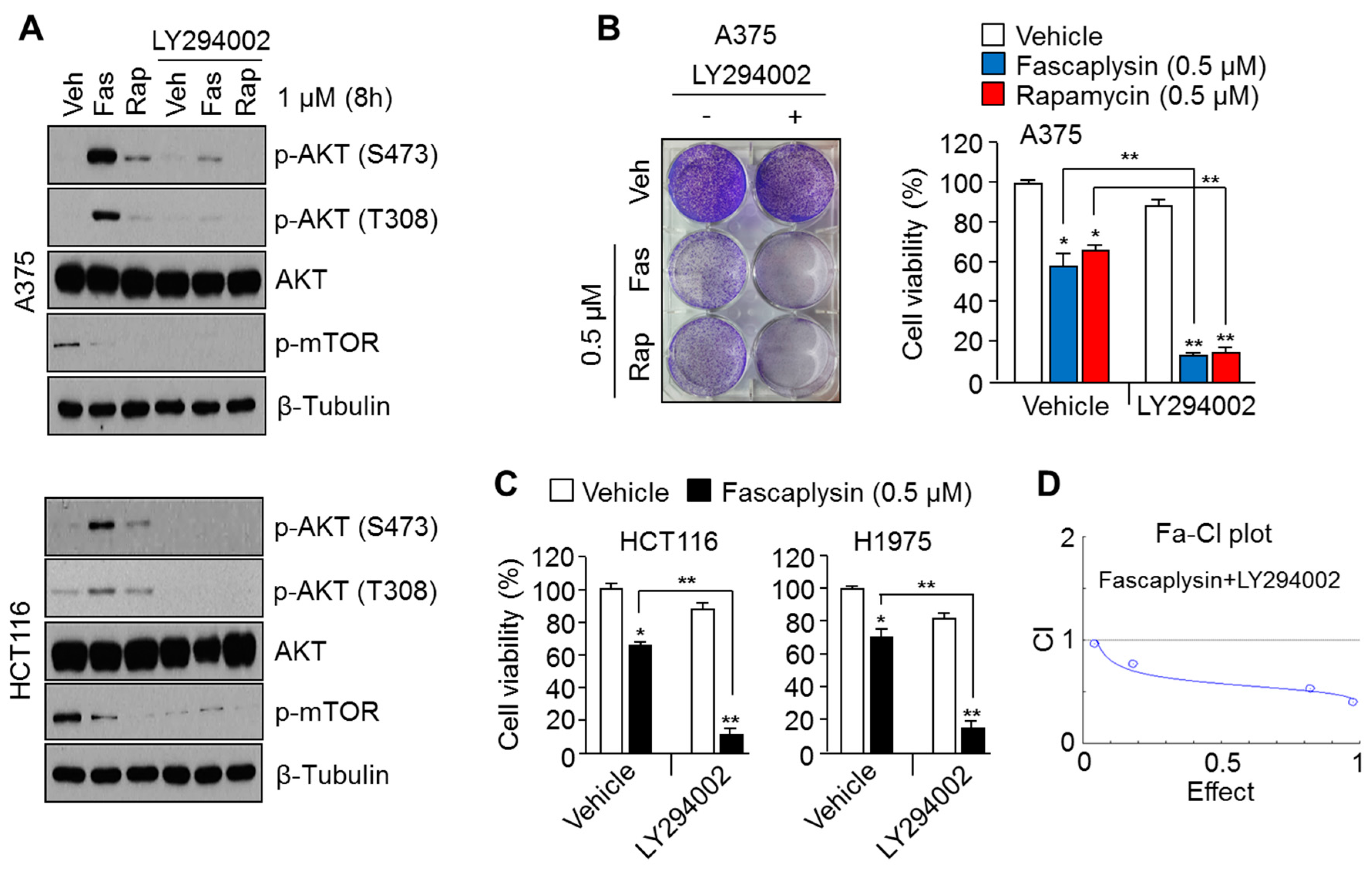
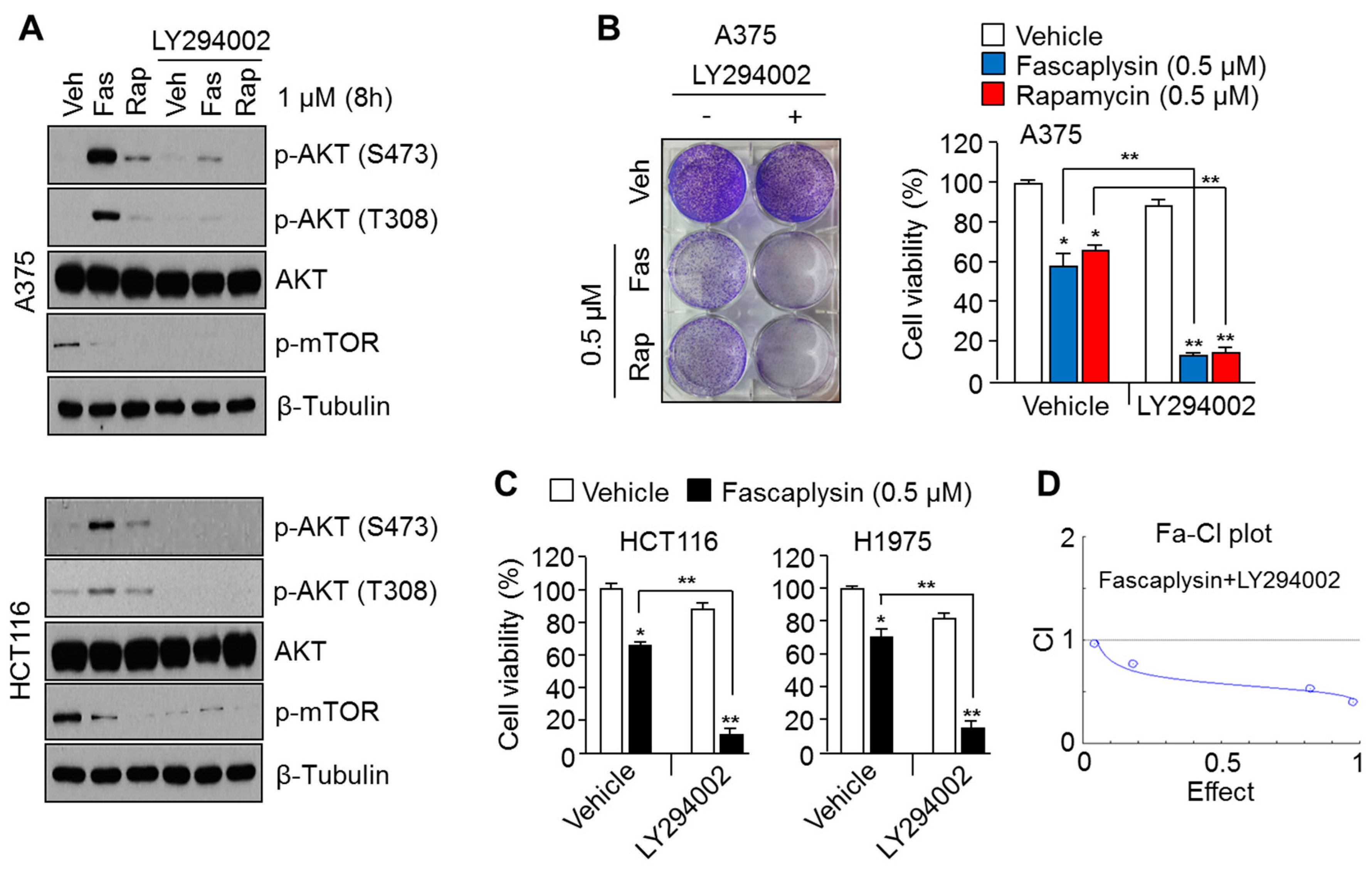
2.2. Suppression of AKT Activation Synergizes the Anti-Cancer Effects of Fascaplysin
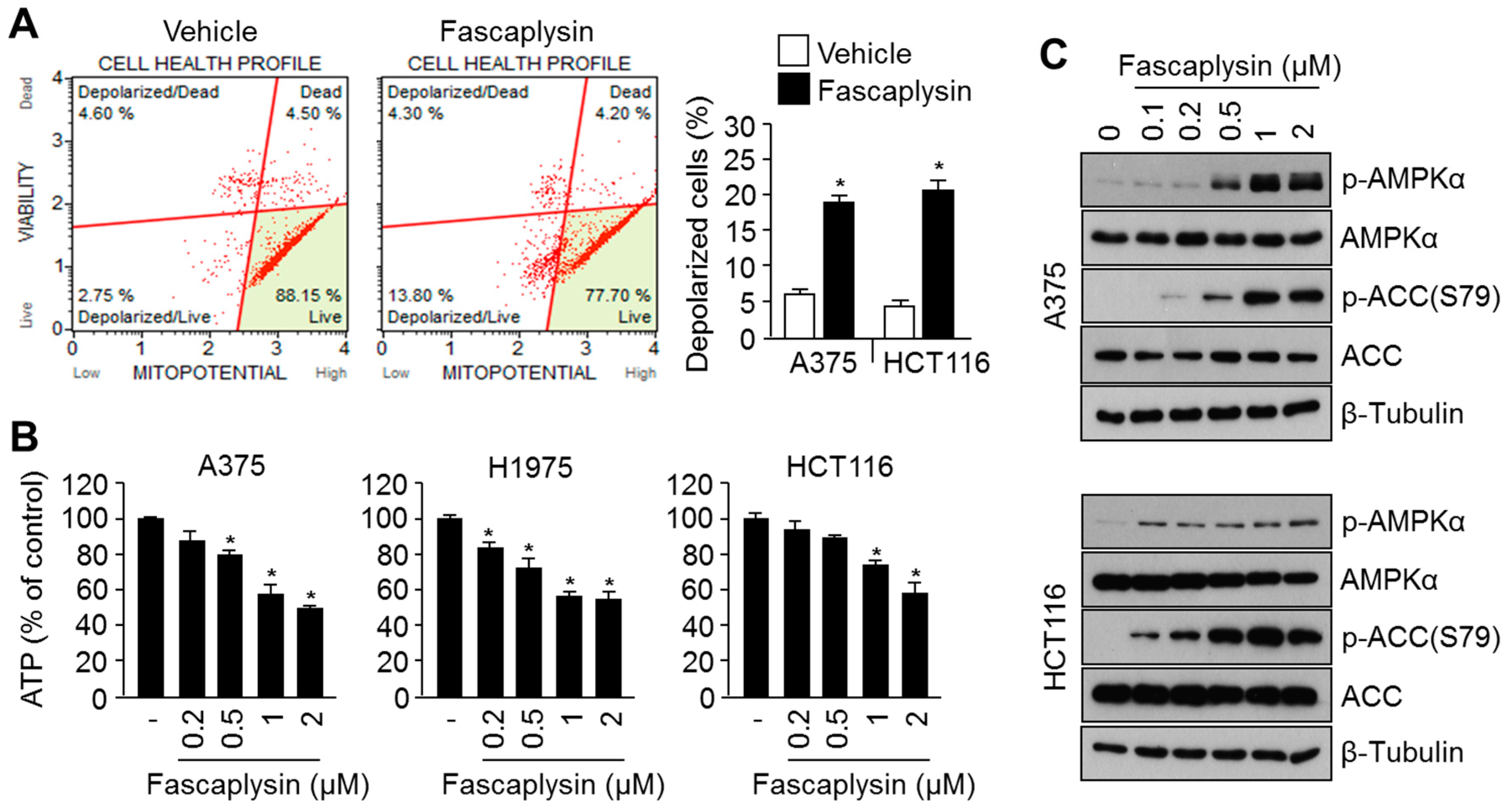
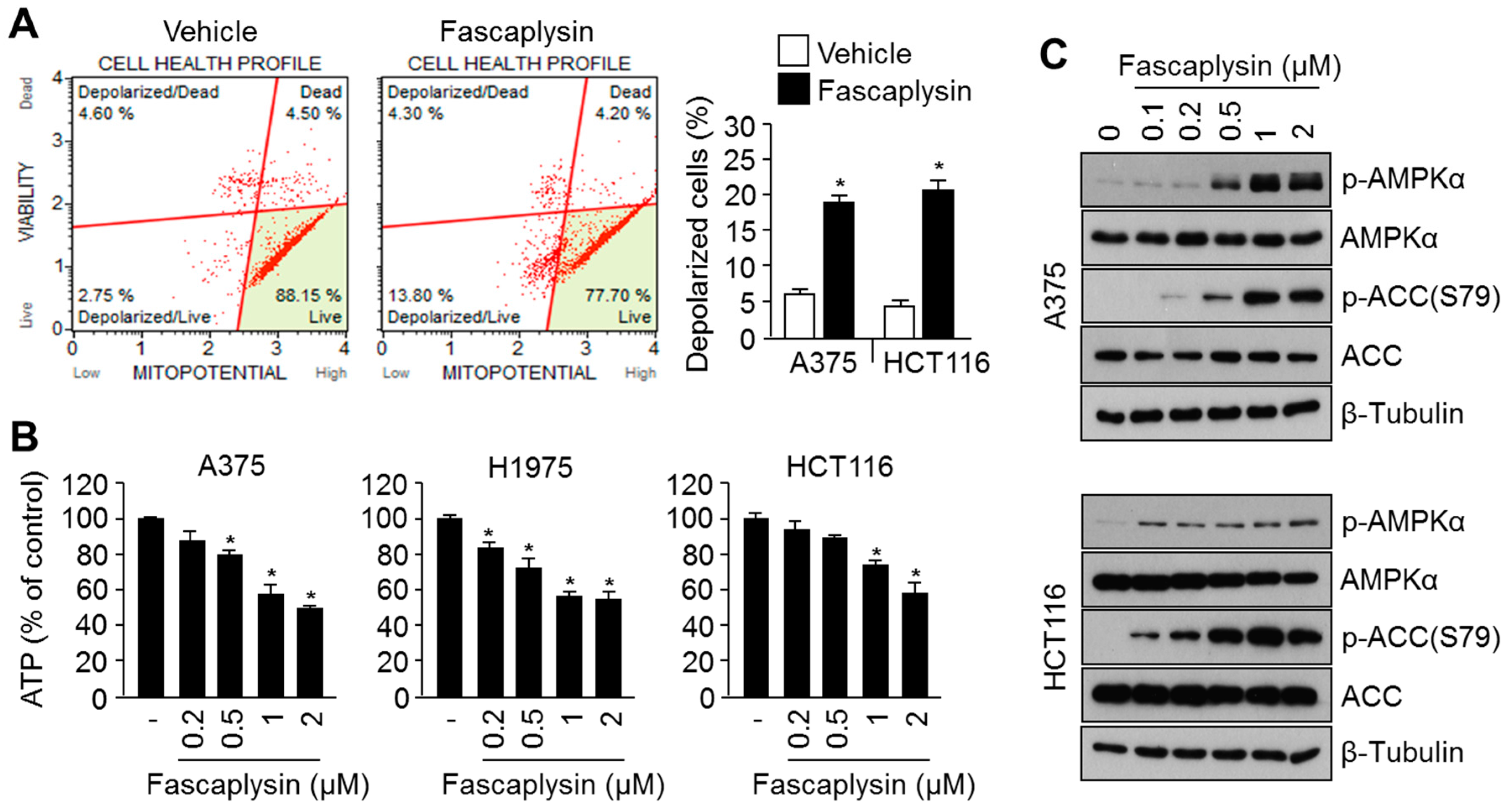
2.3. Fascaplysin Causes Metabolic Stress and Increases AMPK Signaling
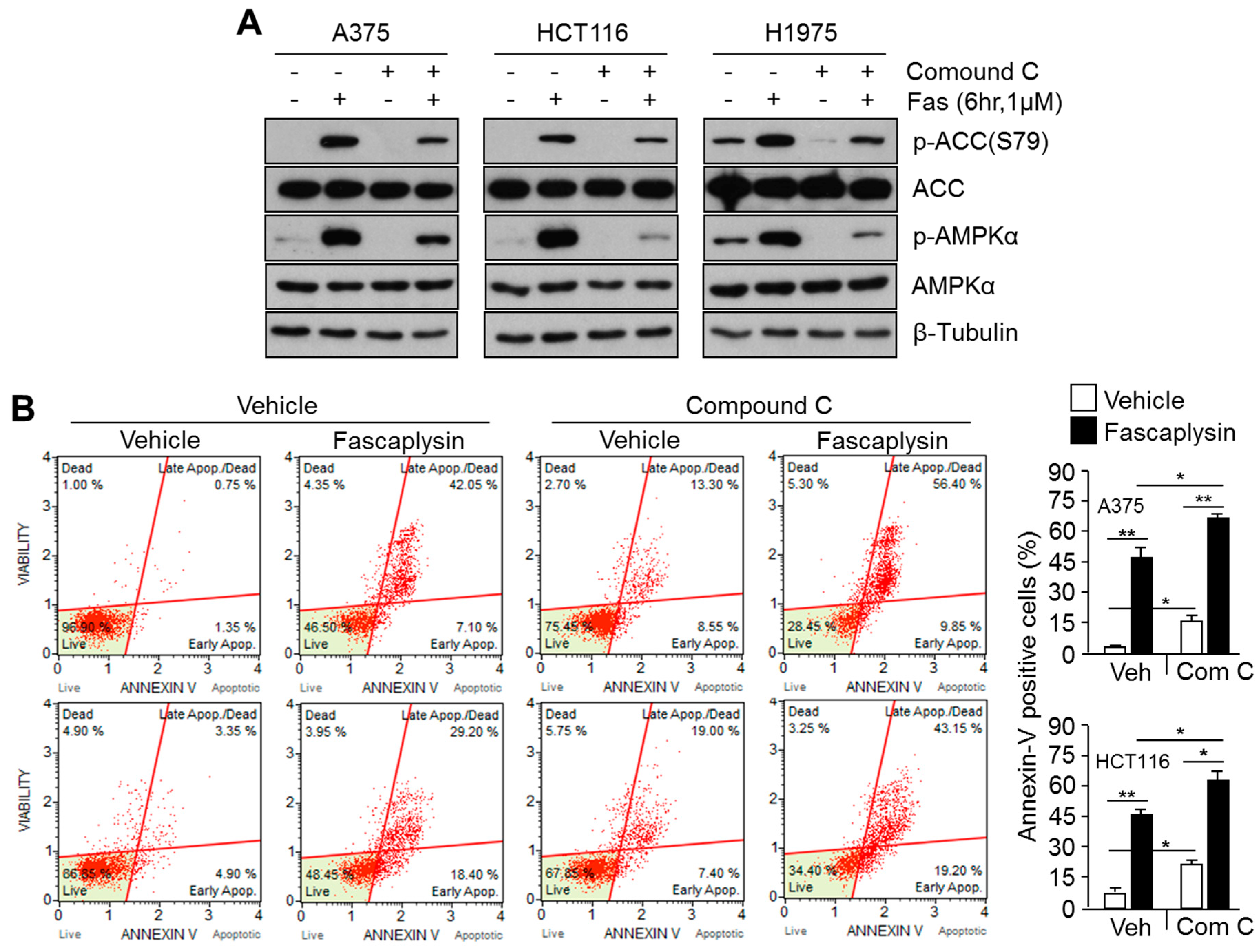
2.4. Suppression of Fascaplysin-Induced AMPK Activation Synergizes Anti-Cancer Efficacy of Fascaplysin
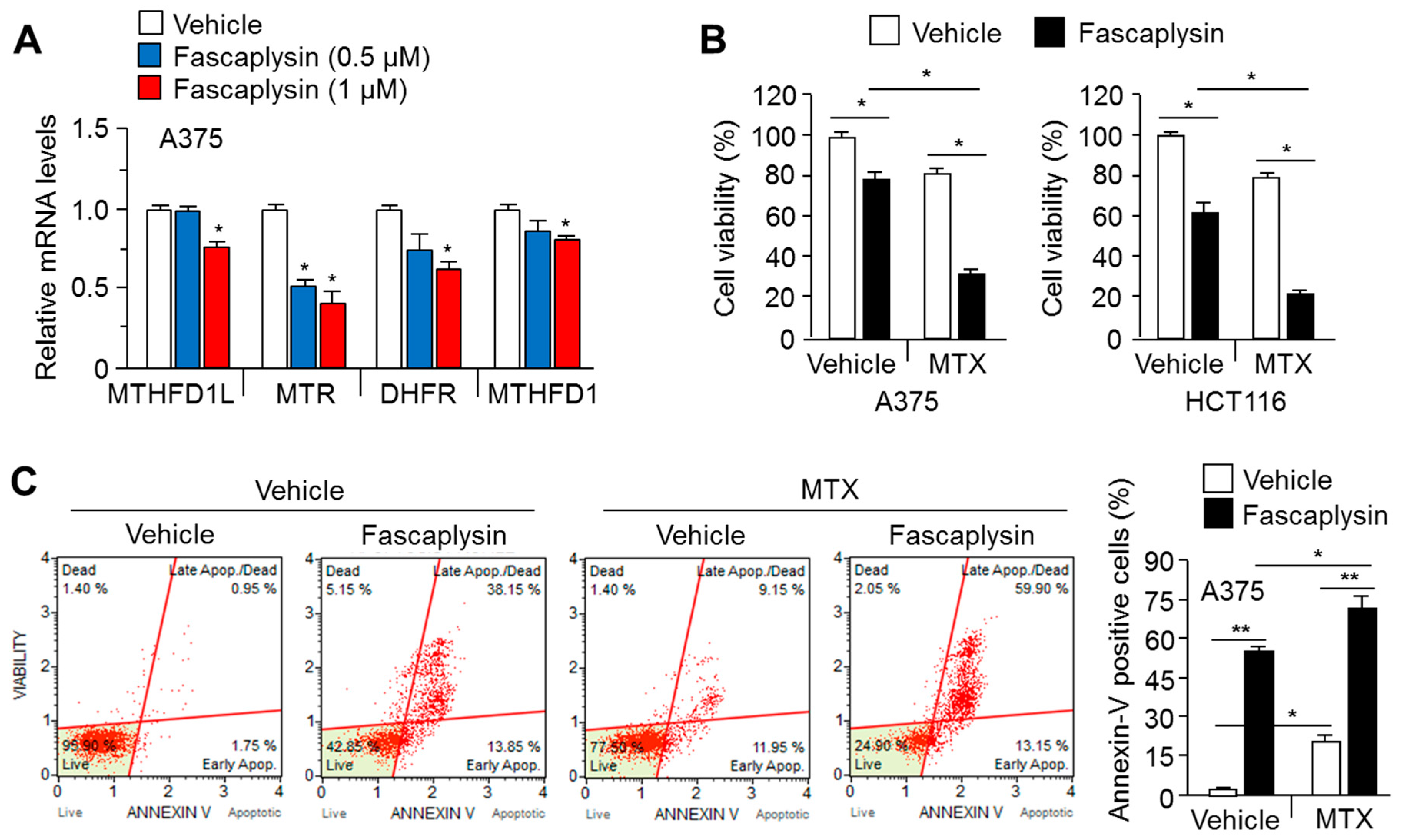
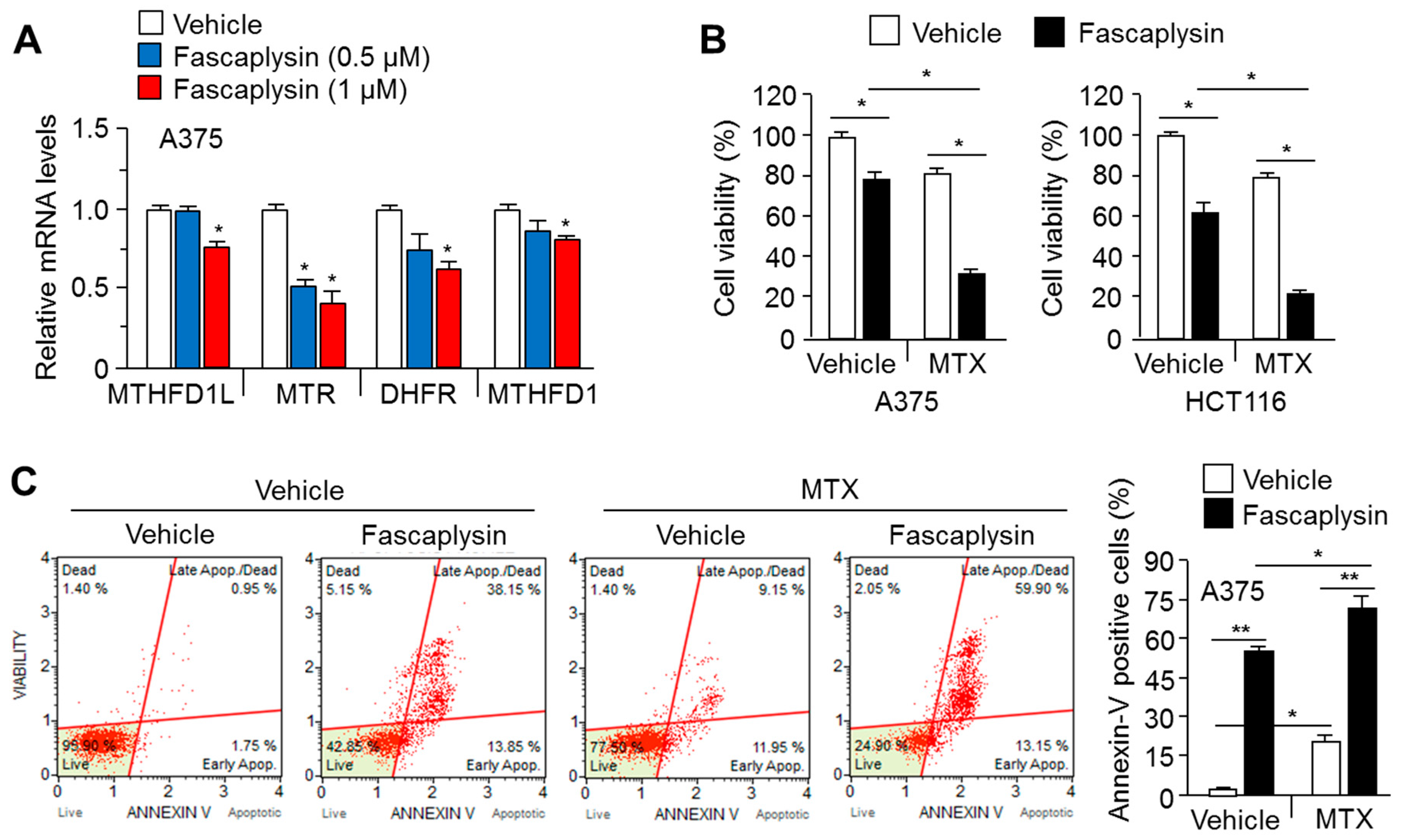
2.5. MTX Enhances the Anti-Cancer Efficacy of Fascaplysin
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Cell Viability Assay
4.3. Western Blotting
4.4. Quantitative Real-Time PCR
4.5. Proteome Profiler Array
4.6. Apoptosis Assays
4.7. Mitochondrial Membrane Potential Assay
4.8. ATP Assay
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guan, X.; Wang, L.L.; Li, B.; Sang, X.B.; Liu, Y.; Zhao, Y. Fascaplysin inhibit ovarian cancer cell proliferation and metastasis through inhibiting CDK4. Gene 2017, 635, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yan, X.J.; Chen, H.M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 59, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. Fascaplysin as a Specific Inhibitor for CDK4: Insights from Molecular Modelling. PLoS ONE 2012, 7, e42612. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.I.; Lee, Y.M.; Nam, T.J.; Ko, Y.S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin exerts anti-cancer effects through the downregulation of survivin and HIF-1alpha and inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Q.; Yu, J.-P.; Yu, H.-G.; Lv, P.; Chen, H.-L. Activation of Akt and ERK signalling pathways induced by etoposide confer chemoresistance in gastric cancer cells. Dig. Liver Dis. 2006, 38, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Hafsi, S.; Pezzino, F.M.; Candido, S.; Ligresti, G.; Spandidos, D.A.; Soua, Z.; McCubrey, J.A.; Travali, S.; Libra, M. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review). Int. J. Oncol. 2012, 40, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-G.; Ai, Y.-W.; Yu, L.-L.; Zhou, X.-D.; Liu, J.; Li, J.-H.; Xu, X.-M.; Liu, S.; Chen, J.; Liu, F.; et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int. J. Cancer 2007, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Page, C.; Lin, H.J.; Jin, Y.; Castle, V.P.; Nunez, G.; Huang, M.; Lin, J. Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res. 2000, 20, 407–416. [Google Scholar] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Walker, C.L. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011, 585, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jung, J.Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.T.; Kim, S.H.; Choi, Y.D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2017, 13, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.M.; Ngan, H.Y.; Chan, D.W. Targeting AMPK signaling in combating ovarian cancers: Opportunities and challenges. Acta Biochim. Biophys. Sin. 2016, 48, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Papadopoli, D.J.; Gravel, S.-P.; Yee, T.; Bridon, G.; Caron, M.; Bourque, G.; Giguère, V.; St-Pierre, J. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep. 2016, 14, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, J.N.; Leclerc, G.J.; Leclerc, G.M.; Barredo, J.C. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol. Cancer Ther. 2011, 10, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-G.; Liu, F.; Song, X.-F.; Wang, Z.-H.; Dong, Z.-Q.; Hu, Z.-Q.; Lan, R.-Z.; Guan, W.; Zhou, T.-G.; Xu, X.-M.; et al. Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol. Carcinog. 2010, 49, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Fu, J.; Chou, T. Synergistic combination of microtubule targeting anticancer fludelone with cytoprotective panaxytriol derived from panax ginseng against MX-1 cells in vitro: experimental design and data analysis using the combination index method. Am. J. Cancer Res. 2016, 6, 97–104. [Google Scholar] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK–ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin. Cancer Res. 2016, 23, 2781–2794. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Cheong, J.W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, T.; Eustace, A.J.; Collins, D.M.; Walsh, N.; O’donovan, N.; Crown, J. Kinase inhibitor screening indentifies CDK4 as a potential therapeutic target for melanoma. Int. J. Oncol. 2015, 47, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Bryukhovetskiy, I.; Lyakhova, I.; Mischenko, P.; Milkina, E.; Zaitsev, S.; Khotimchenko, Y.; Bryukhovetskiy, A.; Polevshchikov, A.; Kudryavtsev, I.; Khotimchenko, M.; et al. Alkaloids of fascaplysin are effective conventional chemotherapeutic drugs, inhibiting the proliferation of C6 glioma cells and causing their death in vitro. Oncol. Lett. 2016, 13, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin induces caspase mediated Crosstalk between apoptosis and autophagy through the inhibition of PI3K/AKT/mTOR signaling cascade in human leukemia HL-60 cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.F.; Wu, D.S.; Zhang, L.; Yu, Y.H.; Yuan, X.Y.; Li, W.J.; Chen, X.P.; Zhao, X.L.; Chen, F.P.; Zeng, H. Inactivation of PTEN increases ABCG2 expression and the side population through the PI3K/Akt pathway in adult acute leukemia. Cancer Lett. 2013, 336, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E Survival Pathways by Rapamycin-Mediated Mammalian Target of Rapamycin Inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed]
- Hörmann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Mahale, S.; Bharate, S.B.; Manda, S.; Joshi, P.; Jenkins, P.R.; Vishwakarma, R.A.; Chaudhuri, B. Antitumour potential of BPT: A dual inhibitor of cdk4 and tubulin polymerization. Cell Death Dis. 2015, 6, e1743. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Dominy, J.E.; Choi, Y.J.; Jurczak, M.; Tolliday, N.; Camporez, J.P.; Chim, H.; Lim, J.-H.; Ruan, H.-B.; Yang, X.; et al. Cyclin D1–Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014, 510, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Bertino, J.R.; Göker, E.; Gorlick, R.; Li, W.W.; Banerjee, D. Resistance mechanisms to methotrexate in tumors. Stem Cells 1996, 14, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Healey, J.H.; Meyers, P.A.; Ladanyi, M.; Huvos, A.G.; Bertino, J.R.; Gorlick, R. Mechanisms of methotrexate resistance in osteosarcoma. Clin. Cancer Res. 1999, 5, 621–627. [Google Scholar] [PubMed]
Sample Availability: Not available. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, T.-I.; Lee, J.H.; Kim, S.; Nam, T.-J.; Kim, Y.-S.; Kim, B.M.; Yim, W.J.; Lim, J.-H. Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK. Molecules 2018, 23, 42. https://doi.org/10.3390/molecules23010042
Oh T-I, Lee JH, Kim S, Nam T-J, Kim Y-S, Kim BM, Yim WJ, Lim J-H. Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK. Molecules. 2018; 23(1):42. https://doi.org/10.3390/molecules23010042
Chicago/Turabian StyleOh, Taek-In, Jun Ho Lee, Seongman Kim, Taek-Jin Nam, Young-Seon Kim, Byeong Mo Kim, Woo Jong Yim, and Ji-Hong Lim. 2018. "Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK" Molecules 23, no. 1: 42. https://doi.org/10.3390/molecules23010042