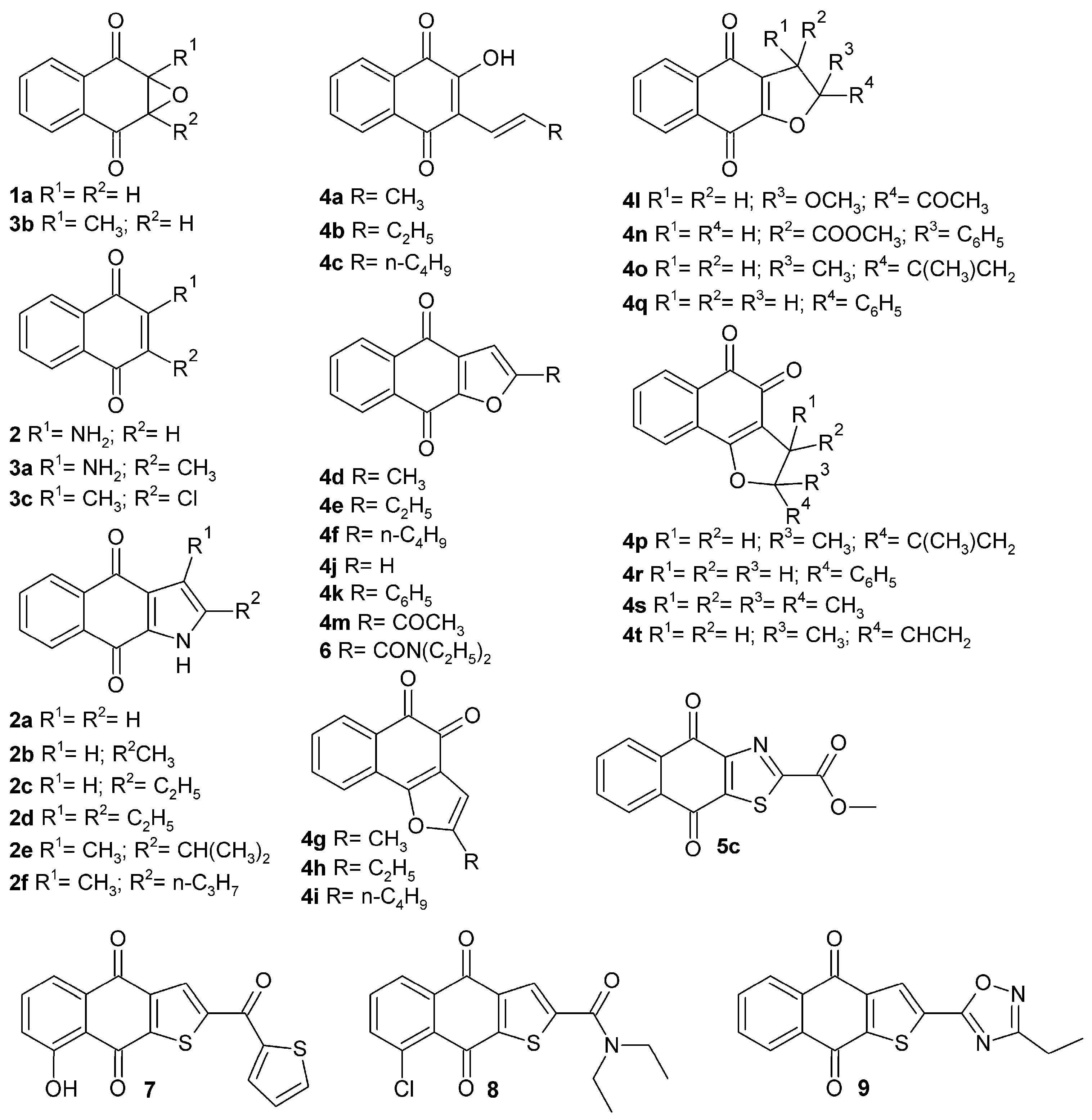
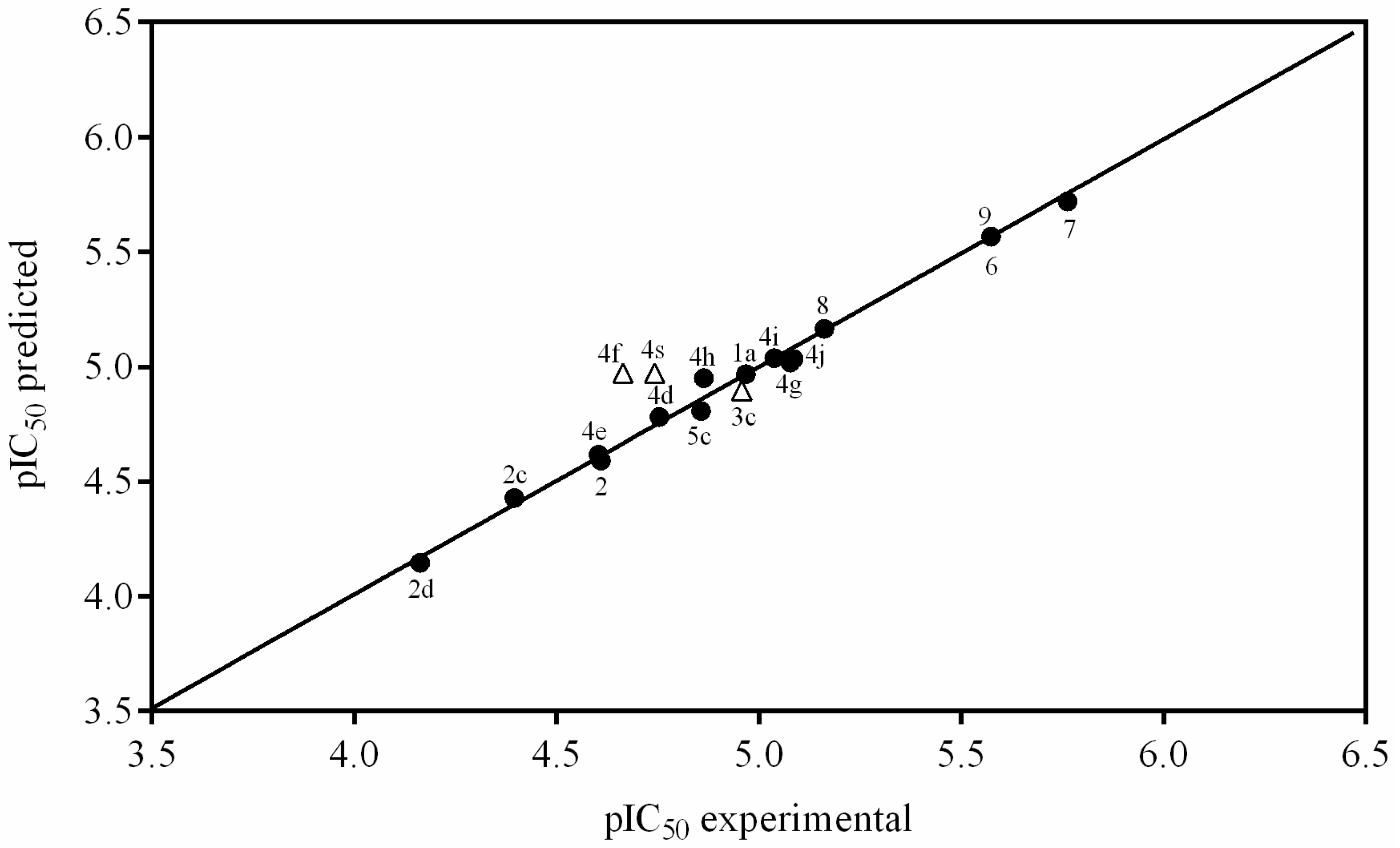
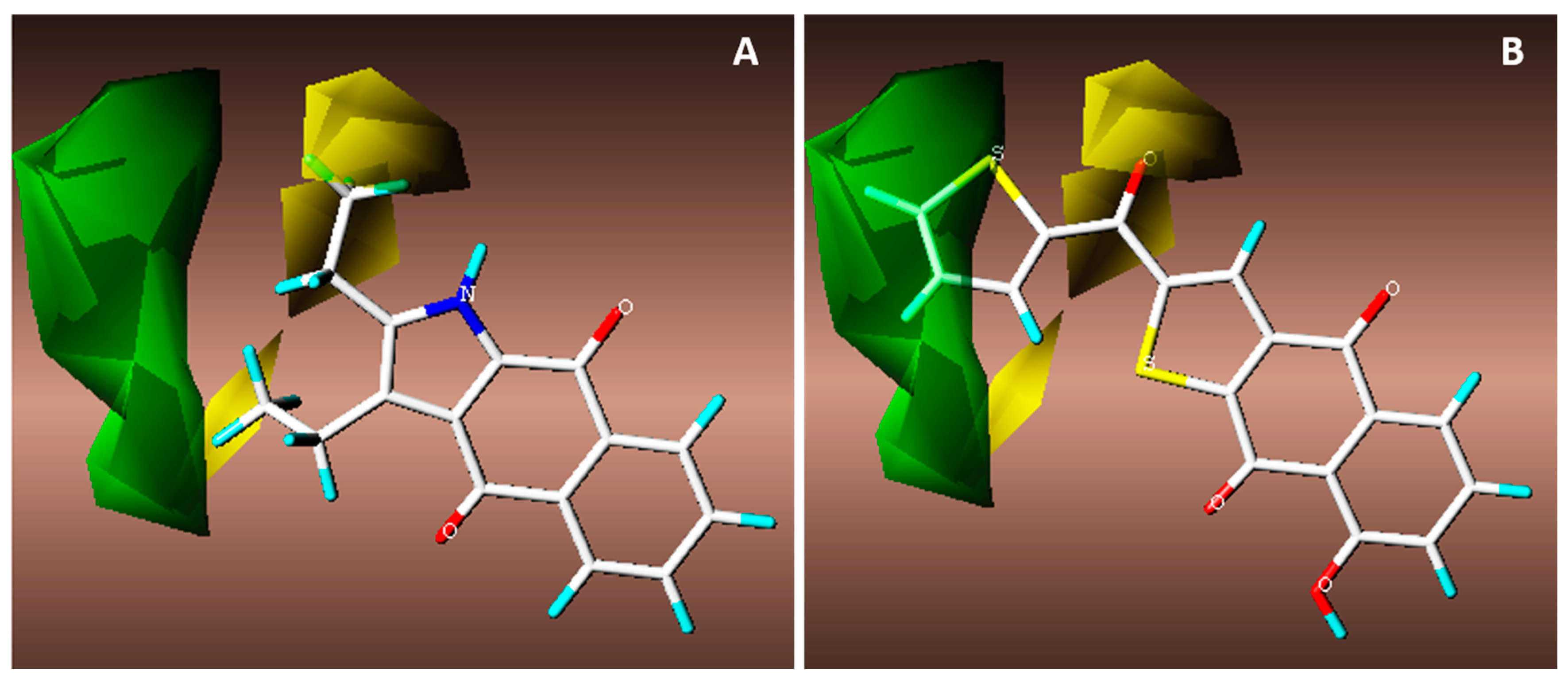
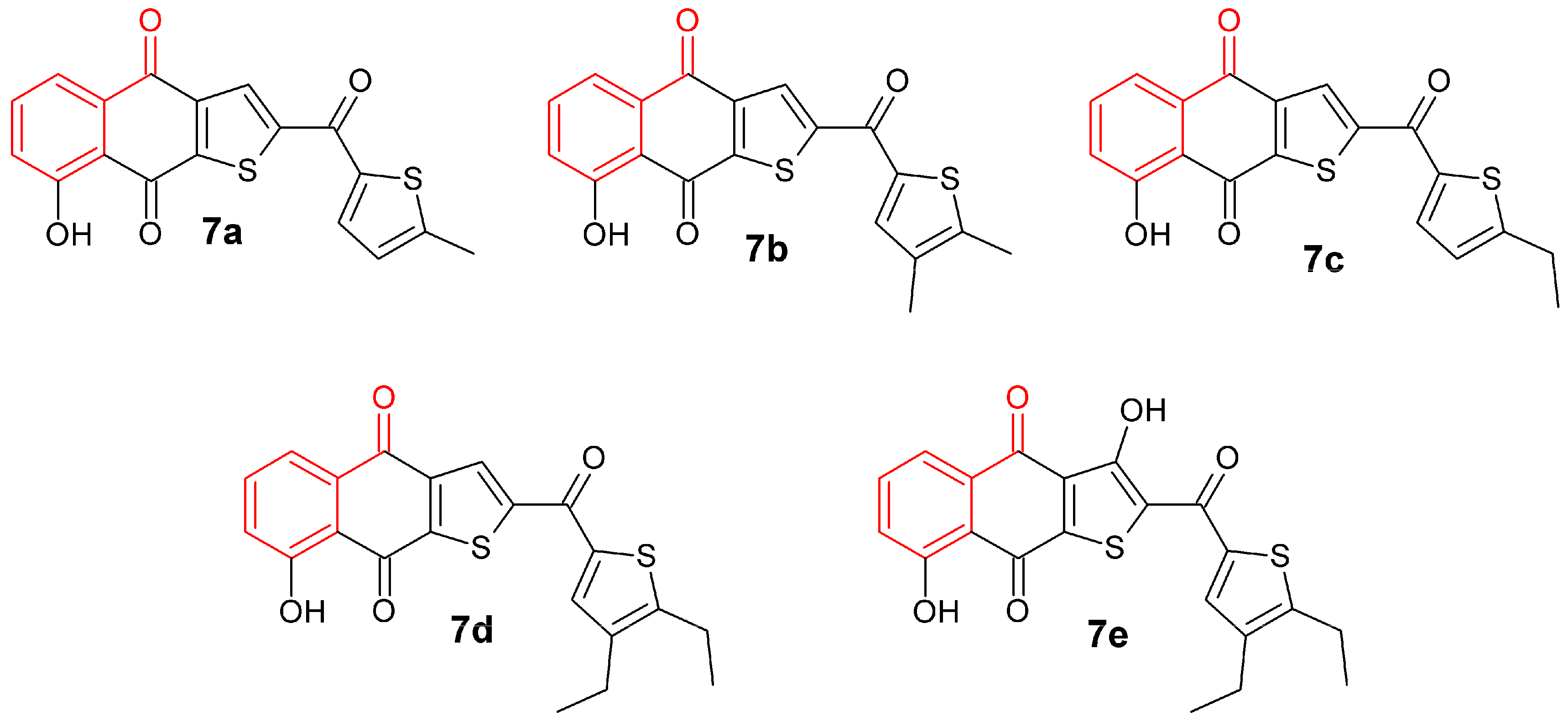
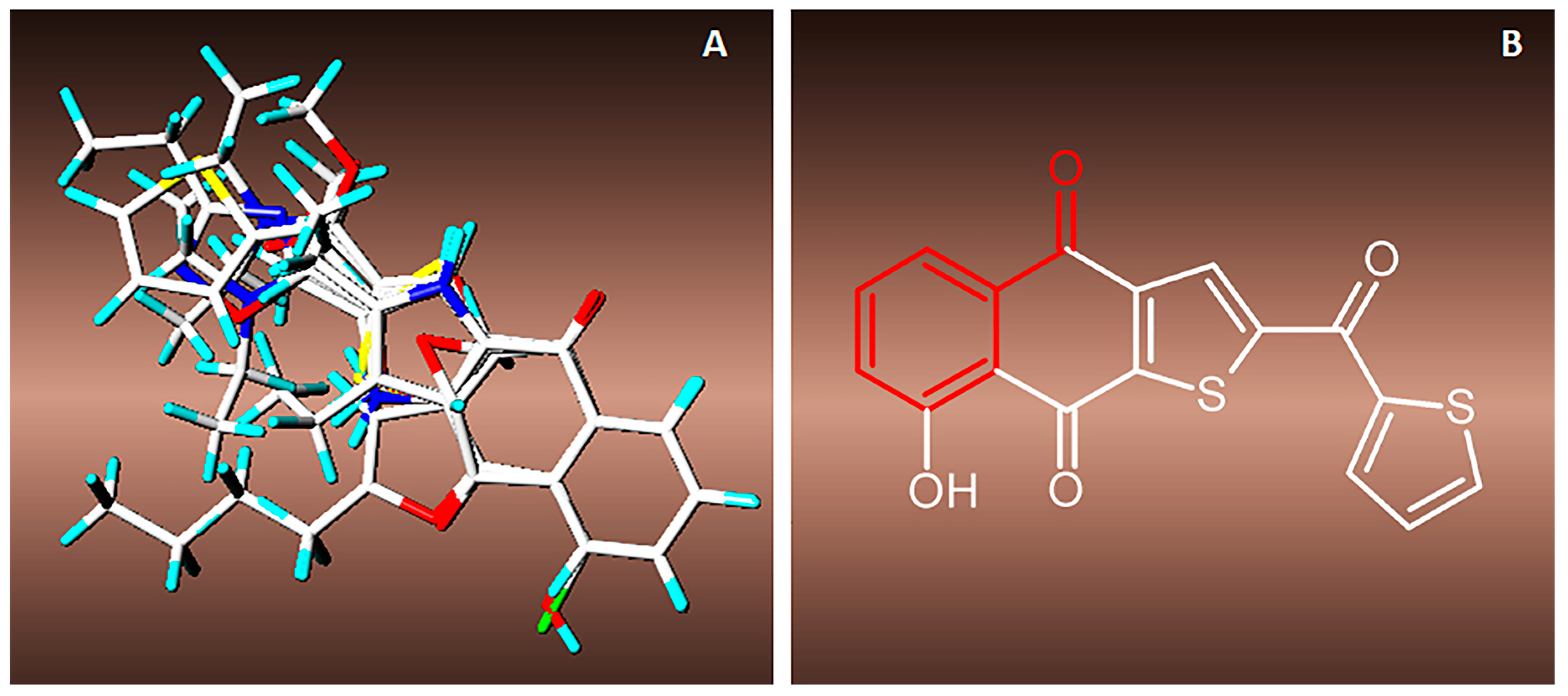
3.2. Synthesis of Naphthoquinone Derivatives
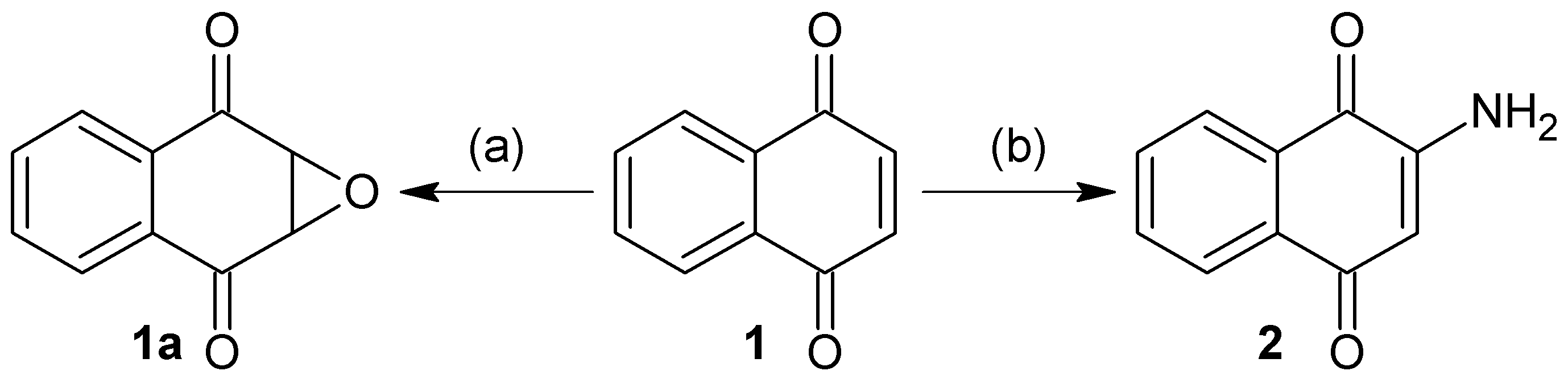
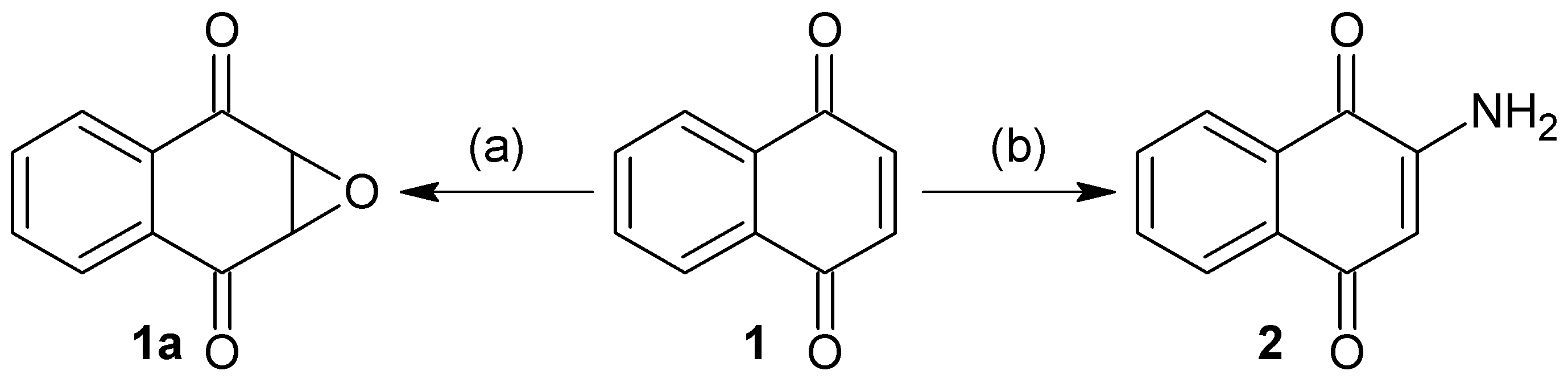
3.2.1. Synthesis of 1a,7a-Dihydronaphtho[2,3-b]oxirene-2,7-dione (1a)
To a solution of 200 mg of 1 in 10 mL of hot ethanol was added 3 mL of H2O2 (30%) with 0.2 g of anhydrous sodium carbonate; the reaction mixture was allowed to cool until the yellow color disappeared completely. Then, 50 mL of cold water was added and the mixture was immersed in an ice bath to form a white precipitate, which was filtered in vacuum to afford white needles; 22.63% yield; m.p. 125 °C; FTIR (KBr) ν: 3000, 2968, 1680, 1590, 1260 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.20 (m, 2H), 7.80 (m, 2H), 3.90 (s, 2H) ppm; 13C-NMR (600 MHz, CDCl3): δ 191.150 (C-1; C-4), 135.162 (C-9; C-10), 134.72 (C-6; C-7); 127.673 (C-5; C-8), 55.74 ppm (C-2; C-3); MS m/z 174 [M+, C10H6O3].
3.2.2. Synthesis of 2-Aminonaphthalene-1,4-dione (2)
To a previously stirred solution of 2 g of 1 in 100 mL of ethanol was added 4.74 g of sodium azide in 34 mL of distilled water, then adjusted to pH 4 with HCl and allowed to stir for 15 h at reflux temperature. After completion of the reaction time, extraction was performed with ethyl acetate (3 × 100 mL). The organic extracts were washed with saline solution, dried over anhydrous Na2SO4 and concentrated. The residue was crystallized from a mixture of diethyl ether/hexane to afford an orange powder; 90% yield; m.p. 207.5 °C; FTIR (KBr) ν: 3381, 3191, 3065, 2923, 1685, 1613 cm−1; 1H-NMR (300 MHz, CDCl3): δ 8.10 (m, 2H), 7.67 (m, 2H), 6.0 (s, 1H), 5.12 (s, 2H) ppm; 13C-NMR (300 MHz, CDCl3): δ 186.20 (C-4), 184.01 (C-1), 151.52 (C-2), 136.34 (C-9), 135.31 (C-10), 133.80 (C-6), 132.52 (C-7), 126.90 (C-8), 126.71 (C-5), 104.90 (C-3) ppm; MS m/z 173 [M+, C10H7O2N].
3.2.3. General Synthesis of Benzo[f]indole-4,9-diones (2a–2f)
A mixture of 109 mg (0.64 mmol) of 2, 6.4 mmol of ketone (or aldehyde) and 1.04 g (3.88 mmol) of manganese (III) acetate in 10 mL of acetonitrile was heated at 80 °C for 16 h; at this point, 1.04 g (3.88 mmol) of manganese (III) acetate was added again and heated for another 23 h, then the mixture reaction was diluted with 100 mL of ethyl acetate, washed with 50 mL of saturated aqueous sodium bisulfate, three time with 50 mL of water, and dried over Na2SO4. The solvent was evaporated under reduce pressure and the crude product was purified by column chromatography (SiO2; CH2Cl2/hexane, 1/1), followed by crystallization (ethyl acetate–hexane).
1H-Benzo[f]indole-4,9-dione (2a). Yellow powder; 44% yield; m.p. 281–283 °C; FTIR (KBr) ν: 3424, 3292, 1655, 1387 cm−1; 1H-NMR (300 MHz, DMSO-d): δ 8.01 (m, 2H), 7.75 (m, 2H), 6.42 (s, 1H), 2.3 (s, 3H) ppm; 13C-NMR (300 MHz, DMSO-d): δ 180.76 (C-4), 175.16 (C-9), 134.08 (C-12), 133.95 (C-10), 133.86 (C-11), 133.55 (C-7), 132.73 (C-6), 128.39 (C-2), 127.53 (C-8), 126.73 (C-5), 126.42 (C-13), 108.55 (C-3) ppm; MS m/z 197.19 [M+, C12H7O2N].
2-Methyl-1H-benzo[f]indole-4,9-dione (2b). Yellow powder; 34% yield; m.p. 302–304 °C; FTIR (KBr) ν: 3454, 3218,1648, 1223 cm−1; 1H-NMR (300 MHz, DMSO-d): δ 8.03 (m, 2H), 7.7 (m, 2H), 7.35 (s, 1H), 6.69 (s, 1H) ppm; 13C-NMR (300 MHz, DMSO-d): δ 180.80 (C-4), 174.24 (C-9), 139.45 (C-12), 133.92 (C-10), 133.79 (C-11), 133.68 (C-7), 133.64 (C-6), 131.89 (C-2), 128.17 (C-5), 126.57, 126.26 (C-8), 126.0 (C-13), 107.04 (C-3), 13.13 (C-14) ppm; MS m/z 211.21 [M+, C13H9O2N].
2-Ethyl-1H-benzo[f]indole-4,9-dione (2c). Yellow powder; 53% yield; m.p. 200–207 °C; FTIR (KBr) ν: 3485, 3201, 1652, 1393 cm−1; 1H NMR (300 MHz, DMSO- d) δ 8.01 (m, 2H), 7.74 (m, 2H), 7.15 (s, 1H), 2.76 (q, J = 7.4 Hz, 2H), 1.19 (t, J = 7.4 Hz, 3H) ppm; 13C-NMR (300 MHz, DMSO-d): δ 181.47 (C-4), 175.06 (C-9), 134.59 (C-12), 133.72 (C-10), 133.65 (C-11), 133.52 (C-7), 133.47 (C-6), 129.08 (C-2), 126.95 (C-5), 126.49 (C-8), 126.16 (C-13), 124.13 (C-3), 19.28 (C-14), 14.79 (C-15); MS m/z 225.24 [M+, C14H11O2N].
2,3-Diethyl-1H-benzo[f]indole-4,9-dione (2d). Orange powder; 36% yield; m.p. 205–209 °C; FTIR (KBr) ν: 3446, 3214, 1655, 1467 cm−1; 1H NMR (300 MHz, DMSO-d) δ 13.17 (s, 1H), 8.06 (d, J = 4.5 Hz, 2H), 7.81 (m, 2H), 3.03 (q, J = 7.1 Hz, 2H), 2.70 (q, J = 7.4 Hz, 2H), 1.1 (m, 6H) ppm; 13C-NMR (300 MHz, DMSO-d): δ 180.91 (C-4), 175.36 (C-9), 146.74 (C-12), 134.80 (C-10), 134.25 (C-11), 133.99 (C-7), 132.69 (C-6), 131.73 (C-2), 127.10 (C-5), 126.19 (C-8), 124.32 (C-13), 121.34 (C-3), 19.91 (C-14), 19.02 (C-16), 14.74 (C-15), 8.96 (C-17); MS m/z 253.29 [M+, C16H15O2N].
3-Methyl-2-(propan-2-yl)-1H-benzo[f]indole-4,9-dione (2e). Orange powder; 42% yield; m.p. 245–248 °C; FTIR (KBr) ν: 3423, 3198, 1655, 1378 cm−1; 1H NMR (300 MHz, DMSO-d) δ 7.99 (m, 2H), 7.74 (m, 2H), 2.65 (m, 1H), 2.22 (s, 3H), 1.01 (m, 6H) ppm; 13C-NMR (300 MHz, DMSO-d): δ 181.62 (C-4), 173.85 (C-9), 136.24 (C-12), 134.29 (C-10), 133.61 (C-11), 133.52 (C-7), 130.74 (C-6), 126.42 (C-2), 126.01 (C-5), 125.22 (C-8), 124.64 (C-13), 106.24 (C-3), 17.75 (C-14), 15.18 (C-15; C-16), 10.82 (C-17); MS m/z 253.31 [M+, C16H15O2N].
3-Methyl-2-propyl-1H-benzo[f]indole-4,9-dione (2f). Orange powder; 41% yield; m.p. 266–270 °C; FTIR (KBr) ν: 3424, 1655 cm−1; 1H NMR (300 MHz, DMSO-d) δ 7.99 (m, 2H), 7.72 (m, 2H), 2.60 (m, 2H), 2.26 (s, 3H), 1.64 (m, 2H), 1.15 (t, 3H) ppm; 13C-NMR (300 MHz, DMSO-d): δ 181.91 (C-4), 173.95 (C-9), 143.12 (C-12), 134.43 (C-10), 133.87 (C-11), 133.47 (C-7), 133.38 (C-6), 131.25 (C-2), 126.34 (C-5), 126.03 (C-8), 125.34 (C-13), 117.84 (C-3), 25.54 (C-14), 18.80 (C-15), 14.32 (C-17), 10.07 (C-16); MS m/z 253.29 [M+, C16H15O2N].
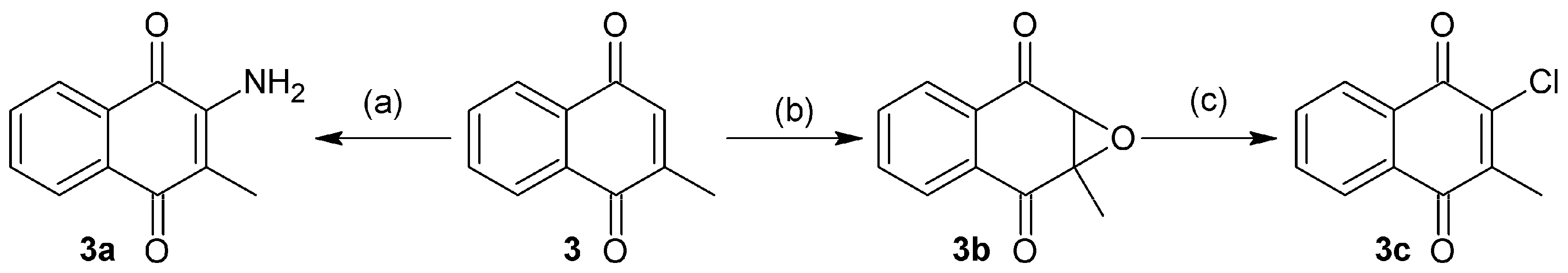
3.2.4. Synthesis of 2-Amino-3-methylnaphthalene-1,4-dione (3a)
Compound 3a was obtained with the same procedure for the synthesis of 2, using 2-methyl-1,4-naphthoquinone as starting material and 36 h for reaction time. This afforded a red powder; 68.66% yield; m.p. 158 °C; FTIR (KBr) ν: 3455, 3411, 2940, 1670, 1628, 1590 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.1 (m, 2H), 7.7 (m, 2H), 5.15 (s, 2H), 2.01 (s, 3H) ppm; 13C-NMR (600 MHz, CDCl3): δ 182.95 (C-1), 181.22 (C-4), 145.32 (C-2), 134.56 (C-10), 133.22 (C-9), 132.06 (C-7), 130.48 (C-6), 126.27 (C-8), 125.82 (C-5), 113.29 (C-3), 12.981 (C-11) ppm; MS m/z 187.19 [M+, C11H9O2N].
3.2.5. Synthesis of 1a-Methyl-1a,7a-dihydronaphtho[2,3-b]oxirene-2,7-dione (3b)
To a solution of 1.25 g of 3 in 20 mL of hot ethanol was added 7.5 mL of H2O2 (30%) with 0.5 g of sodium carbonate. The reaction mixture was allowed to reach room temperature (rt) until the yellow color of the quinone completely disappeared. Then, 125 mL of cold water was added and the mixture was immersed in an ice bath to form a white precipitate which was filtered in vacuum to afford white solid; 65.5% yield; m.p. 138 °C; FTIR (KBr) ν: 3095, 3039, 2995, 2939, 1680, 1590, 1260 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.04 (m, 2H), 7.76 (m, 2H), 3.86 (s, 1H), 1.75 (s, 1H) ppm; 13C-NMR (600 MHz, CDCl3): δ 192.36 (C-1), 192.19 (C-4), 134.99 (C-9), 134.80 (C-10), 132.56 (C-7), 132.44 (C-6), 127.89 (C-8), 127.25 (C-5), 61.82 (C-2), 59.67 (C-3), 15.13 (C-11) ppm; MS m/z 188.18 [M+, C11H8O3].
3.2.6. Synthesis of 2-Chloro-3-methylnaphthalene-1,4-dione (3c)
To 100 mL of HCl (5%) in methanol was added 280 mg of 3b and refluxed at 70 °C for 3h. The obtained mixture was extracted with ethyl acetate (3 × 100 mL), then the organic phase was filtered in the presence of anhydrous Na2SO4, the solvent was removed by rotaevaporation. The obtained residue was purified by column chromatography (SiO2; CH2Cl2) to afford yellow needles; 25.5% yield; m.p. 158 °C; FTIR (KBr) ν: 3095, 3038, 3000, 2957, 1669, 1587, 705 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.10 (m, 2H), 7.71 (m, 2H), 2.28 (s, 3H) ppm; 13C-NMR (600 MHz, CDCl3): δ 183.05 (C-1), 178.01 (C-4), 145.32 (C-3), 143.77 (C-2), 134.59 (C-6), 134.35 (C-7), 132.14 (C-10), 131.83 (C-9), 127.62 (C-5), 127.44 (C-8), 14.92 (C-11) ppm; MS m/z 206.63 [M+, C11H7O2Cl].
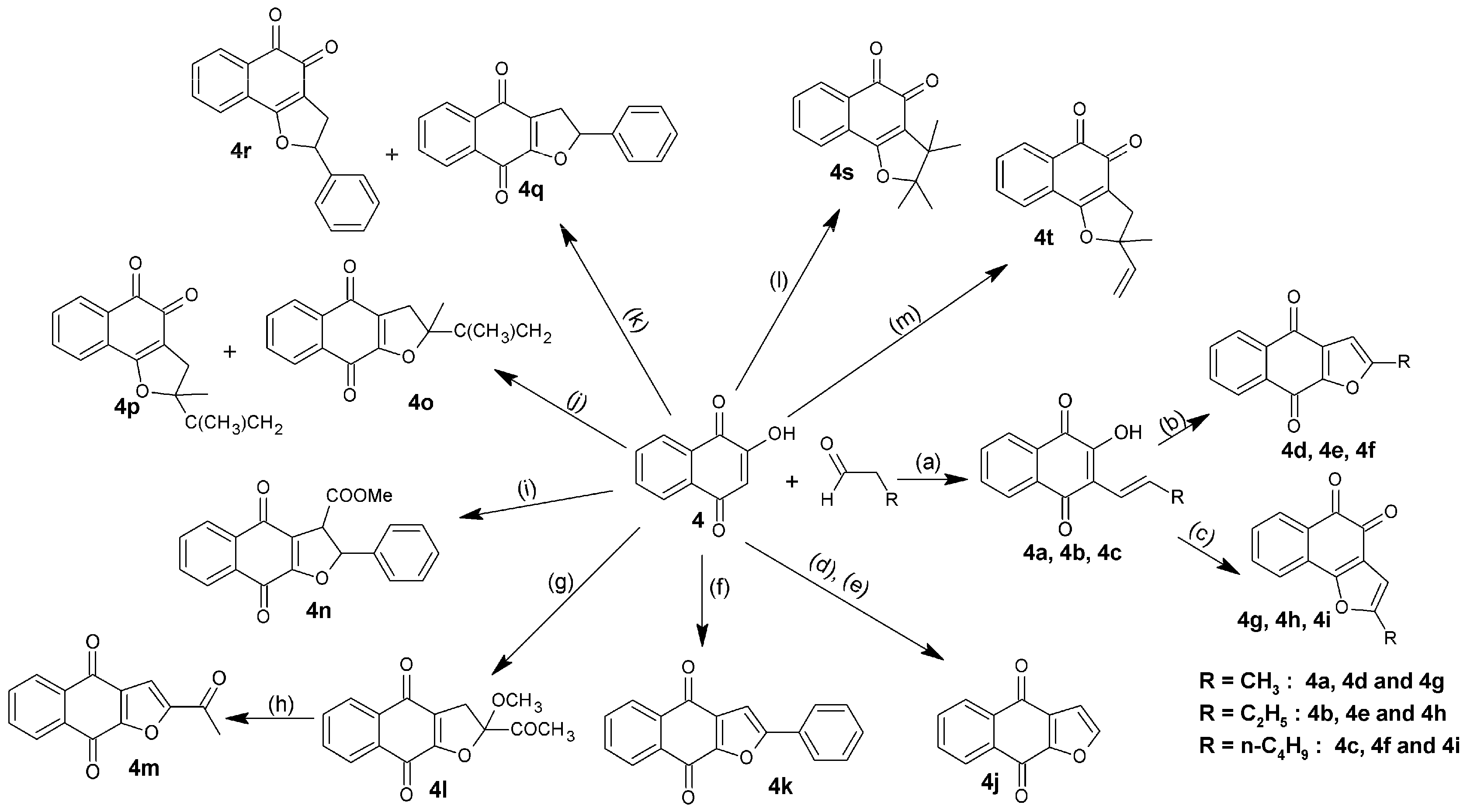
3.2.7. General Synthesis of Hydroxynaphthalene-1,4-diones (4a–4c)
Two grams of 4 were dissolved in 30 mL of glacial acetic acid, and then 2 mL of hydrochloric acid and 7 mL of aldehyde (propanal, butanal or hexanal) were added. The mixture was heated at 60 °C until disappearance of the starting compounds, monitored by analytical TLC. The obtained mixture was mixed with 200 mL of water and kept in the dark overnight, affording a dark oily phase and a clear aqueous layer, which was separated by filtration. Subsequently, the oil phase was dissolved in diethyl ether and extraction was carried out with NaOH (1%), obtaining an intense purple alkaline solution. HCl diluted was then added dropwise until purple color of the solution turned to orange, causing the formation of a precipitate. The solid formed was filtered, washed, extracted with hexane and purified by crystallization from ethanol/water.
2-Hydroxy-3-[(1E)-prop-1-en-1-yl]naphthalene-1,4-dione (4a). Orange scales; 41.5% yield; m.p. 135 °C; r.t.: 90 min; FTIR (KBr) ν: 3231.84, 2962.76, 1654.01, 1614.47, 1573 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.11 (dd, J = 24.9, 7.4 Hz, 2H), 7.73 (m, 3H), 7.09 (dd, J = 15.9, 7.0 Hz, 1H), 6.65 (d, J = 16.1 Hz, 1H), 2.01 (d, J = 6.6 Hz, 3H); 13C-NMR (600 MHz, CDCl3): δ 187.55 (C-1), 181.49 (C-4), 149.84 (C-2), 133.97 (C-12), 133.69 (C-7), 133.06 (C-6), 131.96 (C-10), 131.34 (C-9), 128.24 (C-8), 126.99 (C-5), 123.67 (C-11), 116.49 (C-3), 17.20 (C-13) ppm; MS m/z 214 [M+, C13H10O3].
2-[(1E)-But-1-en-1-yl]-3-hydroxynaphthalene-1,4-dione (4b). Orange powder; 47.5% yield; m.p. 107 °C; r.t.: 75 min; FTIR (KBr) ν: 3329.25, 3069.81, 3039.91, 2962.76, 2921.29, 2880.78, 1649.19, 1593.25 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.11 (dd, J = 24.8, 7.5 Hz, 2H), 7.74 (m, 3H), 7.13 (dt, J = 16.1, 6.7 Hz, 1H), 6.63 (d, J = 16.2 Hz, 1H), 2.24 (m, 2H), 1.67 (s, 14H), 1.14 (t, J = 7.4 Hz, 3H); 13C-NMR (600 MHz, CDCl3): δ 187.48 (C-1), 182.11 (C-4), 150.44 (C-3), 142.36 (C-12), 133.97 (C-7), 133.06 (C-6), 131.96 (C-9), 131.34 (C-10), 128.24 (C-8), 126.99 (C-5), 123.28 (C-11), 116.37 (C-2), 23.89 (C-13), 13.84 (C-14) ppm; MS: m/z 228 [M+, C14H12O3].
2-[(1E)-Hex-1-en-1-yl]-3-hydroxynaphthalene-1,4-dione (4c). Orange powder; 40.1% Yield; m.p. 85 °C; r.t.: 40 min; FTIR (KBr) ν: 3268.49, 2958.9, 2925.15, 2869.56, 1653.06, 1620.26, 1568.18 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.10 (dd, J = 25.1, 7.5 Hz, 2H), 7.77 (m, 3H), 7.09 (m, 1H), 6.63 (d, J = 16.2 Hz, 1H), 2.32 (q, J = 7.2 Hz, 2H), 1.44 (m, 4H), 0.93 (m, 3H); 13C-NMR (600 MHz, CDCl3): δ 187.48 (C-1), 182.11 (C-4), 150.44 (C-3), 140.39 (C-12), 133.97 (C-7), 133.06 (C-6), 131.96 (C-9), 131.34 (C-10), 128.24 (C-8), 127.81 (C-5), 126.99 (C-11), 116.37 (C-2), 31.63 (C-13), 31.33 (C-14), 21.86 (C-15), 14.02 (C-16) ppm; MS: m/z 256 [M+, C16H16O3].
3.2.8. General Synthesis of Naphtho[2,3-b]furan-4,9-diones and Naphtho[1,2-b]furan-4,5-diones (4d–4i)
Two grams of 4a, 4b or 4c were added in a solution of 6 g of mercuric acetate in 60 mL of acetic acid. The mixture was allowed to stir continuously for 30 min at r.t., subsequently warmed to 65 °C and allowed to cool to r.t., then the solvent was removed by rotaevaporation and ethyl ether was added to the concentrate. This solution was filtered to remove the non-soluble impurities and the ether was allowed to evaporate to form a very viscous, deep red liquid which was diluted in 30 mL of ethanol and 10 mL of concentrated HCl, then heated to 65 °C for 2 h with constant stirring. Once the reaction was completed and r.t. was reached, a pinch of activated carbon was added and filtered. Finally, 250 mL of water was added to the solution with caution, precipitating the compound (4d, 4e or 4f) as a yellow powder which was filtered, washed and dried. Compounds 4g, 4h and 4i were obtained as an orange powder, following the same procedure described above, with changes in the amount of ethanol (40 mL instead of 30 mL), HCl (1 mL instead of 10 mL) and reaction time (15 min/65 °C instead of 2 h/65 °C).
2-Methylnaphtho[2,3-b]furan-4,9-dione (4d). Yellow powder; 21.3% yield; m.p. 174 °C; FTIR (KBr) ν: 2924.18, 1676.2, 1585.5, 1533.46, 1206.51 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.11 (m, 2H), 7.67 (m, 2H), 6.54 (t, J = 2.4 Hz, 1H), 2.44 (d, J = 0.7 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 180.85 (C-4), 173.11 (C-9), 160.48 (C-2), 151.67 (C-13), 133.82 (C-6), 133.57 (C-7), 133.09 (C-12), 132.51 (C-10), 131.93 (C-11), 126.88 (C-8), 126.79 (C-5), 105.00 (C-3), 14.14 (C-14) ppm; MS: m/z 212 [M+, C13H8O3].
2-Ethylnaphtho[2,3-b]furan-4,9-dione (4e). Yellow powder; 42.3% yield; m.p. 143 °C; FTIR (KBr) ν: 3117.07, 2978.19, 2930.93, 1673.3, 1530.57, 1202.66 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.18 (m, 2H), 7.74 (p, J = 7.6 Hz, 2H), 6.63 (s, 1H), 2.86 (q, J = 7.5 Hz, 2H), 1.37 (t, J = 7.6 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 180.91 (C-4), 173.17 (C-9), 165.88 (C-2), 151.56 (C-13), 133.78 (C-6), 133.53 (C-7), 133.09 (C-12), 132.57 (C-10), 131.78 (C-11), 126.85 (C-8), 126.78 (C-5), 103.51 (C-3), 21.80 (C-14), 11.60 (C-15) ppm; MS: m/z 226 [M+, C14H10O3].
2-Butylnaphtho[2,3-b]furan-4,9-dione (4f). Yellow powder; 39.2% yield; m.p. 104 °C; FTIR (KBr): ν: 3121.09, 3068.24, 2936.72, 2863.42, 1668.48, 1530.57, 1212.3 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.2 (m, 2H), 7.75 (m, 2H), 6.63 (s, 1H), 2.83 (t, J = 7.6 Hz, 2H), 1.76 (m, 2H), 1.43 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 180.97 (C-4), 173.14 (C-9), 164.85 (C-2), 151.56 (C-13), 133.80 (C-6), 133.53 (C-7), 133.11 (C-12), 132.60 (C-10), 131.79 (C-11), 126.85 (C-8), 126.79 (C-5), 104.16 (C-3), 29.51 (C-14), 28.04 (C-15), 22.18 (C-16), 13.68 (C-17) ppm; MS: m/z 254 [M+, C16H14O3].
2-Methylnaphtho[1,2-b]furan-4,5-dione (4g). Orange powder; 12.3% Yield; m.p. 124 °C; FTIR (KBr) ν: 3122.86, 2923.22, 1673.3, 1554.68, 1215.19 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.04 (m, 1H), 7.63 (m, 2H), 7.43 (ddd, J = 7.7, 6.8, 2.0 Hz, 1H), 6.45 (d, J = 1.1 Hz, 1H), 2.43 (d, J = 1.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 180.71 (C-5), 174.38 (C-4), 159.62 (C-2), 155.90 (C-13), 135.34 (C-8), 130.45 (C-10), 129.78 (C-6), 128.69 (C-7), 128.57 (C-11), 122.67 (C-9), 121.95 (C-12), 104.52 (C-3), 13.61 (C-14) ppm; MS: m/z 212 [M+, C13H8O3].
2-Ethylnaphtho[1,2-b]furan-4,5-dione (4h). Orange powder; 35.6% yield; m.p. 136 °C; FTIR (KBr): ν: 3119.96, 2976.26, 2927.26, 1668.48, 1549.86, 1208.44 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.03 (m, 1H), 7.63 (m, 2H), 7.42 (ddd, J = 7.7, 7.1, 1.7 Hz, 1H), 6.43 (m, 1H), 2.76 (qd, J = 7.5, 1.0 Hz, 2H), 1.32 (t, J = 7.5 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 180.74 (C-5), 174.43 (C-4), 161.46 (C-2), 159.55 (C-13), 135.33 (C-8), 130.42 (C-10), 129.77 (C-6), 128.74 (C-7), 128.60 (C-11), 122.48 (C-9), 121.99 (C-12), 103.07 (C-3), 21.31 (C-14), 11.67 (C-15) ppm; MS: m/z 226 [M+, C14H10O3].
2-Butylnaphtho[1,2-b]furan-4,5-dione (4i). Orange powder; 32.7% Yield; m.p. 64 °C; FTIR (KBr): ν: 3109.36, 2943.47, 2861.49, 1668.48, 1552.75, 1206.51 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.03 (d, J = 7.5 Hz, 1H), 7.63 (qd, J = 7.6, 1.2 Hz, 2H), 7.42 (td, J = 7.7, 1.7 Hz, 1H), 6.44 (s, 1H), 2.72 (t, J = 7.5 Hz, 2H), 1.72 (tt, J = 15.3, 7.7 Hz, 2H), 1.44 (dq, J = 14.6, 7.4 Hz, 2H), 0.97 (m, 3H); 13C-NMR (101 MHz, CDCl3): δ 180.77 (C-5), 174.47 (C-4), 160.32 (C-2), 159.54 (C-13), 135.33 (C-8), 130.43 (C-10), 129.76 (C-6), 128.78 (C-7), 128.59 (C-11), 122.50 (C-9), 122.01 (C-12), 103.69 (C-3), 29.55 (C-14), 27.56 (C-15), 22.13 (C-16), 13.73 (C-17) ppm; MS: m/z 254 [M+, C16H14O3].
3.2.9. Synthesis of Naphtho[2,3-b]furan-4,9-dione (4j)
Vinyl acetate and 1 in a molar relation 1:1, were dissolved in acetone. The mixture was irradiated with a mercury lamp of 450 W of high pressure at r.t. for 7 h, to yield a yellow precipitate. Subsequently, precipitate was reacted with p-TsOH in dry benzene at reflux temperature to afford yellow crystals; 65.6% yield; m.p. 229.4 °C; FTIR (KBr) ν: 3069, 1679, 1646, 1206 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.16 (m, 2H), 8.14 (d, 1H), 7.88 (m, 2H), 7.08 (d, 1H) ppm; 13C-NMR (600 MHz, CDCl3): δ 180.65 (C-4), 180.26 (C-9), 158.36 (C-13), 156.01 (C-2), 136.34 (C-6), 135.16 (C-7), 134.07 (C-12), 133.24 (C-10), 133.36 (C-11), 129.05 (C-8), 128.27 (C-5), 110.11 (C-3); MS m/z 198 [M+, C12H6O3].
3.2.10. Synthesis of 2-Phenylnaphtho[2,3-b]furan-4,9-dione (4k)
Phenylacetylene (2.93 g) and 1 (500 mg) were dissolved in 115 mL of acetonitrile by stirring at r.t. This solution was then cooled to 0 °C and 3.16 g of CAN was added in portions to the yellow solution, which immediately turned orange, this solution was kept at 0 °C for 20 min until the reaction was quenched with saturated solution of NH4Cl. The reaction mixture was extracted with ethyl acetate (4 × 20 mL), washed with saturated solution of NaHCO3, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by open column chromatography (SiO2; CH2Cl2) to afford yellow crystals; 18.2% yield, m.p. 220 °C; FTIR (KBr) ν: 3046, 1665, 1580, 1530, 1252 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.10 (m, 2H), 7.80 (m, 2H), 7.70 (m, 2H), 7.40 (m, 3H), 7.10 (s, 1H) ppm; 13C-NMR (600 MHz, CDCl3): δ 181.27 (C-4), 173.49 (C-9), 160.79 (C-2), 152.01 (C-13), 134.43 (C-6), 134.06 (C-7), 133.53 (C-10), 133.30 (C-11), 132.86 (C-12), 130.72 (C-1’), 129.54 (C-4’), 128.75 (C-3’; C-5’), 127.40 (C-8), 127.35 (C-5), 125.99 (C-2’; C-6’), 103.37 (C-3) ppm; MS m/z 274.27 [M+, C18H10O3].
3.2.11. Synthesis of 2-Acetyl-2-methoxy-2,3-dihydronaphtho[2,3-b]furan-4,9-dione (4l)
To a solution of 1 g of 1 and 1.5 mL of 2,3-dimethoxy-1,3-butadiene in 60 mL of acetonitrile was added 9.58 g of CAN. The reaction was carried out at 0 °C for 6 h. The obtained mixture was diluted with water and extracted with ethyl acetate which was subsequently evaporated. The residue was purified by chromatography (SiO2; hexane/ethyl acetate, 5/1) to afford yellow powder; 51.43% yield; m.p. 145 °C; FTIR (KBr): ν: 3069, 2946, 2843, 1690, 1673, 1634, 1560, 1260 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.11 (m, 2H), 7.75 (m, 2H), 3.66 (d, 1H), 307 (d, 1H), 3.36 (s, 3H), 3.30 (s, 3H); 13C-NMR (600 MHz, CDCl3): δ 182.00 (C-14), 178.00 (C-4), 172.00 (C-9), 158.97 (C-13), 134.89 (C-6), 134.75 (C-7), 133.52 (C-10), 126.80 (C-11), 126.51 (C-8), 124.65 (C-5), 118.20 (C-12), 102.32 (C-2), 51.27 (C-16), 27.33 (C-3), 17.03 (C-15) ppm; MS m/z 272.25 [M+, C15H12O5].
3.2.12. Synthesis of 2-Acetylnaphtho[2,3-b]furan-4,9-dione (4m)
To a solution of 266 mg of 4l in 15 mL of benzene was added 0.3 mL of 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU), the mixture was allowed to react at r.t. for 5 h. The reaction was quenched with aqueous ammonium chloride solution and extracted with ethyl acetate. After drying the solvent, the residue was purified by chromatography (SiO2; hexane/ethyl acetate, 4/1) initially, following by CH2Cl2 to afford yellow solid; 31% yield; m.p. 209 °C; FTIR (KBr): ν: 3113, 3017, 2940, 1690, 1672, 1582, 1262 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.20 (m, 2H), 7.77 (m, 2H), 7.53 (s, 1H), 2.59 (s, 1H) ppm; 13C-NMR (600 MHz, CDCl3): δ 188.23 (C-14), 180.29 (C-4), 174.44 (C-9), 155.76 (C-2), 153.33 (C-13), 134.96 (C-6), 134.83 (C-7), 133.45 (C-10), 132.94 (C-11), 131.10 (C-12), 127.76 (C-8), 127.62 (C-5), 112.95 (C-3), 27.04 (C-15) ppm; MS m/z 240.21 [M+, C14H8O4].
3.2.13. Synthesis of Methyl 4,9-Dioxo-2-phenyl-2,3,4,9-tetrahydronaphtho[2,3-b]furan-3-carboxylate (4n)
To a solution of 200 mg of 1 in 46 mL of acetone was added 931 mg of methyl (2E) 2-phenylpropionate, and irradiated with pyrex® glass filtered ultraviolet light at r.t. for 22 h, then the solvent was removed by evaporation under reduced pressure and the crude reaction product was purified by chromatography (SiO2; CH2Cl2) to afford yellow crystals; 20% yield; m.p. 167 °C; FTIR (KBr): ν: 3066, 3030, 2953, 1741, 1680, 1649, 1587, 1203 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.20 (m, 2H), 7.60 (m, 2H), 7.30 (s, 5H), 6.00 (dd, 1H), 4.30 (dd, 1H), 3.80 (s, 3H) ppm; 13C-NMR (600 MHz, CDCl3): δ 181.57 (C-4), 178.01 (C-9), 171.33 (C-14), 160.86 (C-13), 138.55 (C-1’), 134.95 (C-6), 133.71 (C-7), 133.36 (C-10), 132.02 (C-11), 129.72 (C-8), 129.51 (C-4’), 126.99 (C-3’; C-5’), 126.80 (C-2’; C-6’), 125.98 (C-5), 122.06 (C-12), 90.06 (C-2), 54.57 (C-3), 53.67 (C-15) ppm; MS m/z 334.32 [M+, C20H14O5].
3.2.14. General Synthesis of Dihydronaphtho[2,3-b]furan-4,9-diones and Dihydronaphtho[1,2-b]furan-4,5-diones (4o–4p)
To a solution of 174 mg of 1 and 0.6 mL of 2,3-dimethyl-1,3-butadiene in 25 mL of acetonitrile, 1.206 g of CAN and sodium bicarbonate were added at 0 °C, allowing to react for 3 h. The obtained mixture was extracted with ethyl acetate (3 × 100 mL), the solvent was dried and the residue purified by chromatography (SiO2; CH2Cl2).
2-Methyl-2-(prop-1-en-2-yl)-2,3-dihydronaphtho[2,3-b]furan-4,9-dione (4o). Yellow powder; 47.63% yield; m.p. 135 °C; FTIR (KBr): ν: 3097, 3070, 2985, 2929, 2860, 1680, 1643, 1590, 1254 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.0 (m, 2H), 7.62 (m, 2H), 5.05 (d, 1H), 4.86 (d, 1H), 3.14 (d, 1H), 2,96 (d, 1H), 1.78 (s, 3H), 1.59 (s, 3H) ppm; 13C-NMR (600 MHz, CDCl3): δ 182.91 (C-4), 178.49 (C-9), 159.38 (C-13), 145.99 (C-14), 134.54 (C-6), 133.35 (C-7), 132.09 (C-10), 126.76 (C-11), 126.75 (C-8), 123.81 (C-5), 117.72 (C-12), 111.73 (C-15), 95.01 (C-2), 39.14 (C-3), 26.67 (C-17), 18.86 (C-16) ppm; MS m/z 254.28 [M+, C16H14O3].
2-Methyl-2-(prop-1-en-2-yl)-2,3-dihydronaphtho[1,2-b]furan-4,5-dione (4p). Red powder; 40.55% yield; m.p. 147 °C; FTIR (KBr): ν: 3066, 2980, 2932, 1700, 1647, 1625, 1590, 1254 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.03 (d, 1H), 7.6 (m, 3H), 5.11 (s, 1H), 4.95 (s, 1H), 3.12 (d, 1H), 2.86 (d, 1H), 1.78 (s, 3H), 1.61 (s, 3H) ppm; 13C-NMR (600 MHz, CDCl3): δ 181.59 (C-4), 176.03 (C-5), 169.03 (C-13), 146.17 (C-14), 134.96 (C-8), 132.38 (C-10), 131.34 (C-6), 129.94 (C-7), 129.91 (C-9), 128.18 (C-11), 115.35 (C-12), 111.55 (C-15), 96.63 (C-2), 38.36 (C-3), 26.77 (C-17), 18.91 (C-16) ppm; MS m/z 254.3 [M+, C16H14O3].
3.2.15. General Synthesis of Dihydronaphtho[2,3-b]furan-4,9-diones and Dihydronaphtho[1,2-b]furan-4,5-diones (4q–4r)
Compound 1 (500 mg) and styrene (3 g) were dissolved in 115 mL of acetonitrile. This solution was then cooled to 0 °C and 3.16 g (5.74 mmol) of CAN was added in portions to the yellow solution, which immediately turned orange, this solution was kept at 0 °C for 20 m until the reaction was quenched with saturated solution of NH4Cl. After evaporation of the solvent, the crude product was extracted with ethyl acetate. The organic phase was washed successively with saturated sodium bicarbonate solution and saline solution and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the crude product was purified by chromatography (SiO2; CH2Cl2) initially, followed by (SiO2; hexane/ethyl acetate, 4/1).
2-Phenyl-2,3-dihydronaphtho[2,3-b]furan-4,9-dione (4q). Yellow crystals; 27% yield; m.p. 158 °C; FTIR (KBr): ν: 3057, 3010, 1674, 1647, 1594, 1243 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.00 (m, 2H), 7.60 (m, 2H), 7.30 (s, 5H), 6.00 (dd, 1H), 3.60 (dd, 1H), 3.20 (dd, 1H) ppm; 13C-NMR (600 MHz, CDCl3): δ 182.65 (C-4), 178.17 (C-9), 160.28 (C-13), 139.96 (C-1’), 134.66 (C-6), 133.51 (C-7), 132.04 (C-10), 129.35 (C-11), 126.83 (C-8), 126.55 (C-4’), 126.43 (C-3’; C-5’), 124.29 (C-2’; C-6’), 123.64 (C-5), 123.57 (C-12), 87.22 (C-2), 35.72 (C-3) ppm; MS m/z 276.28 [M+, C18H12O3].
2-Phenyl-2,3-dihydronaphtho[1,2-b]furan-4,5-dione (4r). Orange crystals; 26% yield; m.p. 115 °C; FTIR (KBr): ν: 3062, 3035, 1652, 1618, 1588, 1240 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.10 (m, 1H), 7.60 (m, 3H), 7.30 (s, 5H), 6.00 (dd, 1H), 3.50 (dd, 1H), 3.10 (dd, 1H) ppm; 13C-NMR (600 MHz, CDCl3): δ 182.00 (C-4), 175.86 (C-5), 169.93 (C-13), 139.93 (C-1’), 135.03 (C-8), 132.53 (C-10), 131.24 (C-6), 130.04 (C-7), 129.55 (C-4’), 129.49 (C-3’; C-5’), 127.88 (C-2’; C-6’), 126.48 (C-11), 125.11 (C-9), 115.55 (C-12), 88.79 (C-2), 34.93 (C-3) ppm; MS m/z 276.28 [M+, C18H12O3].
3.2.16. Synthesis 2,2,3,3-Tetramethyl-2,3-dihydronaphtho[1,2-b]furan-4,5-dione (4s)
To a solution of 500 mg of 1 and 2.42 g of 2,3-dimethyl-2-butene in 115 mL of acetonitrile was cooled to 0 °C and 3.16 g of CAN were added in portions to the yellow solution which immediately turned orange, this solution was kept at 0 °C for 20 min until the reaction was quenched with saturated solution of NH4Cl. After evaporation of the solvent under reduced pressure, the crude product was extracted with ethyl acetate (3 × 100 mL). The organic phase was washed successively with saturated solution of NaHCO3, then with saline and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the crude product was separated by chromatography (SiO2; CH2Cl2) to afford orange crystals; 2.5% yield; m.p. 91 °C; FTIR (KBr): ν: 3090, 2980, 1963, 1695, 1638, 1614, 1588 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.06 (m, 1H), 7.65 (m, 2H), 7.58 (m, 1H), 1.48 (s, 6H), 1,36 (s, 6H) ppm; 13C-NMR (600 MHz, CDCl3): δ 181.73 (C-4), 175.63 (C-5), 167.40 (C-13), 134.36 (C-8), 131.58 (C-10), 130.91 (C-6), 129.13 (C-7), 128.26 (C-11), 124.44 (C-9), 123.23 (C-12), 97.23 (C-2), 46.35 (C-3), 23.17 (C-14; C-15), 22.01 (C-16; C-17) ppm; MS m/z 256.30 [M+, C16H16O3].
3.2.17. Synthesis of 2-Ethenyl-2-methyl-2,3-dihydronaphtho[1,2-b]furan-4,5-dione (4t)
To a solution of 174 mg of 1 and 0.6 mL of isoprene in 25 mL of acetonitrile was added 1.206 g of CAN and sodium bicarbonate at 0 °C, allowing reacting for 3 h. The obtained mixture was extracted with ethyl acetate, the solvent was dried and the residue purified by flash chromatography (SiO2; CH2Cl2) to afford yellow powder; 48.75% yield; m.p. 150 °C; FTIR (KBr): ν: 3068, 2926, 1693, 1643, 1612, 1586, 1251 cm−1; 1H-NMR (600 MHz, CDCl3): δ 8.09 (d, 1H), 7.69 (m, 3H), 6.10 (dd, 1H), 5.39 (d, 1H), 5.26 (d, 1H), 3.17 (d, 1H), 3.02 (d, 1H), 1.70 (s, 3H), ppm; 13C-NMR (600 MHz, CDCl3): δ 181.11 (C-4), 175.53 (C-5), 168.44 (C-13), 139.79 (C-14), 134.51 (C-8), 132.05 (C-10), 130.09 (C-6), 129.60 (C-7), 127.75 (C-11), 124.68 (C-9), 114.19 (C-15), 93.86 (C-12), 89.96 (C-2), 38.34 (C-3), 26.95 (C-16) ppm; MS m/z 240.25 [M+, C15H12O3].
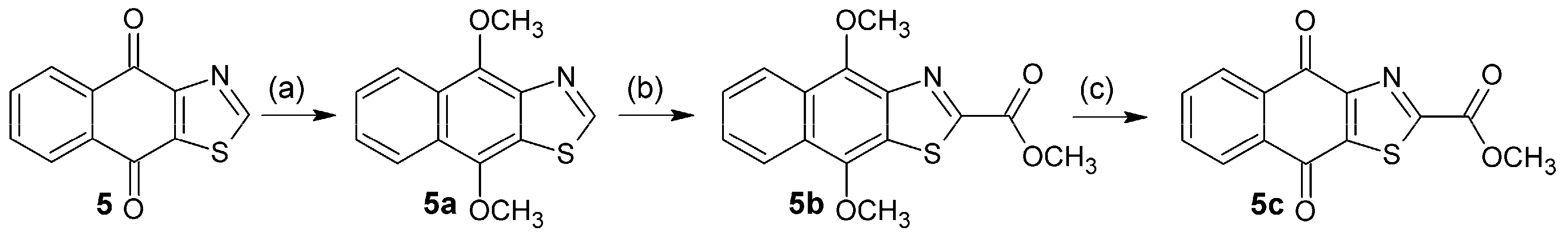
3.2.18. Synthesis of 4,9-Dimethoxy-4,9-dihydronaphtho[2,3-d][1,3]thiazole (5a)
To a solution of 4.30 g (20 mmol) of 5 and 65 mg (0.20 mmol) tetrabutylammonium bromide in 150 mL of dry THF under N2, 34.82 g (200 mmol) sodium dithionite in 100 mL of water was added dropwise. The mixture was vigorously stirred for 30 min and then treated with 22.44 g (400 mmol) KOH in 70 mL of water. After 30 min, it was cooled to 0 °C on an ice-bath, 22.8 mL of dimethyl sulfate (30.27 g, 240 mmol) was added, and the mixture was then stirred for 2 h at 0 °C. Then, it was allowed to warm at rt and stirred for 2 h. Ammonia solution (10%, 50 mL) was added, and the mixture was stirred for 30 min and then extracted with CH2Cl2 (3 × 100 mL). The combined organic phase was washed with water (150 mL), then with a saturated solution of NaCl (150 mL), dried over Na2SO4 and evaporated. The residue was purified by chromatography (SiO2; CH2Cl2) to afford beige crystals; 77% yield; m.p. 85 °C; FTIR (KBr): ν: 2935, 1582 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.93 (s, 1H,), 8.43 (m, 1H), 8.21 (m, 1H), 7.51 (m, 2H), 4.42 (s, 3H), 4.11 (s, 3H); 13C-NMR (400 MHz, CDCl3): δ 153.36 (C-2), 146.14 (C-12), 145.33 (C-4), 142.78 (C-9), 126.51 (C-13), 126.31 (C-10), 125.86 (C-11), 125.49 (C-6), 124.09 (C-7), 123.43 (C-8), 121.17 (C-5), 63.07 (C-14), 60.81 (C-15) ppm; MS m/z 245.29 [M+, C13H11O2NS].
3.2.19. Synthesis of Methyl 4,9-dimethoxy-4,9-dihydronaphtho[2,3-d][1,3]thiazole-2-carboxylate (5b)
Following a procedure reported by Vechorkin et al. [
18], 2.50 g (10.20 mmol)
5a and 4.00 g (12.28 mmol) Cs
2CO
3 were evacuated in a dried Schlenck flask for 1 h. Then, anhydrous DMF (20 mL) was added under N
2, and the reaction vessel was degassed and flushed alternately with N
2 and CO
2 three times each. Then, the reaction mixture was heated at 125 °C under an overpressure of CO
2 (400 mbar) for 16 h. Thereafter, the reaction mixture was allowed to reach 60 °C, methyl iodide (1.87 mL, 4.26 g, 30 mmol) was added with a syringe through a septum, and the mixture was stirred for an additional 2 h at 60 °C. Then, it was cooled to r.t., water (20 mL) was added, and the mixture was extracted with CH
2Cl
2 (3 × 30 mL). The combined organic phase was washed with a saturated solution of NaCl (40 mL) and dried over Na
2SO
4. The solvent was evaporated and the residue purified by chromatography (SiO
2; CH
2Cl
2/hexane, 9/1) to afford orange crystals; 64% yield; m.p. 150 °C; FTIR (KBr): ν: 2970, 1713, 1574 cm
−1;
1H-NMR (600 MHz, CDCl
3): δ 8.46 (d,
3J = 8.7 Hz, 1H), 8.20 (d,
3J = 8.6 Hz, 1H), 7.60 (dd,
3J1 = 8.7 Hz,
3J2 = 6.7 Hz, 1H), 7.54 (dd,
3J1 = 8.6 Hz,
3J2 = 6.7 Hz, 1H), 4.51 (s, 3H), 4.11 (s, 3H), 4,09 (s, 3H);
13C-NMR (600 MHz, CDCl
3): δ 161.35 (C-14), 156.64 (C-2), 148.40(C-9), 144.71 (C-12), 142.02 (C-4), 127.42 (C-13), 127.18 (C-10), 126.55 (C-11), 126.22 (C-6), 125.75 (C-7), 124.00 (C-8), 121.24 (C-5), 63.51 (C-16), 60.93 (C-17), 53.99 (C-15) ppm; MS
m/
z 318.36 [M
+, C
16H
16O
4NS].
3.2.20. Synthesis of Methyl 4,9-Dioxo-4,9-dihydronaphtho[2,3-d][1,3]thiazole-2-carboxylate (5c)
To a suspension of 5b (0.10 g, 0.33 mmol) in MeCN (1 mL) and water (4 mL) at 0 °C was added dropwise within 20 min (NH4)2[Ce(NO3)6] (0.51 g, 0.92 mmol) in MeCN (4 mL) and water (4 mL) under vigorous stirring. The mixture was allowed to react for 30 min under the same conditions. Then it was poured onto ice-water (20 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic phase was washed with a saturated solution of NaCl (2 × 50 mL), dried over Na2SO4, then concentrated, and purified by chromatography (SiO2; CH2Cl2) to afford a yellow solid; 43% yield; m.p. 175–176 °C; FTIR (KBr): ν: 2966, 1720, 1678, 1585 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 8.34 (dd, 3J = 7.5 Hz, 4J = 1.6 Hz, 1H), 8.24 (dd, 3J = 7.3 Hz, 4J = 1.8 Hz, 1H), 7.83 (m, 2H), 4.08 (s, 3H,); 13C-NMR (CDCl3, 600 MHz) δ 178.50 (C-9), 177.22 (C-4), 164.25 (C-14), 159.96 (C-2), 155.12 (C-12), 145.76 (C-10), 135.33 (C-11), 134.69 (C-7), 133.26 (C-6), 133.20 (C-13), 128.46 (C-8), 127.67 (C-5), 54.38 (C-15) ppm; MS m/z 288.3 [M+, C14H10O4NS].

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}