Characterization of Polyphenolic Content in the Aquatic Plants Ruppia cirrhosa and Ruppia maritima —A Source of Nutritional Natural Products



Abstract
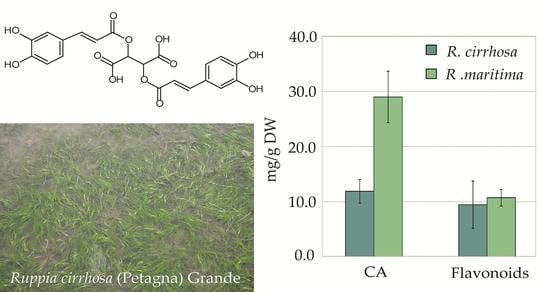
:
1. Introduction
2. Results and Discussion
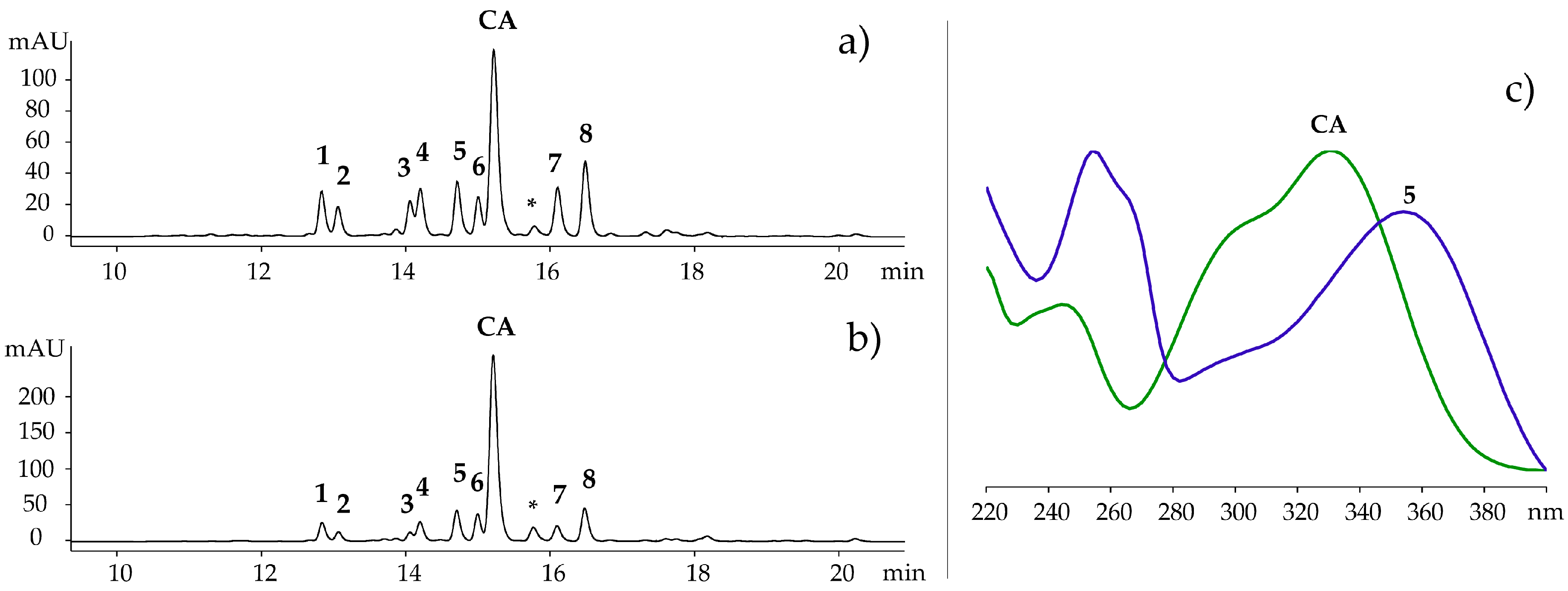
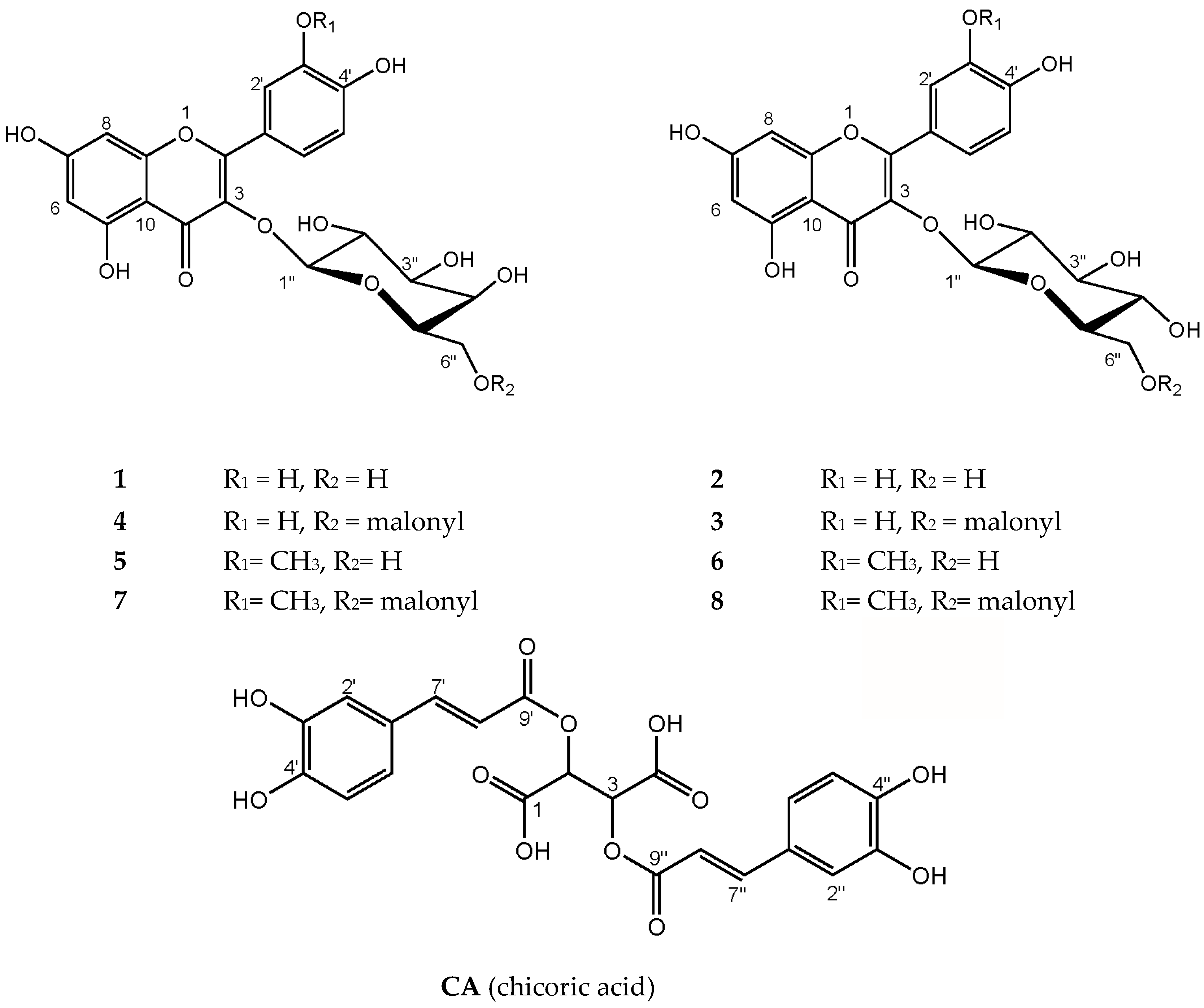
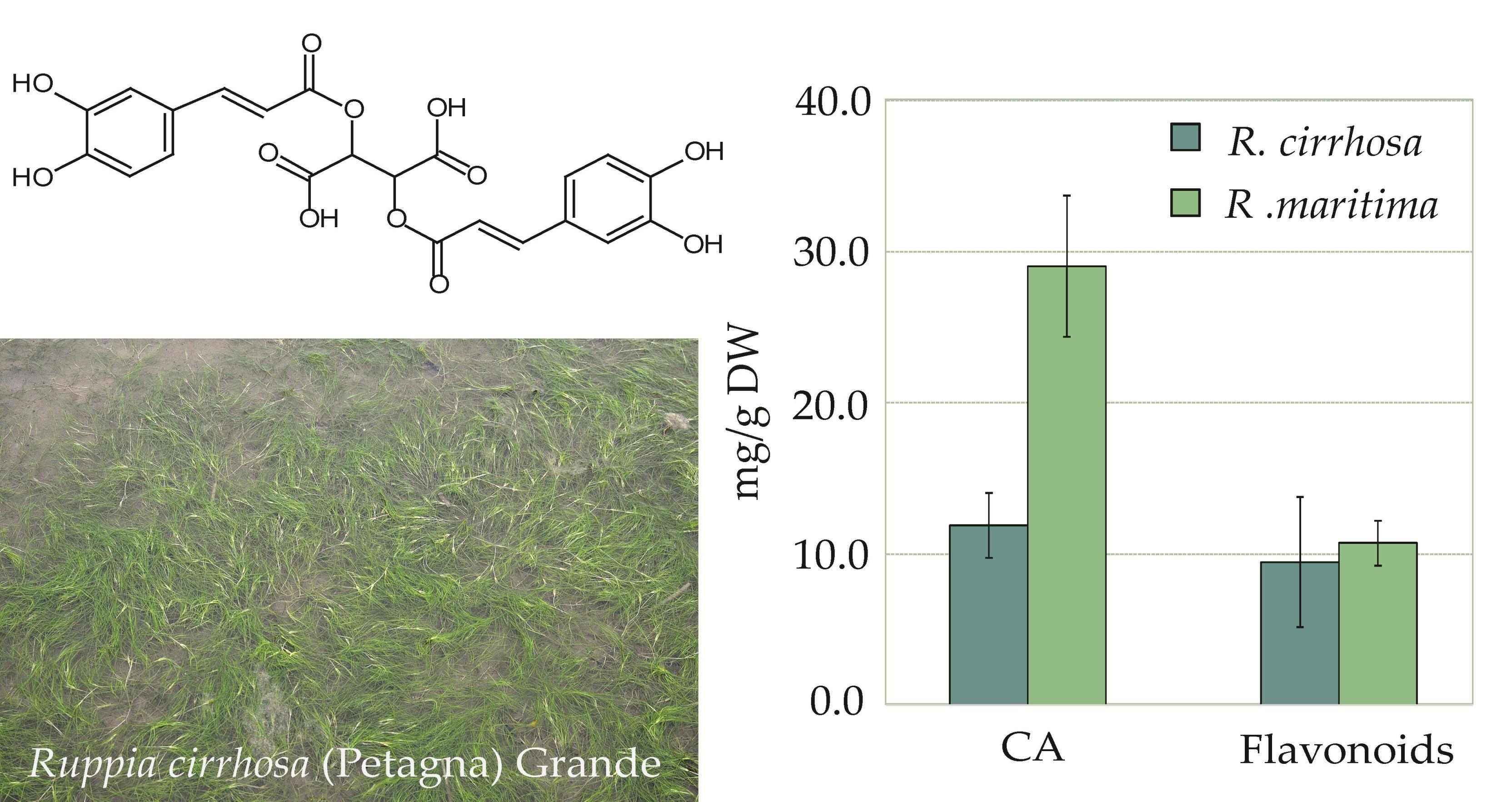
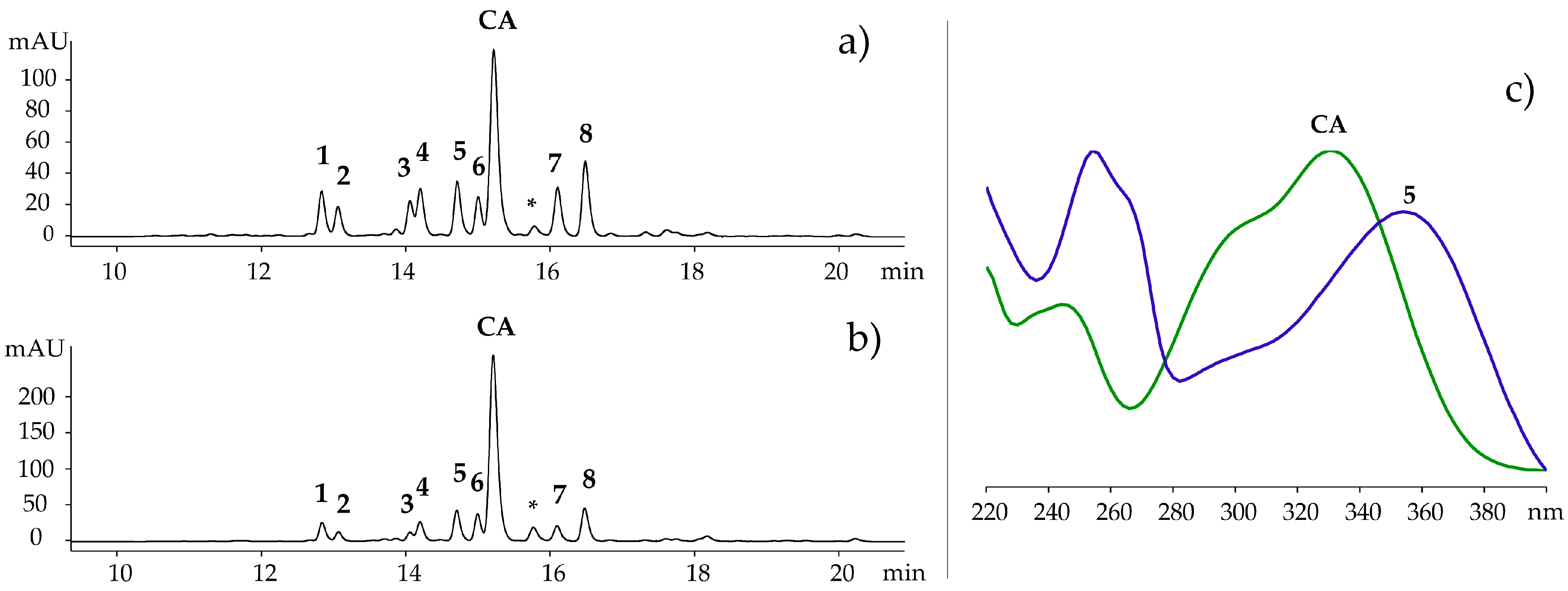
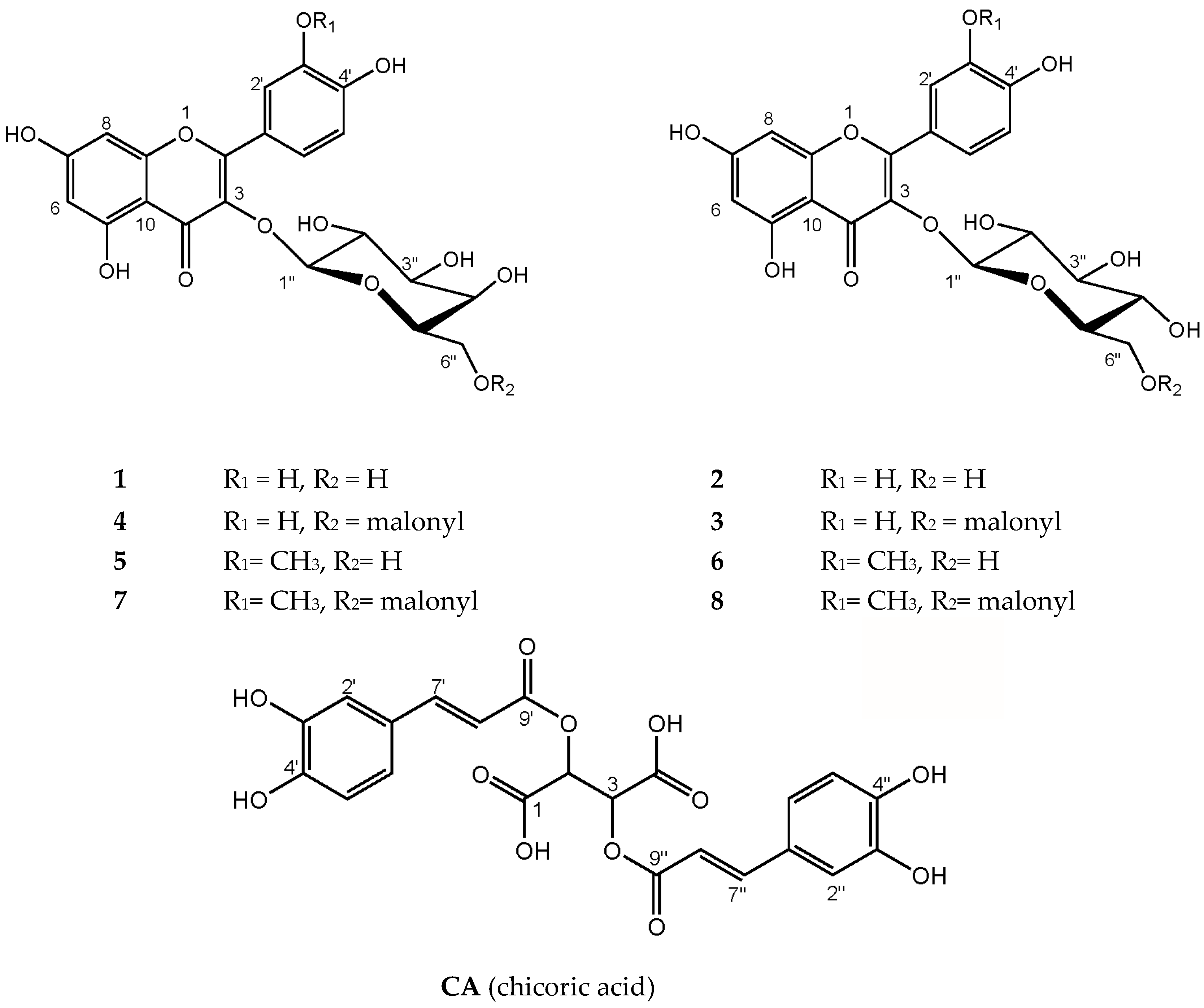
2.1. Characterization of Polyphenolic Compounds in Ruppia cirrhosa
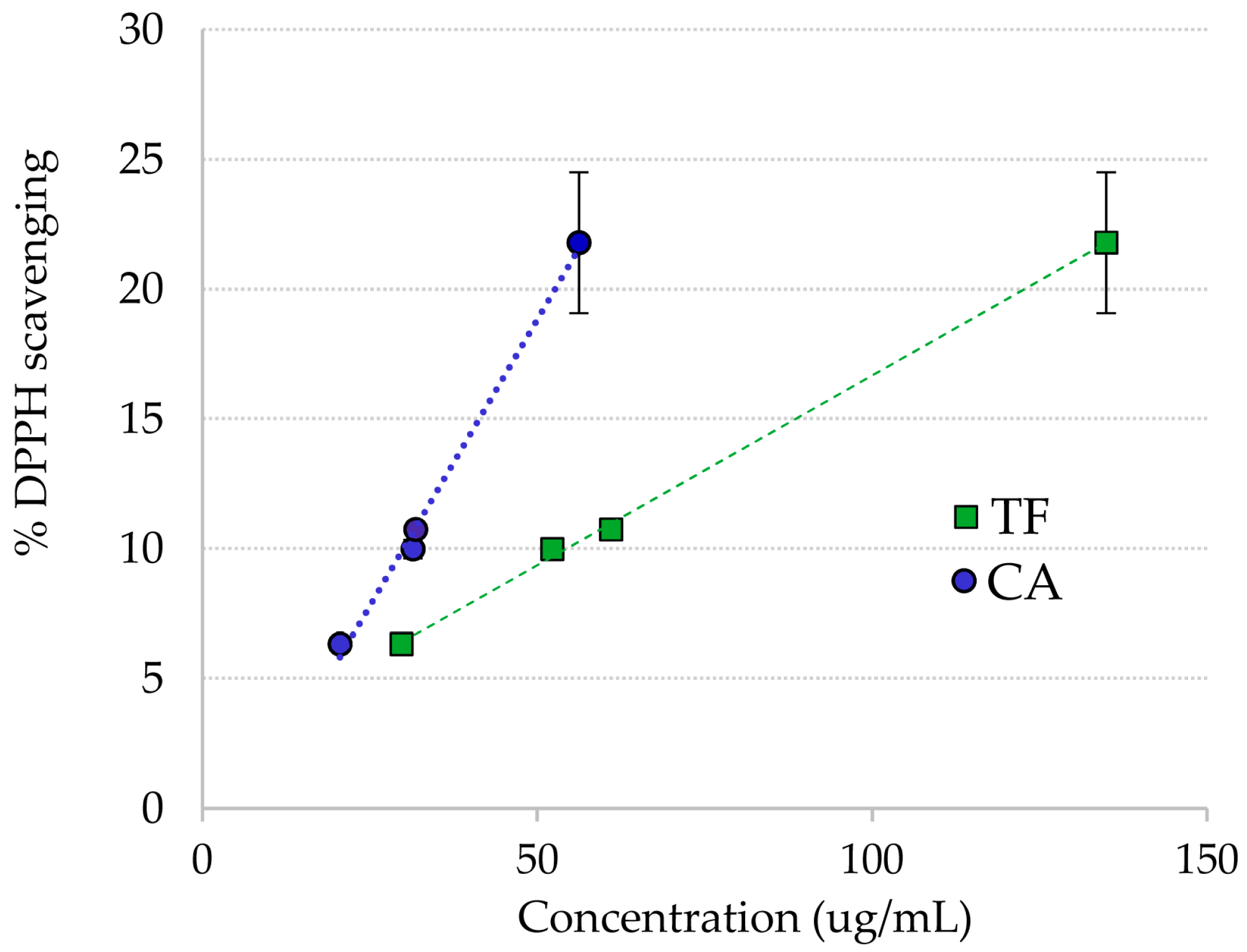
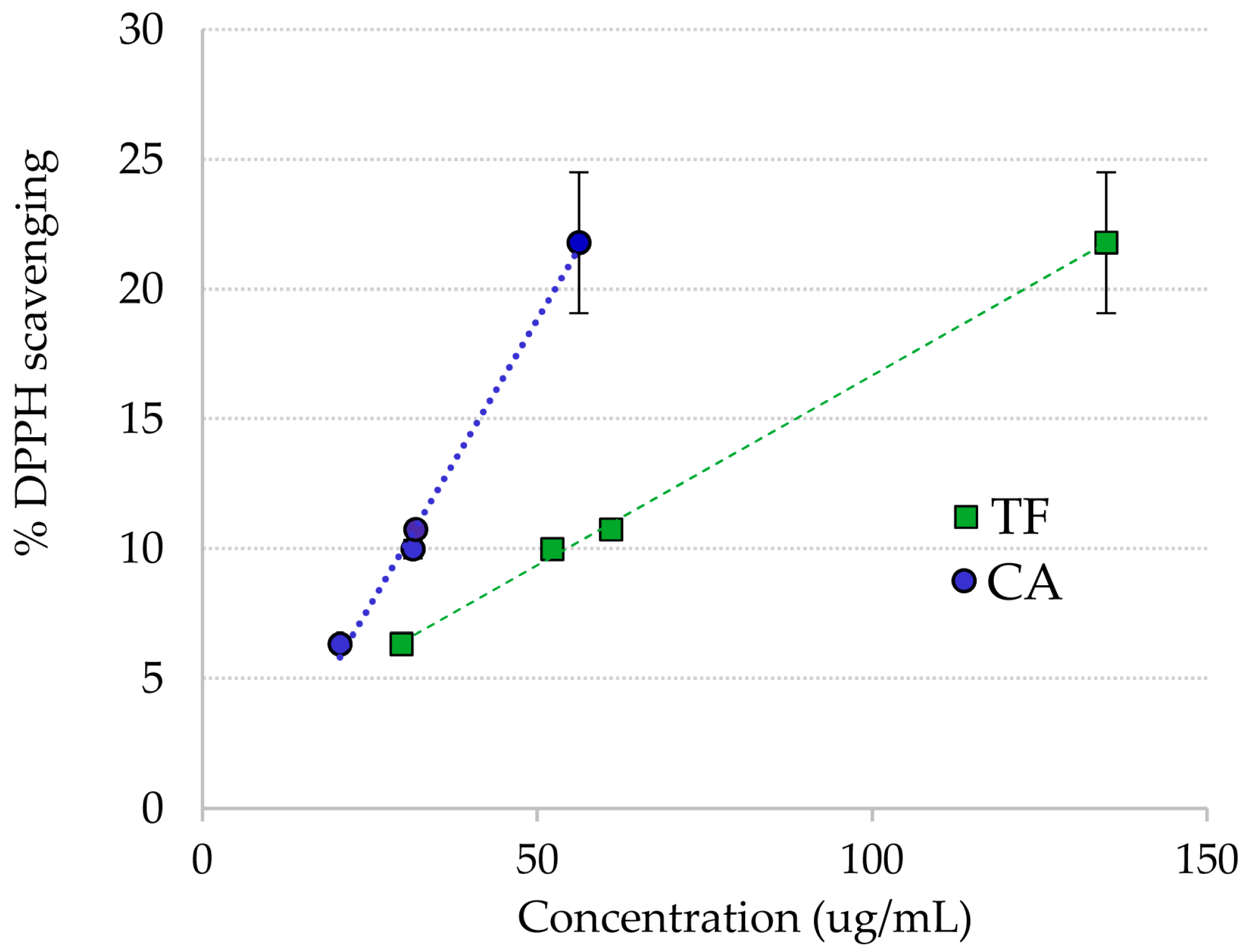
2.2. DPPH Radical Scavenging of Ruppia Polyphenols
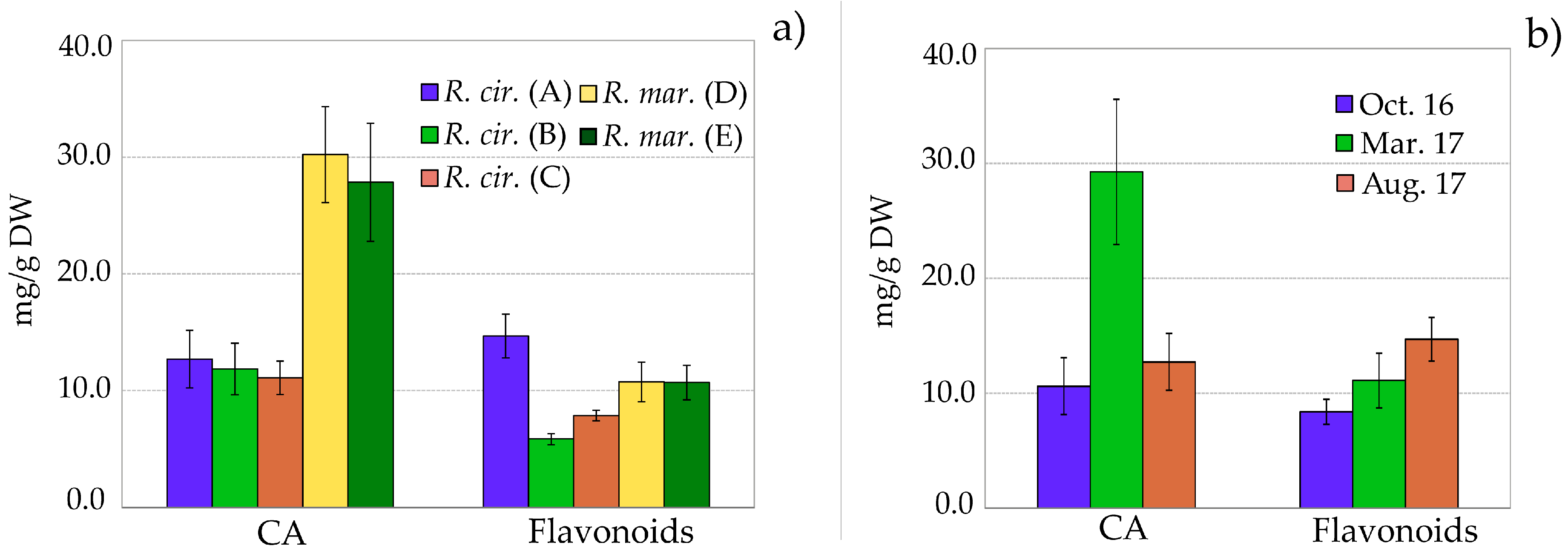
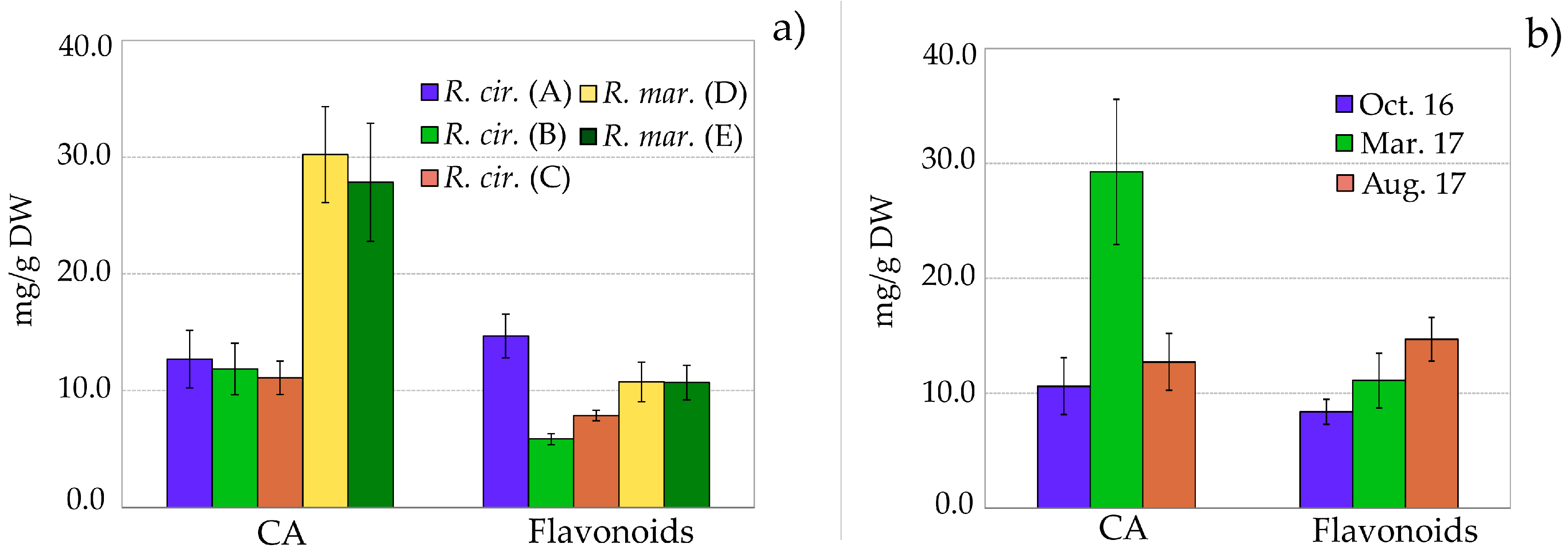
2.3. Quantitative Analysis of Polyphenolic Content in Ruppia
3. Experimental
3.1. General Instrumentation
3.1.1. Analytical HPLC
3.1.2. Preparative HPLC
3.1.3. LC–MS
3.1.4. NMR-Spectroscopy
3.2. Plant Material and Study Sites
3.3. Extraction, Purification and Identification
3.4. Quantitative Determination
3.5. Method Validation
3.6. DPPH Radical Scavenging
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cardozo, K.H.M.; Guaratini, T.; Barros, M.P.; Falcão, V.R.; Tonon, A.P.; Lopes, N.P.; Campos, S.; Torres, M.A.; Souza, A.O.; Colepicolo, P.; et al. Metabolites from algae with economical impact. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2007, 146, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Subhashini, P.; Dilipan, E.; Thangaradjou, T.; Papenbrock, J. Bioactive natural products from marine angiosperms: Abundance and functions. Nat. Prod. Bioprospect. 2013, 3, 129–136. [Google Scholar] [CrossRef]
- Zidorn, C. Secondary metabolites of seagrasses (Alismatales and Potamogetonales; Alismatidae): Chemical diversity, bioactivity, and ecological function. Phytochemistry 2016, 124, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Achamlale, S.; Rezzonico, B.; Grignon-Dubois, M. Rosmarinic acid from beach waste: Isolation and HPLC quantification in Zostera detritus from Arcachon lagoon. Food Chem. 2009, 113, 878–883. [Google Scholar] [CrossRef]
- Agostini, S.; Desjobert, J.-M.; Pergent, G. Distribution of phenolic compounds in the seagrass Posidonia oceanica. Phytochemistry 1998, 48, 611–617. [Google Scholar] [CrossRef]
- Bitam, F.; Ciavatta, M.L.; Carbone, M.; Manzo, E.; Mollo, E.; Gavagnin, M. Chemical analysis of flavonoid constituents of the seagrass Halophila stipulacea: First finding of malonylated derivatives in marine phanerogams. Biochem. Syst. Ecol. 2010, 38, 686–690. [Google Scholar] [CrossRef]
- Boutard, B.; Bouillant, M.-L.; Chopin, J.; Lebreton, P. Chimiotaxinomie flavonique des fluviales. Biochem. Syst. Ecol. 1973, 1, 133–140. [Google Scholar] [CrossRef]
- Enerstvedt, K.H.; Jordheim, M.; Andersen, Ø.M. Isolation and Identification of Flavonoids Found in Zostera marina Collected in Norwegian Coastal Waters. Am. J. Plant Sci. 2016, 7, 1163–1172. [Google Scholar] [CrossRef]
- Grignon-Dubois, M.; Rezzonico, B. First Phytochemical Evidence of Chemotypes for the Seagrass Zostera noltii. Plants 2012, 1, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Harborne, J.B.; Williams, C.A. Occurrence of sulphated flavones and caffeic acid esters in members of the fluviales. Biochem. Syst. Ecol. 1976, 4, 37–41. [Google Scholar] [CrossRef]
- McMillan, C.; Zapata, O.; Escobar, L. Sulphated phenolic compounds in seagrasses. Aquat. Bot. 1980, 8, 267–278. [Google Scholar] [CrossRef]
- Nuissier, G.; Rezzonico, B.; Grignon-Dubois, M. Chicoric acid from Syringodium filiforme. Food Chem. 2010, 120, 783–788. [Google Scholar] [CrossRef]
- Hawas, U.W. A New 8-Hydroxy Flavone O-Xyloside Sulfate and Antibacterial Activity from the Egyptian Seagrass Thalassia hemprichii. Chem. Nat. Compd. 2014, 50, 629–632. [Google Scholar] [CrossRef]
- Richardson, F.D. Ecology of Ruppia maritima L. in New Hampshire (U.S.A) tidal marshes. Rhodora 1980, 82, 403–439. [Google Scholar]
- Den Hartog, C.; Wiegleb, G. Potamogetonaceae, Zosteraceae, and Cymodoceaceae. Flora Males. Ser. 1 Spermatophyta 2002, 16, 167–216. [Google Scholar]
- Haynes, R.R. The Potamogetonaceae in the Southeastern United States. J. Arnold Arbor. 1978, 59, 170–191. [Google Scholar]
- Les, D.H.; Haynes, R.R. Systematics of subclass Alismatidae: A synthesis of approaches. In Monocyledons: Systematics and Evolution; Cutler, D.F., Humphries, C.J., Eds.; Royal Botanic Gardens, Kew: London, UK, 1995; Volume 2. [Google Scholar]
- Lee, J.; Scagel, C.F. Chicoric acid: Chemistry, distribution, and production. Front. Chem. 2013, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Foo, L.Y. Identification and quantification of major polyphenols in apple pomace. Food Chem. 1997, 59, 187–194. [Google Scholar] [CrossRef]
- Sikorska, M.; Matlawska, I. Quercetin and its glycosides in the flowers of Asclepias syriaca L. Acta Pol. Pharm. 2000, 57, 321–324. [Google Scholar] [PubMed]
- Pereira, C.; Barreto Júnior, C.B.; Kuster, R.M.; Simas, N.K.; Sakuragui, C.M.; Porzel, A.; Wessjohann, L. Flavonoids and a neolignan glucoside from Guarea macrophylla (Meliaceae). Quím. Nova 2012, 35, 1123–1126. [Google Scholar] [CrossRef]
- Liu, H.; Mou, Y.; Zhao, J.; Wang, J.; Zhou, L.; Wang, M.; Wang, D.; Han, J.; Yu, Z.; Yang, F. Flavonoids from Halostachys caspica and their antimicrobial and antioxidant activities. Molecules 2010, 15, 7933–7945. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Seo, S.; Nakajima, J.-I. Constituents from leaves of Apocynum venetum L. J. Nat. Med. 2008, 62, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.A.M.; Mansour, R.M.A.; Markham, K.R. An acylated isorhamnetin glycoside from Aerva javanica. Phytochemistry 1990, 29, 1344–1345. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Complete 1H NMR spectral analysis of ten chemical markers of Ginkgo biloba. Magn. Reson. Chem. 2012, 50, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Hassanean, H.A.; Desoky, E.K. An acylated isorhamnetin glucoside from Zygophyllum simplex. Phytochemistry 1992, 31, 3293–3294. [Google Scholar] [CrossRef]
- Wald, B.; Wray, V.; Galensa, R.; Herrmann, K. Malonated flavonol glycosides and 3,5-dicaffeoylquinic acid from pears. Phytochemistry 1989, 28, 663–664. [Google Scholar] [CrossRef]
- Cheminat, A.; Zawatzky, R.; Becker, H.; Brouillard, R. Caffeoyl conjugates from Echinacea species: Structures and biological activity. Phytochemistry 1988, 27, 2787–2794. [Google Scholar] [CrossRef]
- Grignon-Dubois, M.; Rezzonico, B. The economic potential of beach-cast seagrass—Cymodocea nodosa: A promising renewable source of chicoric acid. Bot. Mar. 2013, 56, 303–311. [Google Scholar] [CrossRef]
- Cariello, L.; Zanetti, L. Distribution of Chicoric Acid during Leaf Development of Posidonia oceanica. Bot. Mar. 1979, 22, 359–360. [Google Scholar] [CrossRef]
- Haznedaroglu, M.Z.; Zeybek, U. HPLC Determination of Chicoric Acid in Leaves of Posidonia oceanica. Pharm. Biol. 2007, 45, 745–748. [Google Scholar] [CrossRef]
- Grignon-Dubois, M.; Rezzonico, B. Phenolic fingerprint of the seagrass Posidonia oceanica from four locations in the Mediterranean Sea: First evidence for the large predominance of chicoric acid. Bot. Mar. 2015, 58, 379–391. [Google Scholar] [CrossRef]
- Qi, S.-H.; Huang, L.-S.; He, F.; Zhang, S.; Dong, J.-D. Phytochemical and chemotaxonomic investigation of seagrass Thalassia hemprichii (Ehrenb.) Aschers (Hydrocharitaceae). Biochem. Syst. Ecol. 2012, 43, 128–131. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant. Songklanakarin J. Sci. Technol. 2004, 26, 211–219. [Google Scholar]
- Yuvaraj, N.; Kanmani, P.; Satishkumar, R.; Paari, A.; Pattukumar, V.; Arul, V. Seagrass as a potential source of natural antioxidant and anti-inflammatory agents. Pharm. Biol. 2012, 50, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Santoso, J.; Anwariyah, S.; Rumiantin, R.O.; Putri, A.P.; Ukhty, N.; Yoshie-Stark, Y. Phenol content, antioxidant activity and fibers profile of four tropical Seagrasses from Indonesia. J. Coast. Dev. 2012, 15, 189–196. [Google Scholar]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant and Prooxidant Behavior of Flavonoids: Structure-Activity Relationships. Free Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar] [CrossRef]
- Nimmi, O.S.; Philomena, G. Evaluation of the antioxidant potential of a newly developed polyherbal formulation for antiobesity. Int. J. Pharm. Pharm. Sci. 2012, 4, 505–510. [Google Scholar]
- Ghasemi, K.; Ghasemi, Y.; Ebrahimzadeh, M.A. Antioxidant activity, phenol and flavonoid contents of 13 citrus species peels and tissues. Pak. J. Pharm. Sci. 2009, 22, 277–281. [Google Scholar] [PubMed]
- Majewska, M.; Skrzycki, M.; Podsiad, M.; Czeczot, H. Evaluation of antioxidant potential of flavonoids: An in vitro study. Acta Pol. Pharm. 2011, 68, 611–615. [Google Scholar] [PubMed]
- Nagy, M.; Križková, L.; Mučaji, P.; Kontšeková, Z.; Šeršeň, F.; Krajčovič, J. Antimutagenic Activity and Radical Scavenging Activity of Water Infusions and Phenolics from Ligustrum Plants Leaves. Molecules 2009, 14, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Zuo, A.; Yu, Y.; Li, J.; Xu, B.; Yu, X.; Qiu, Y.; Cao, S. Study on the relation of structure and antioxidant activity of isorhamnetin, quercetin, phloretin, silybin and phloretin isonicotinyl hydrazone. Free Radic. Antioxid. 2011, 1, 39–47. [Google Scholar] [CrossRef]
- Lu, Y.; Khoo, T.J.; Wiart, C. Antioxidant Activity Determination of Citronellal and Crude Extracts of Cymbopogon citratus by 3 Different Methods. Sci. Res. 2014, 5, 395–400. [Google Scholar] [CrossRef]
- Siatka, T.; Kašparová, M. Seasonal Variation in Total Phenolic and Flavonoid Contents and DPPH Scavenging Activity of Bellis perennis L. Flowers. Molecules 2010, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-L.; Fang, X.-Y.; Wang, J.-Q.; Zhao, L.-G.; Li, Y.; Tang, F.; Yue, Y.-D. Structures and bioactivities of seven flavonoids from Osmanthus fragrans ‘Jinqiu’ essential oil extraction residues. Nat. Prod. Res. 2017, 32, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, M.; Aaby, K.; Fossen, T.; Skrede, G.; Andersen, Ø.M. Molar Absorptivities and Reducing Capacity of Pyranoanthocyanins and Other Anthocyanins. J. Agric. Food Chem. 2007, 55, 10591–10598. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Reilly, K.; Kerry, J.P.; Gaffney, M.; Hossain, M.; Rai, D.K. Higher Antioxidant Activity, Total Flavonols, and Specific Quercetin Glucosides in Two Different Onion (Allium cepa L.) Varieties Grown under Organic Production: Results from a 6-Year Field Study. J. Agric. Food Chem. 2017, 65, 5122–5132. [Google Scholar] [CrossRef] [PubMed]
- Slimestad, R.; Fossen, T.; Vagen, I.M. Onions: A source of unique dietary flavonoids. J. Agric. Food Chem. 2007, 55, 10067–10080. [Google Scholar] [CrossRef] [PubMed]
- Hertog, M.G.L.; Hollman, P.C.H.; Venema, D.P. Optimization of a quantitative HPLC determination of potentially anticarcinogenic flavonoids in vegetables and fruits. J. Agric. Food Chem. 1992, 40, 1591–1598. [Google Scholar] [CrossRef]
- Bhagwat, S.; Haytowitz, D.B.; Wasswa-Kintu, S. USDA’s Expanded Flavonoid Database for the Assessment of Dietary Intakes. 2014. Available online: http://www.ars.usda.gov/nutrientdata (accessed 21 December 2017).
- Enerstvedt, K.H.; Lundberg, A.; Sjøtun, I.K.; Fadnes, P.; Jordheim, M. Characterization and seasonal variation of individual flavonoids in Zostera marina and Zostera noltii from Norwegian coastal waters. Biochem. Syst. Ecol. 2017, 74, 42–50. [Google Scholar] [CrossRef]
- Pellati, F.; Benvenuti, S.; Magro, L.; Melegari, M.; Soragni, F. Analysis of phenolic compounds and radical scavenging activity of Echinacea spp. J. Pharm. Biomed. Anal. 2004, 35, 289–301. [Google Scholar] [CrossRef]
- Mølgaard, P.; Johnsen, S.; Christensen, P.; Cornett, C. HPLC Method Validated for the Simultaneous Analysis of Cichoric Acid and Alkamides in Echinacea purpurea Plants and Products. J. Agric. Food Chem. 2003, 51, 6922–6933. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Z.; Abbasi, B.H.; Gao, M.; Murch, S.J.; Saxena, P.K. Caffeic acid derivatives production by hairy root cultures of Echinacea purpurea. J. Agric. Food Chem. 2006, 54, 8456–8460. [Google Scholar] [CrossRef] [PubMed]
- Chaves, N.; Escudero, J.C.; Gutierrez-Merino, C. Role of Ecological Variables in the Seasonal Variation of Flavonoid Content of Cistus ladanifer Exudate. J. Chem. Ecol. 1997, 23, 579–603. [Google Scholar] [CrossRef]
- Venditti, A.; Serrilli, A.M.; Vittori, S.; Papa, F.; Maggi, F.; Di Cecco, M.; Ciaschetti, G.; Bruno, M.; Rosselli, S.; Bianco, A. Secondary metabolites from Pinus mugo Turra subsp. mugo growing in the Majella National Park (Central Apennines, Italy). Chem. Biodivers. 2013, 10, 2091–2100. [Google Scholar] [CrossRef] [PubMed]
- Vergeer, L.H.T.; Develi, A. Phenolic acids in healthy and infected leaves of Zostera marina and their growth-limiting properties towards Labyrinthula zosterae. Aquat. Bot. 1997, 58, 65–72. [Google Scholar] [CrossRef]
- Caprioli, G.; Alunno, A.; Beghelli, D.; Bianco, A.; Bramucci, M.; Frezza, C.; Iannarelli, R.; Papa, F.; Quassinti, L.; Sagratini, G.; et al. Polar Constituents and Biological Activity of the Berry-Like Fruits from Hypericum androsaemum L. Front. Plant Sci. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Malterud, K.E.; Farbrot, T.L.; Huse, A.E.; Sund, R.B. Antioxidant and Radical Scavenging Effects of Anthraquinones and Anthrones. Pharmacology 1993, 47, 77–85. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracts and Compounds | DPPH· 1 IC50 (μg/mL) |
|---|---|
| R. cirrhosa crude extract (October) | 175.7 ± 7.8 |
| R. cirrhosa crude extract (August) | 152.9 ± 8.1 |
| R. cirrhosa purified extract | 31.8 ± 0.7 |
| 3 + 4 | 12.1 ± 2.2 |
| 5 + 6 | 88.4 ±7.0 |
| 7 + 8 | 51.7 ± 6.8 |
| CA | 23.0 ± 3.2 |
| Reference Standard | DPPH· 1 IC50 (μg/mL) |
|---|---|
| quercetin (≥95%) | 5.5 ± 0.3 |
| quercetin 3-O-β-d-glucopyranoside (≥90%) | 11.0 ± 1.0 |
| rutin (≥95%) | 13.9 ± 0.7 |
| Trolox (≥97%) | 6.1 ± 0.4 |
| chicoric acid (≥95%) | 9.7 ± 1.7 |
| Compound | R. cirr. (A) (mg/g) | R. cirr. (B) (mg/g) | R. cirr. (C) (mg/g) | R. mar. (D) (mg/g) | R. mar. (E) (mg/g) |
|---|---|---|---|---|---|
| CA | 12.7 ± 2.5 a | 11.9 ± 2.2 a | 11.1 ± 1.4 a | 30.2 ± 4.3 b | 27.9 ± 5.1 b |
| 1 | 2.2 ± 0.3 d | 0.7 ± 0.1 | 1.1 ± 0.1 g | 2.0 ± 0.5 d | 1.1 ± 0.2 g |
| 2 | 1.3 ± 0.2 | 0.5 ± 0.04 e | 1.0 ± 0.1 f | 1.0 ± 0.2 f | 0.6 ± 0.1 e |
| 3 | 0.9 ± 0.1 | 0.4 ± 0.04 e | 0.7 ± 0.04 f,g | 0.6 ± 0.1 b,f | 0.6 ± 0.1 b,e,g |
| 4 | 1.9 ± 0.3 | 0.7 ± 0.05 a | 0.8 ± 0.04 a | 1.5 ± 0.3 b | 1.4 ± 0.2 b |
| 5 | 2.9 ± 0.4 | 1.0 ± 0.1 a | 1.0 ± 0.1 a | 1.6 ± 0.3 b | 2.0 ± 0.2 b |
| 6 | 2.1 ± 0.2 d | 0.8 ± 0.1 | 1.3 ± 0.1 f | 1.7 ± 0.3 b,d,f | 1.6 ± 0.2 b |
| 7 | 1.1 ± 0.2 c | 0.6 ± 0.1 a,e | 0.5 ± 0.04 a,f | 0.6 ± 0.07 e,f | 1.1 ± 0.2 c |
| 8 | 2.2 ± 0.3 c | 1.1 ± 0.1 | 1.5 ± 0.1 | 1.8 ± 0.2 | 2.3 ± 0.3 c |
| sum flavonoids | 14.7 ± 1.9 | 5.9 ± 0.5 | 7.9 ± 0.5 | 10.7 ± 1.7 b | 10.7 ± 1.5 b |
| sum phenolics | 27.4 ± 4.3 | 17.7 ± 2.1 a | 19.0 ± 1.8 a | 41.0 ± 5.7 b | 38.5 ± 6.3 b |
| Compound | 16 October (mg/g) | 17 March (mg/g) | 17 August(mg/g) |
|---|---|---|---|
| CA | 10.6 ± 2.5 a | 29.2 ± 6.3 | 12.7 ± 2.5 a |
| 1 | 0.8 ± 0.1 | 2.2 ± 0.4 b | 2.2 ± 0.3 b |
| 2 | 0.6 ± 0.1 | 0.8 ± 0.2 | 1.3 ± 0.2 |
| 3 | 0.7 ± 0.1 a | 0.9 ± 0.2 a | 0.9 ± 0.1 a |
| 4 | 1.1 ± 0.2 | 2.5 ± 0.6 b | 1.9 ± 0.3 b |
| 5 | 1.1 ± 0.2 | 1.5 ± 0.3 | 2.9 ± 0.4 |
| 6 | 0.8 ± 0.1 a | 0.7 ± 0.1 a | 2.1 ± 0.2 |
| 7 | 1.2 ± 0.2 a | 1.1 ± 0.2 a | 1.1 ± 0.2 a |
| 8 | 2.0 ± 0.3 | 1.4 ± 0.3 | 2.2 ± 0.3 |
| sum flavonoids | 8.4 ± 1.1 a | 11.1 ± 2.4 a | 14.7 ± 1.9 |
| sum phenolics | 19.0 ± 3.0 | 40.3 ± 8.7 | 27.4 ± 4.3 |
| Calibration Curve (μg/mL) | R2 | Test Range (μg/mL) | LOD (μg/mL) | LOQ (μg/mL) | Spike Recovery % | |
|---|---|---|---|---|---|---|
| quercetin 3-O-β-d-glucopyranoside | y = 36.56x − 11.8 | 0.9998 | 2.5–80 | 2.0 | 6.0 | 94.0 ± 2.0 |
| caffeic acid | y = 102.8x + 12.8 | 0.9994 | 10–80 | 1.1 | 3.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasle Enerstvedt, K.; Lundberg, A.; Jordheim, M. Characterization of Polyphenolic Content in the Aquatic Plants Ruppia cirrhosa and Ruppia maritima —A Source of Nutritional Natural Products. Molecules 2018, 23, 16. https://doi.org/10.3390/molecules23010016
Hasle Enerstvedt K, Lundberg A, Jordheim M. Characterization of Polyphenolic Content in the Aquatic Plants Ruppia cirrhosa and Ruppia maritima —A Source of Nutritional Natural Products. Molecules. 2018; 23(1):16. https://doi.org/10.3390/molecules23010016
Chicago/Turabian StyleHasle Enerstvedt, Kjersti, Anders Lundberg, and Monica Jordheim. 2018. "Characterization of Polyphenolic Content in the Aquatic Plants Ruppia cirrhosa and Ruppia maritima —A Source of Nutritional Natural Products" Molecules 23, no. 1: 16. https://doi.org/10.3390/molecules23010016