Complete Chloroplast Genome of Pinus massoniana (Pinaceae): Gene Rearrangements, Loss of ndh Genes, and Short Inverted Repeats Contraction, Expansion


Abstract
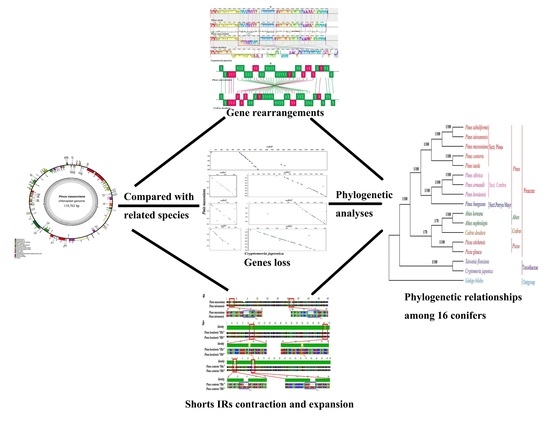
:
1. Introduction
2. Results and Discussion
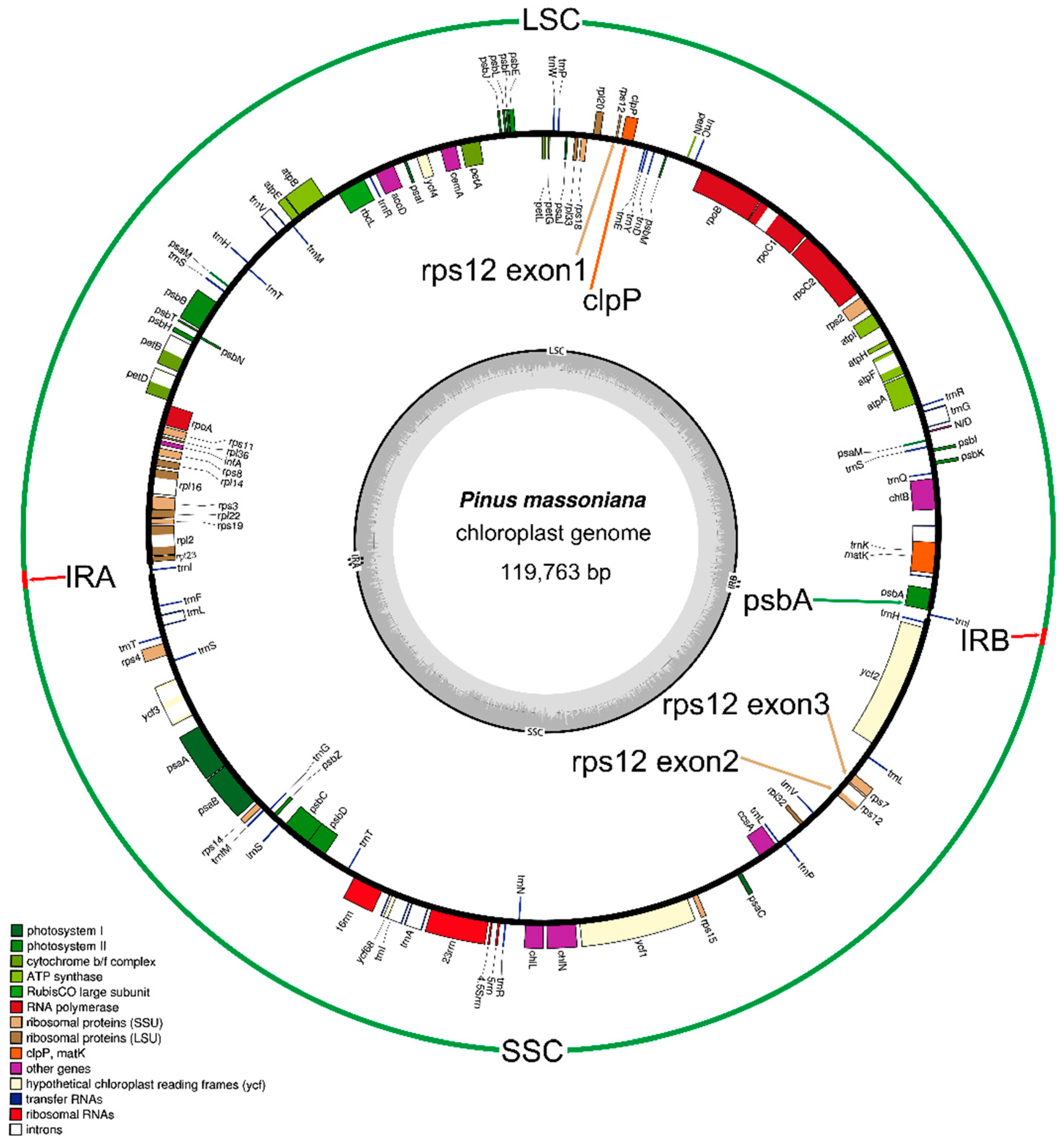
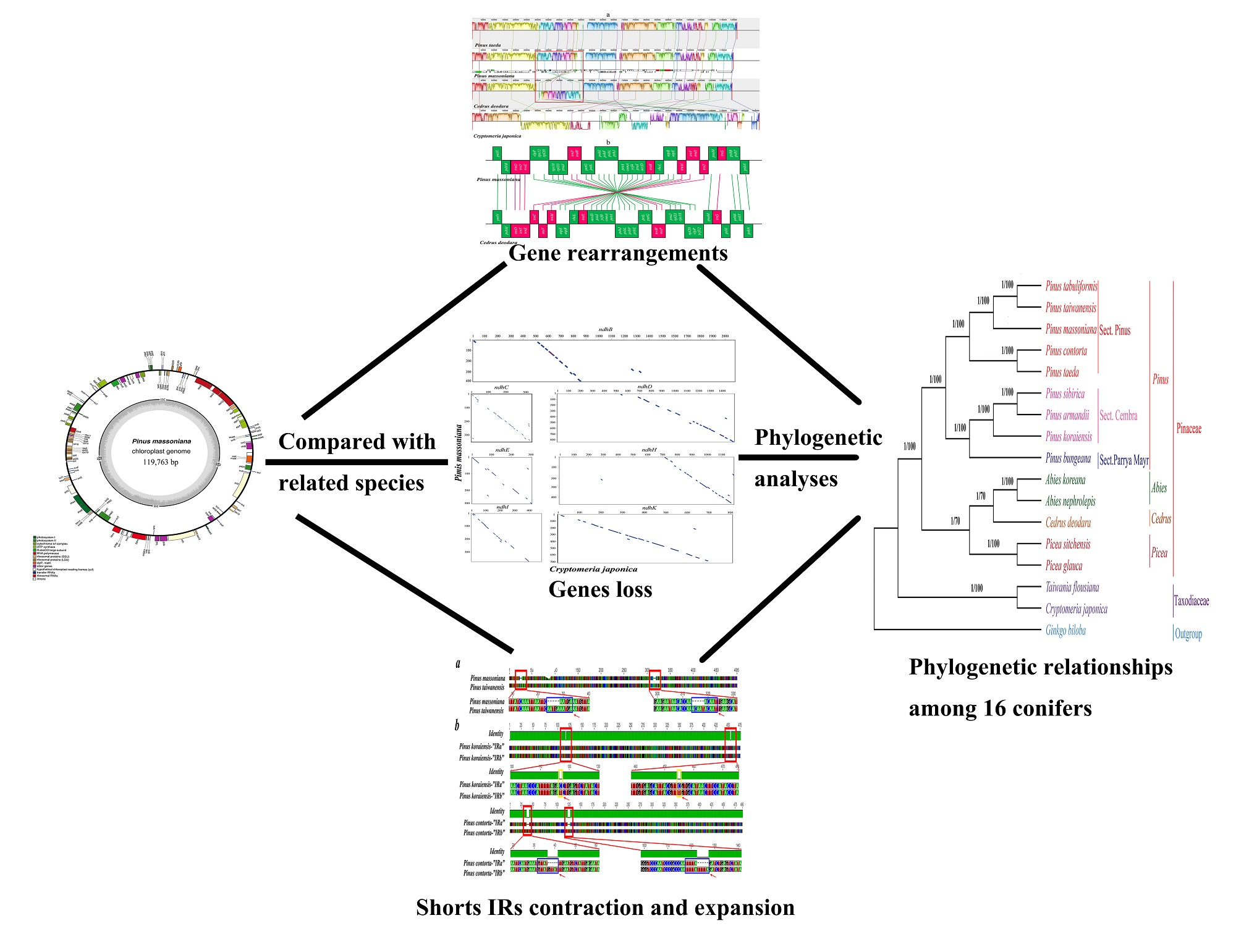
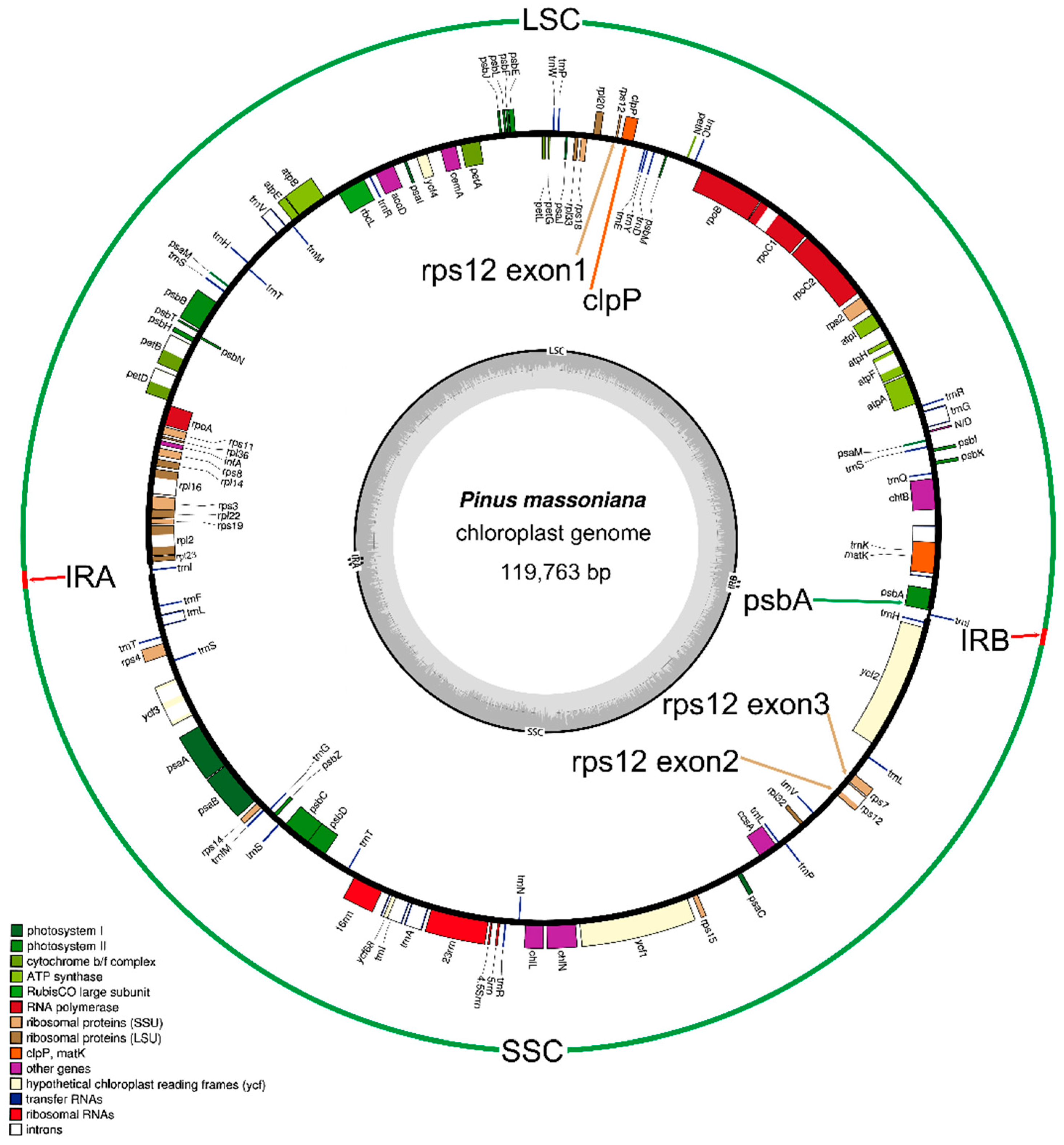
2.1. Genome Organization and Comparison with Other Species
2.2. Gene Contents
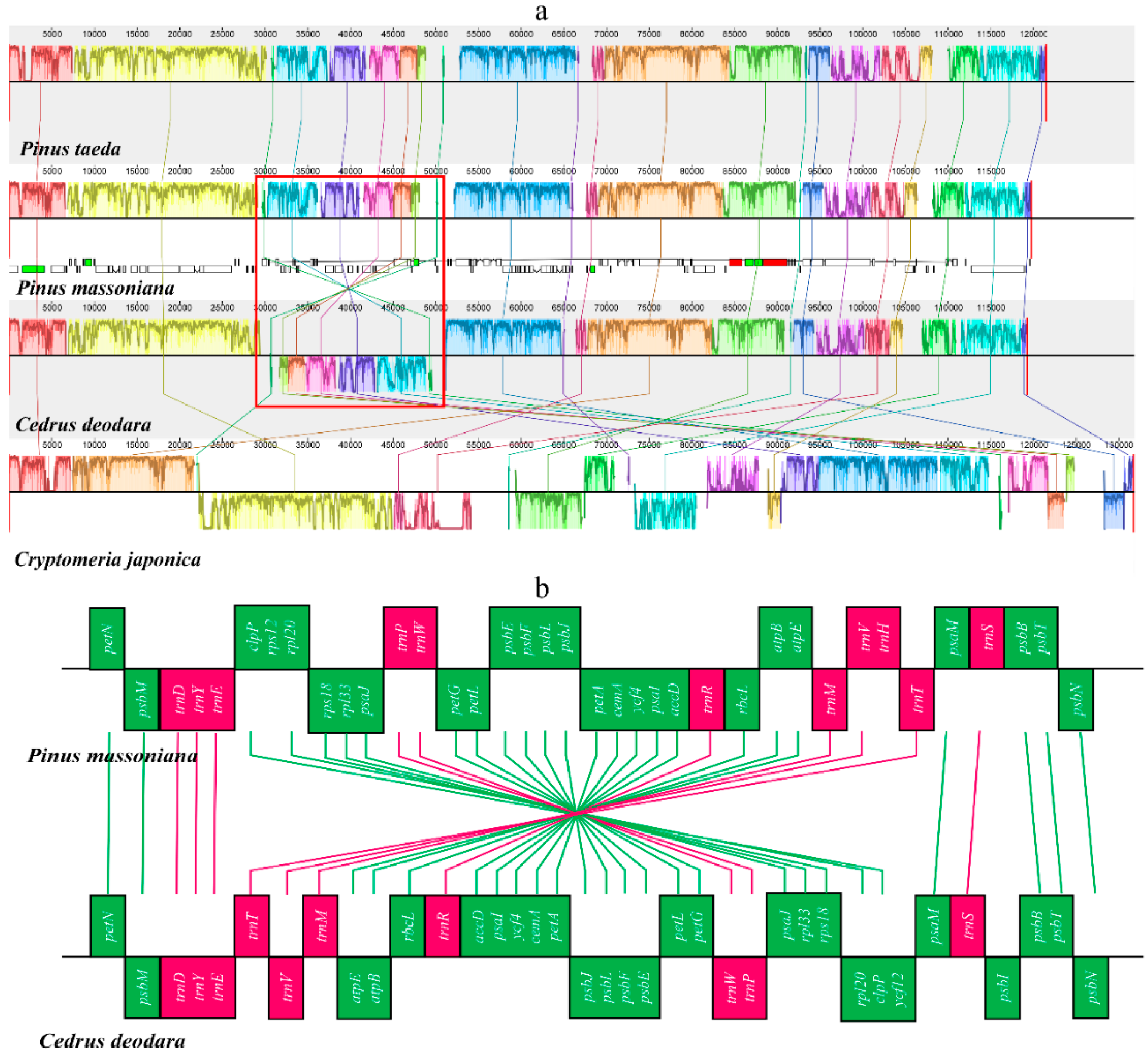
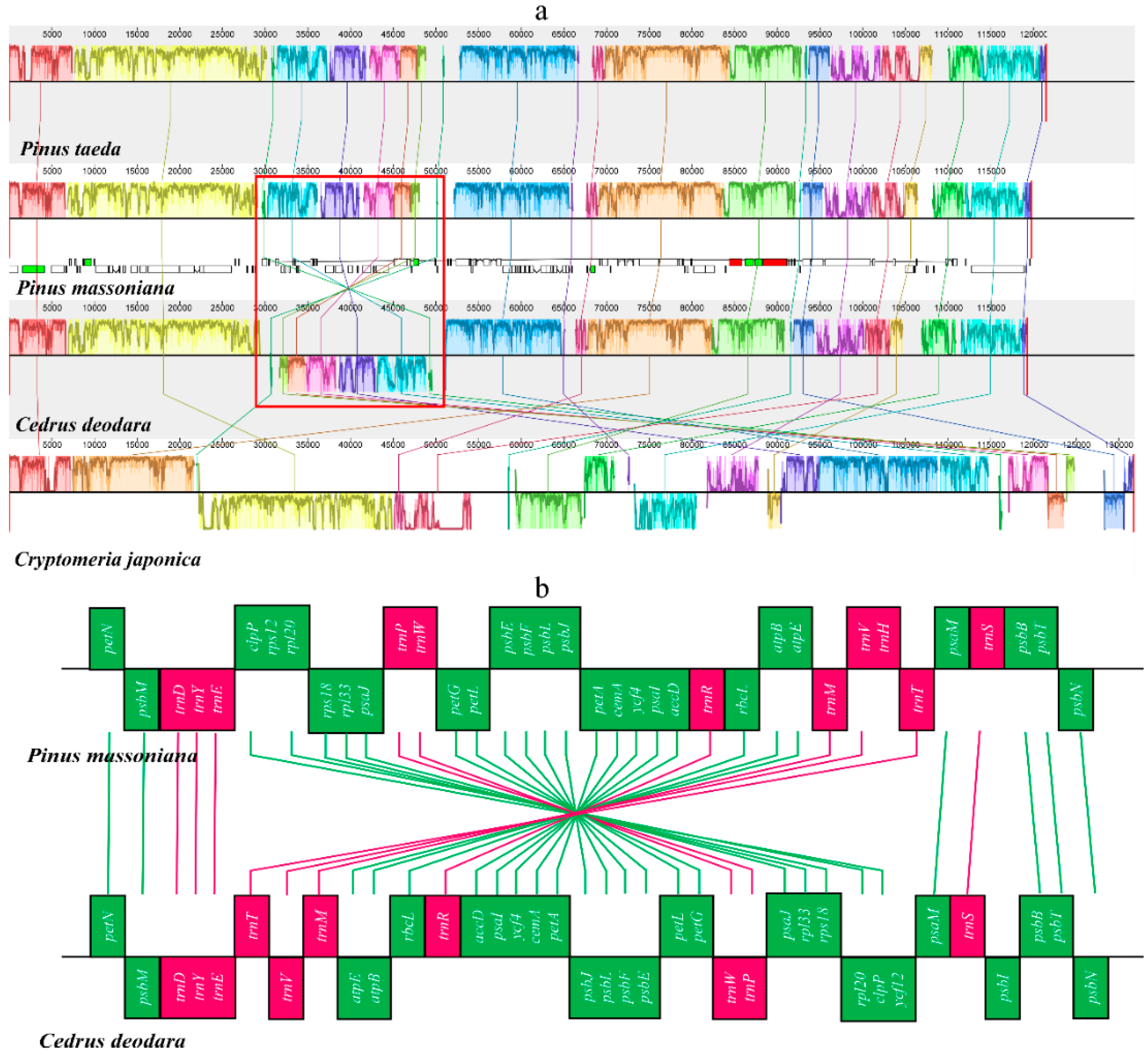
2.3. Structural and Gene Rearrangements
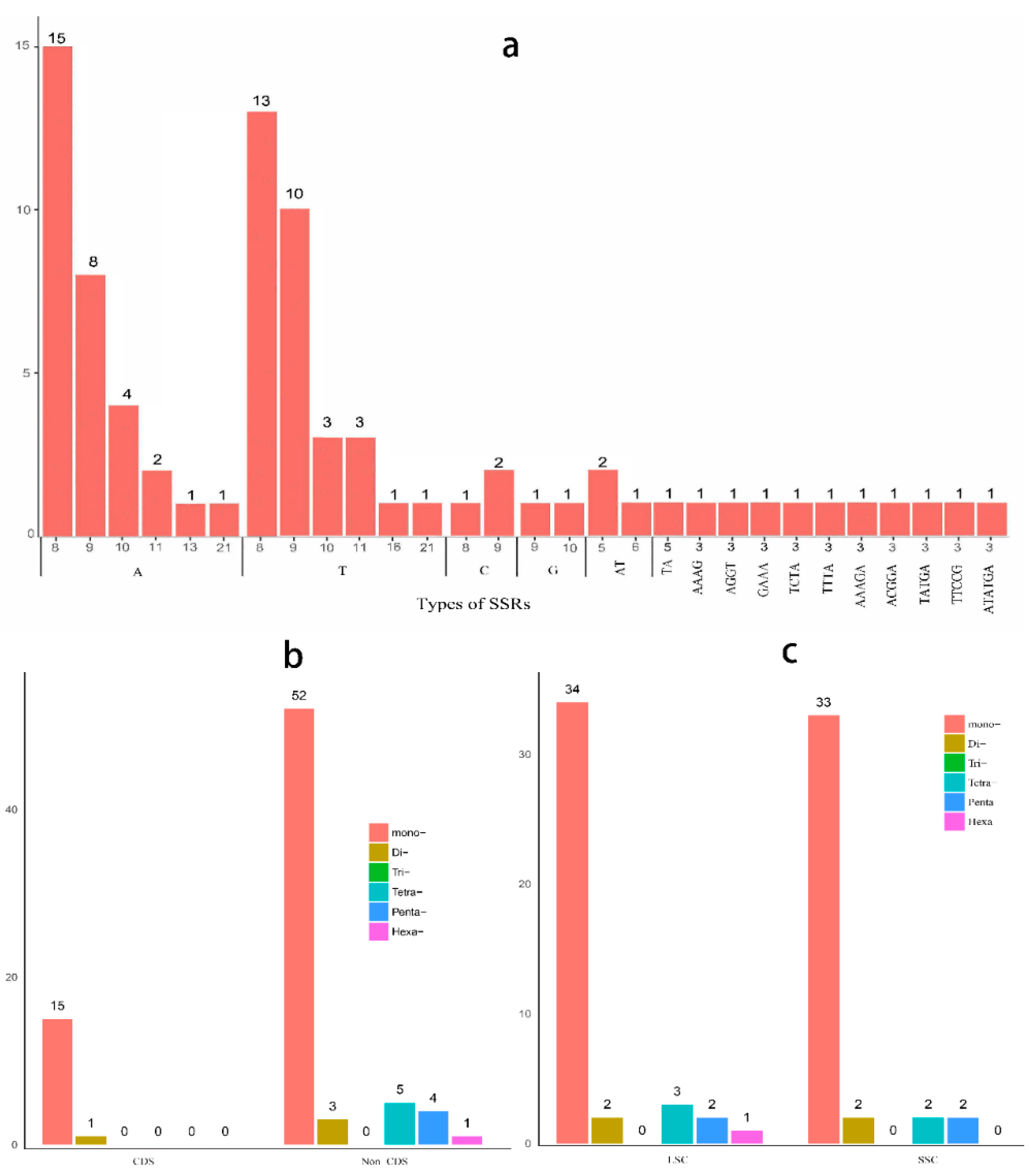
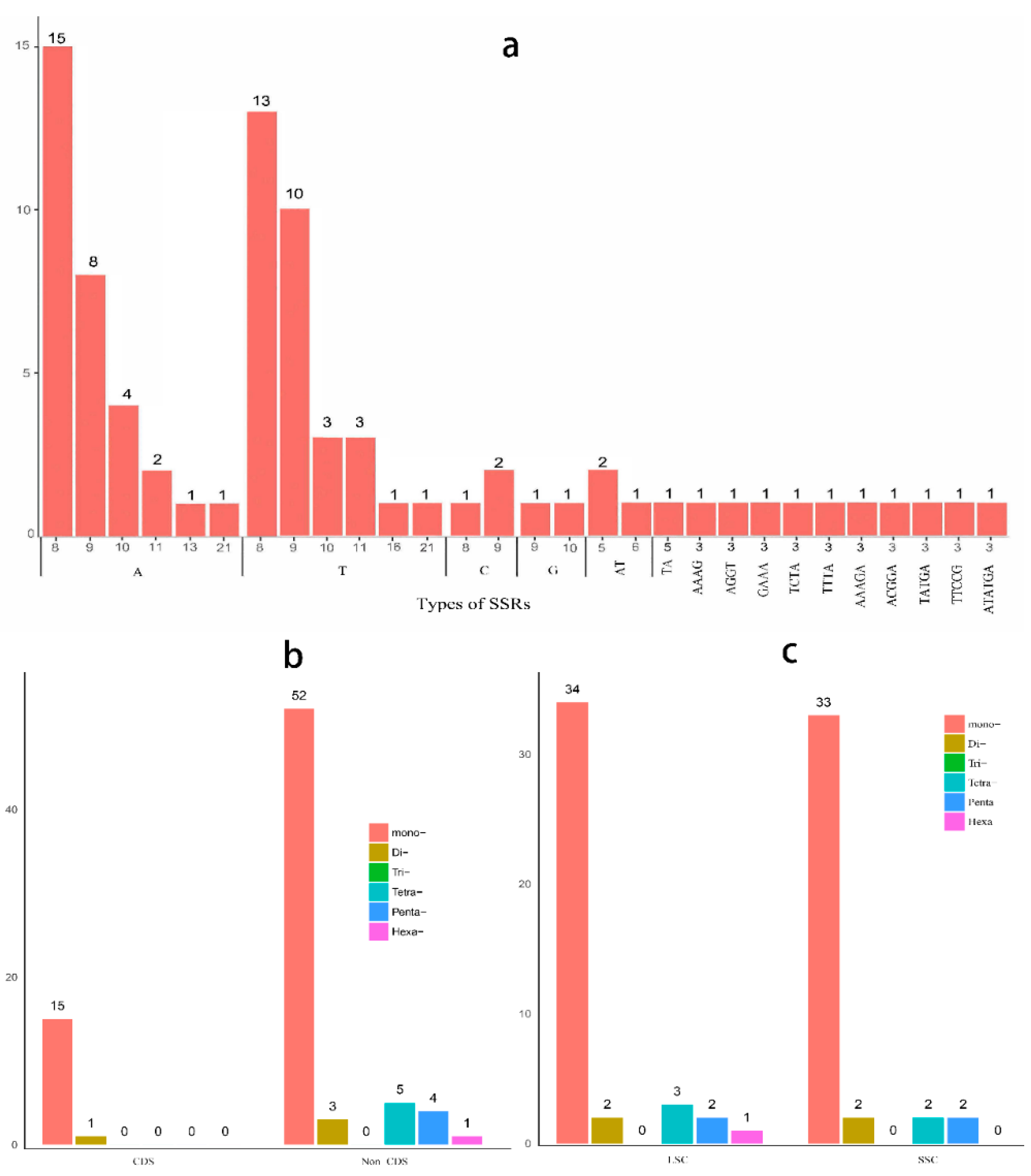
2.4. Microsatellite Polymorphisms
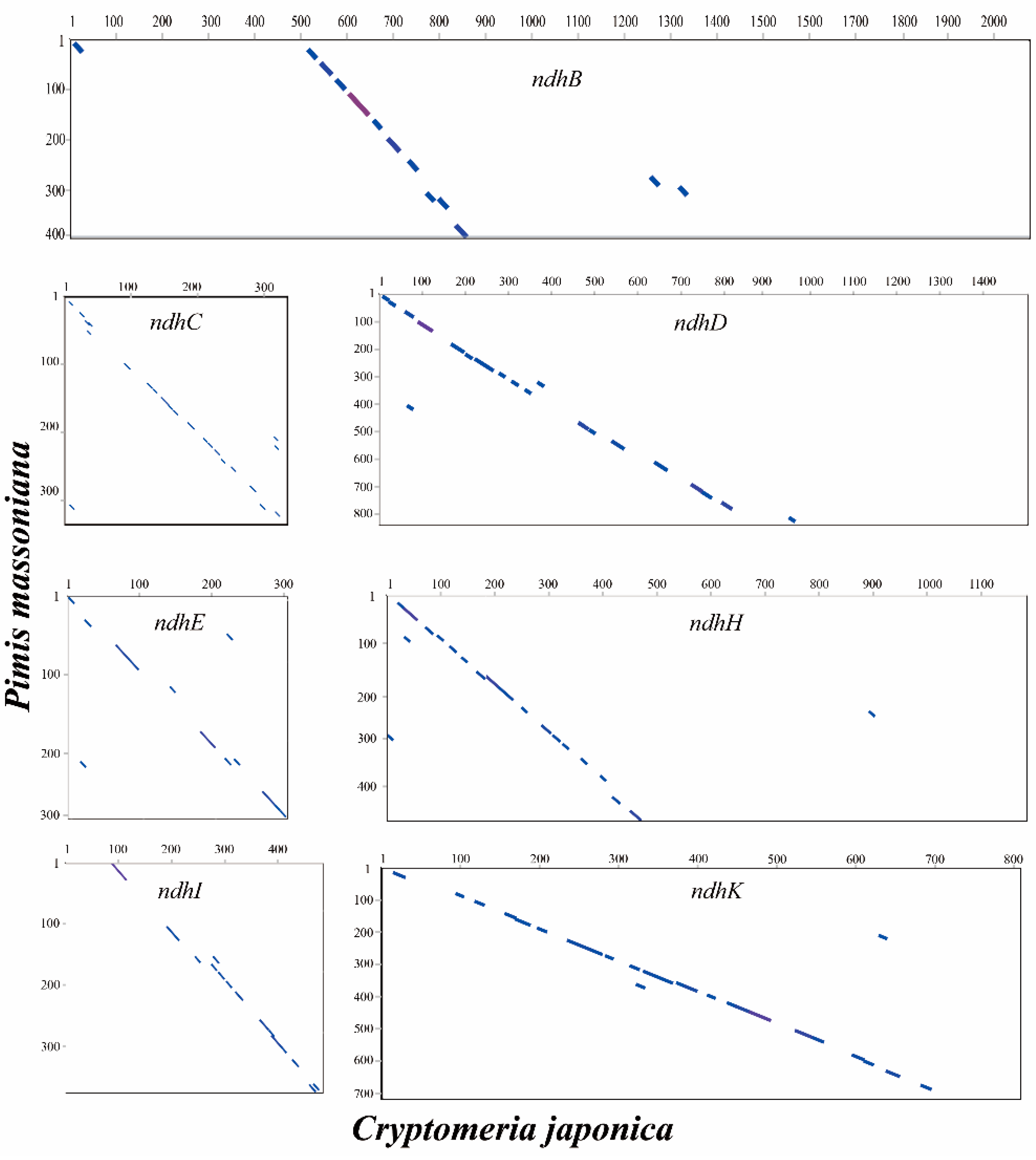
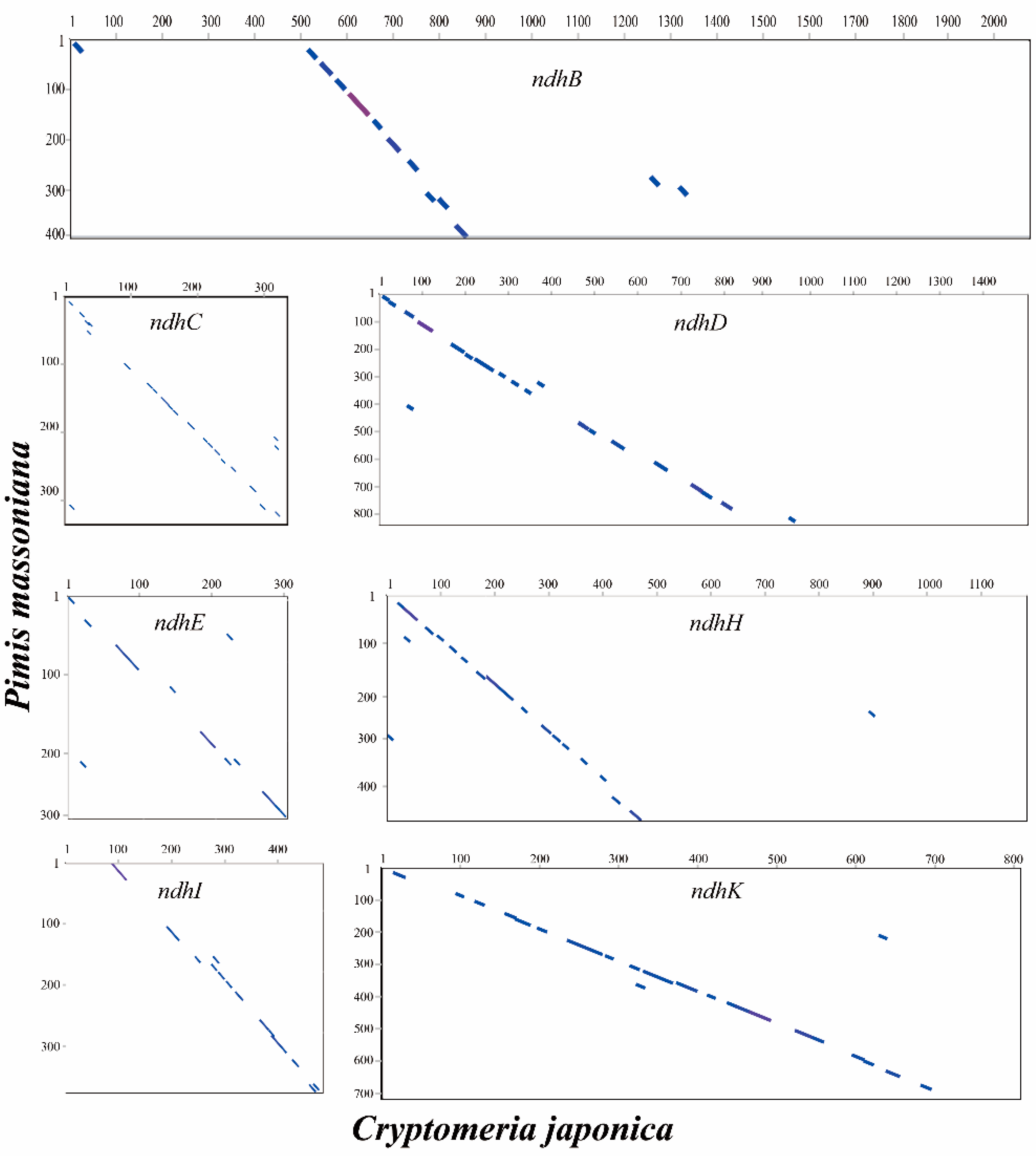
2.5. Loss of ndh Genes
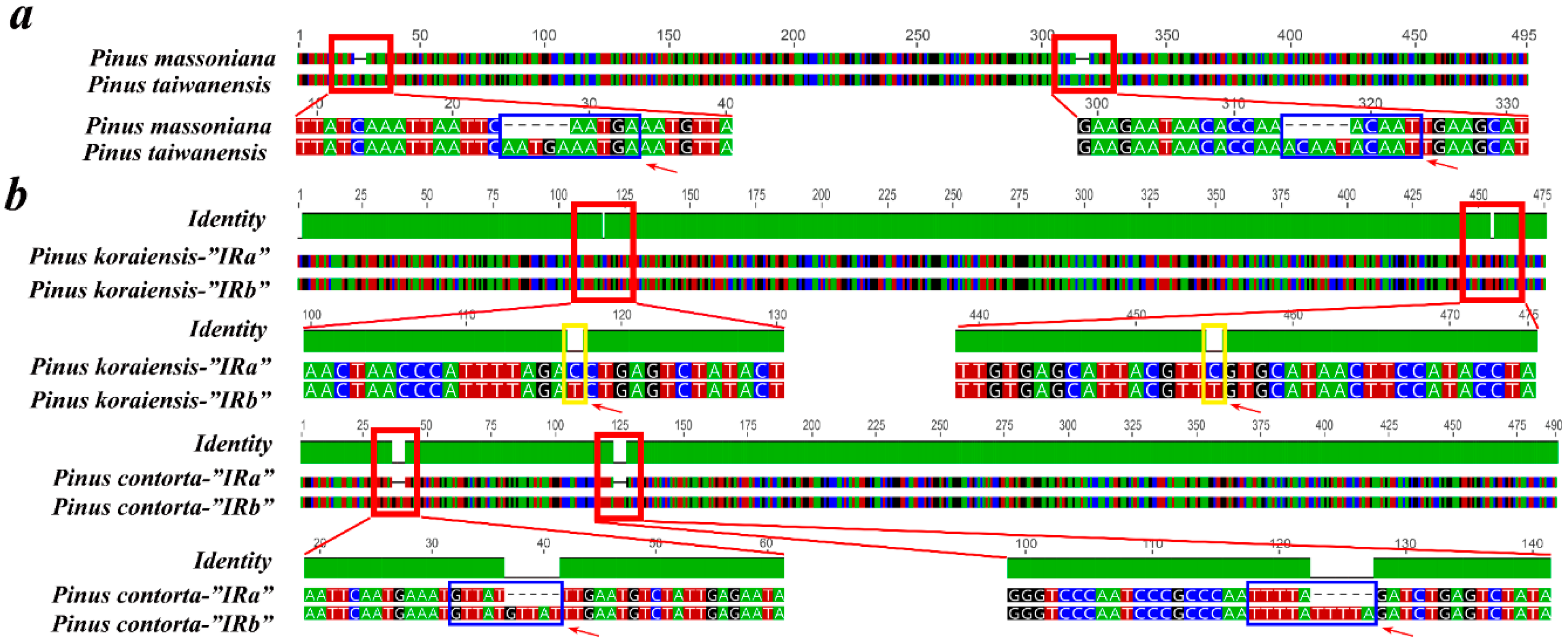
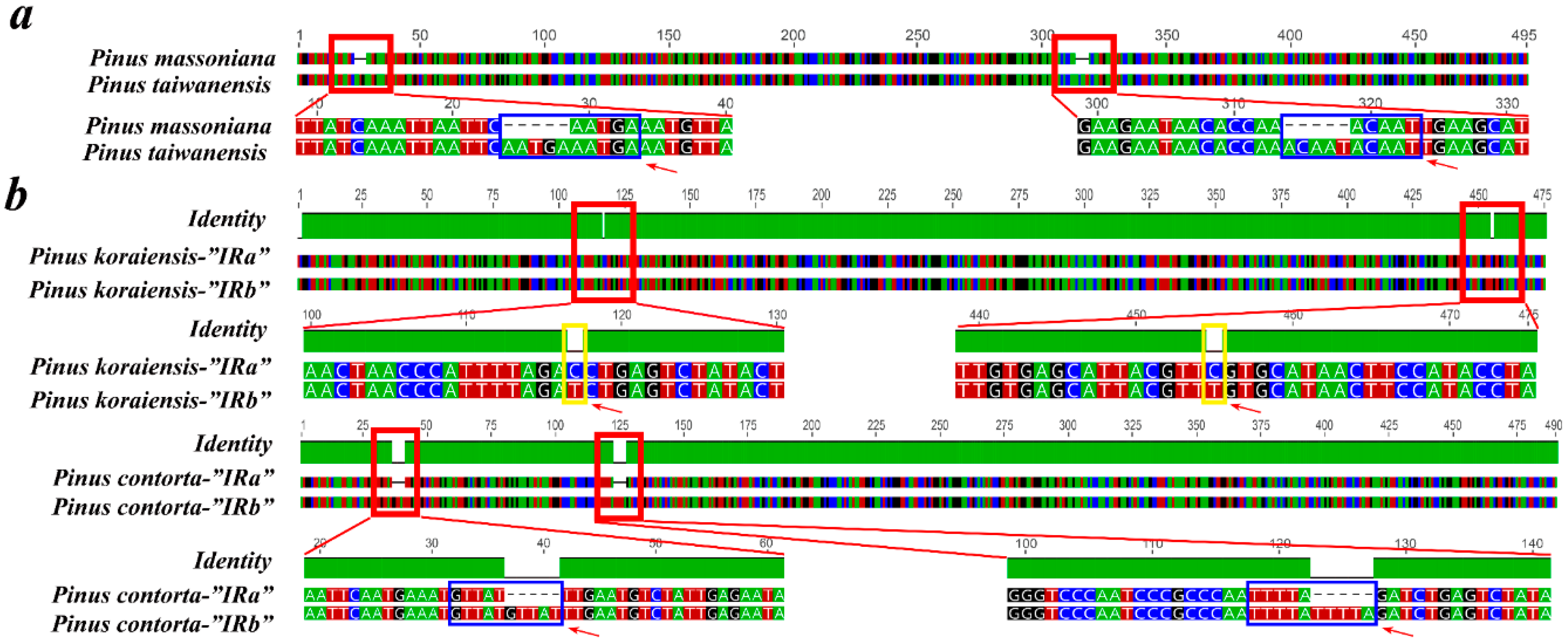
2.6. Contraction and Expansion of Short IRs in Pinus
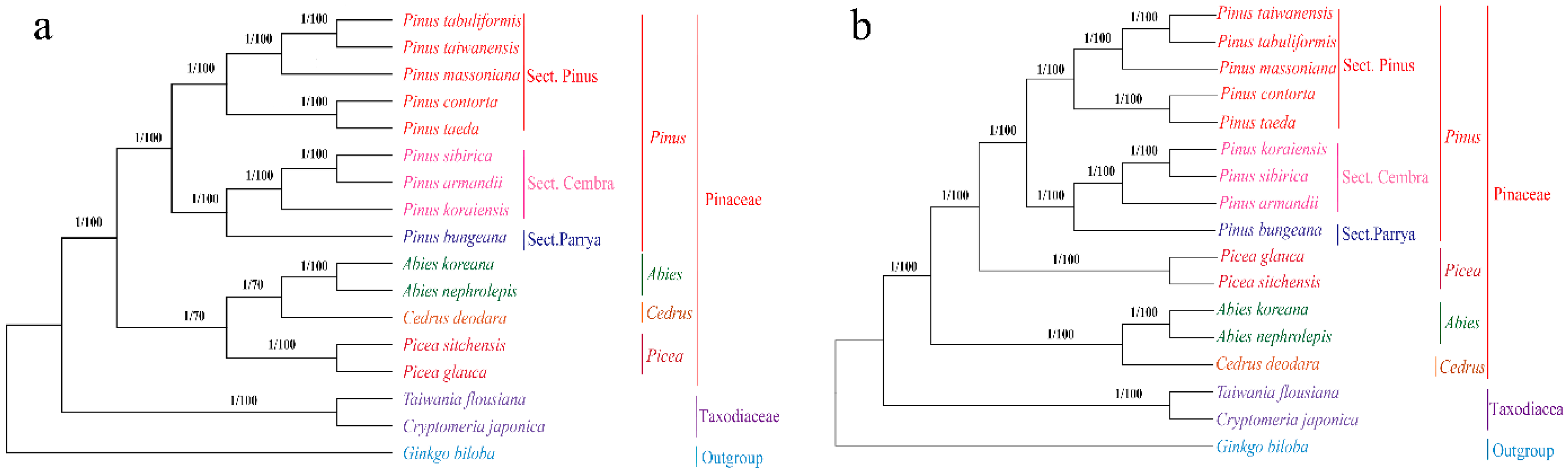
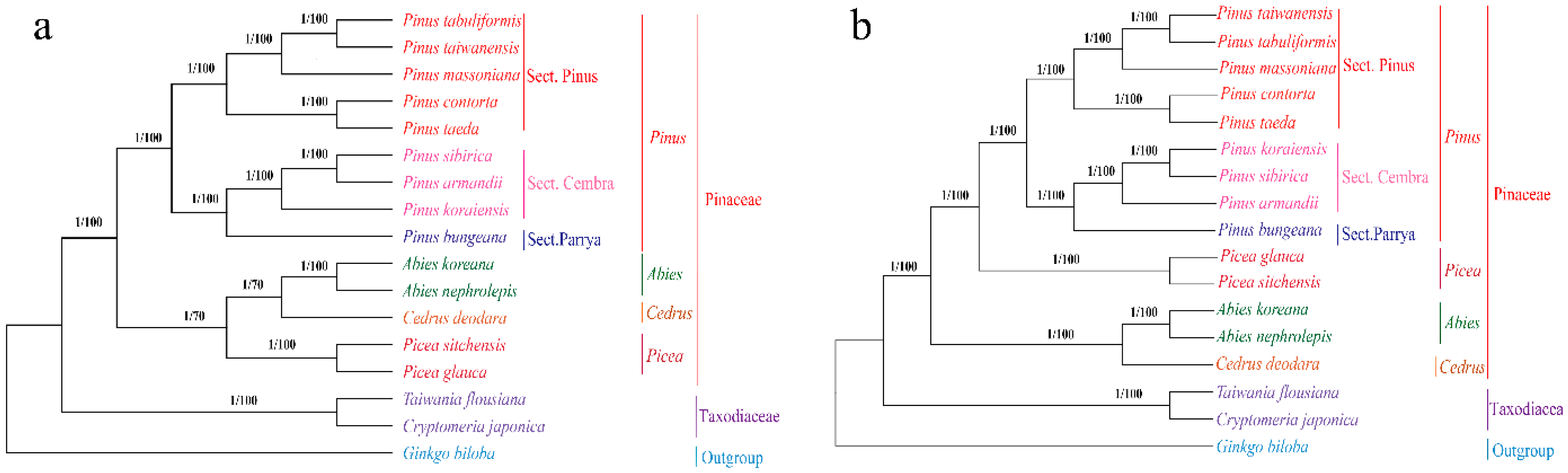
2.7. Phylogenetic Analysis
3. Materials and Methods
3.1. DNA Source, Template Amplification
3.2. Sequencing, Assembly, PCR-Based Gap Filling and Annotation
3.3. Simple Sequence Repeat Analysis
3.4. Sequence Analysis
3.5. Phylogenetic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Asaf, S.; Waqas, M.; Khan, A.L.; Khan, M.A.; Kang, S.-M.; Imran, Q.M.; Shahzad, R.; Bilal, S.; Yun, B.-W.; Lee, I.-J. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Wu, C.-S.; Huang, Y.-Y.; Chaw, S.-M. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol. Evol. 2012, 4, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Cauz-Santos, L.A.; Munhoz, C.F.; Rodde, N.; Cauet, S.; Santos, A.A.; Penha, H.A.; Dornelas, M.C.; Varani, A.M.; Oliveira, G.C.X.; Bergès, H.; et al. The chloroplast genome of Passiflora edulis (passifloraceae) assembled from long sequence reads: Structural organization and phylogenomic studies in malpighiales. Front. Plant Sci. 2017, 8, 334. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Chaw, S.-M. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): Evolution towards shorter intergenic spacers. Plant Biotechnol. J. 2014, 12, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Chen, J.; Hao, Z.; Shi, J. Comparative analysis of the chloroplast genomic information of Cunninghamia lanceolata (Lamb.) Hook with sibling species from the Genera Cryptomeria D. Don, Taiwania Hayata, and Calocedrus Kurz. Int. J. Mol. Sci. 2016, 17, 1084. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. Chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, L.; Chen, S.; Tao, K.; Su, Y.; Wang, T. Evolution of short inverted repeat in cupressophytes, transfer of accd to nucleus in Sciadopitys verticillata and phylogenetic position of Sciadopityaceae. Sci. Rep. 2016, 6, 20934. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Gao, L.; Wang, B.; Su, Y.-J.; Wang, T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. Evol. 2013, 5, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Wang, Y.-N.; Hsu, C.-Y.; Lin, C.-P.; Chaw, S.-M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and Cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Daley, D.O.; Qiu, Y.-L.; Whelan, J.; Palmer, J.D. Repeated, recent and diverse transfers of a mitochondrial gene to the nucleus in flowering plants. Nature. 2000, 408, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Chen, J.J.W.; Chiu, C.-C.; Hsiao, H.C.W.; Yang, C.-J.; Jin, X.-H.; Leebens-Mack, J.; de Pamphilis, C.W.; Huang, Y.-T.; Yang, L.-H.; et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 2017, 90, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Chris Blazier, J.; Guisinger, M.M.; Jansen, R.K. Recent loss of plastid-encoded ndh genes within Erodium (Geraniaceae). Plant Mol. Biol. 2011, 76, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Ranade, S.S.; García-Gil, M.R.; Rosselló, J.A. Non-functional plastid ndh gene fragments are present in the nuclear genome of Norway spruce (Picea abies L. Karsch): Insights from in silico analysis of nuclear and organellar genomes. Mol. Genet. Genomics. 2016, 291, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Sabater, B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010, 48, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Gizaw, A.; Brochmann, C.; Nemomissa, S.; Wondimu, T.; Masao, C.A.; Tusiime, F.M.; Abdi, A.A.; Oxelman, B.; Popp, M.; Dimitrov, D. Colonization and diversification in the African sky ‘islands‘: Insights from fossil-calibrated molecular dating of Lychnis (Caryophyllaceae). New Phytol. 2016, 211, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Group, C.P.W. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Huang, J.-P.; Wu, C.-S.; Hsu, C.-Y.; Chaw, S.-M. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Biol. Evol. 2010, 2, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Cronn, R.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008, 36, e122. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; de Pamphilis, C.W.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 2006, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa(Torr. & Gray). Science 2006, 313, 1596. [Google Scholar] [PubMed]
- Li, Z.H.; Zhu, J.; Yang, Y.X.; Yang, J.; He, J.W.; Zhao, G.F. The complete plastid genome of bunge’s pine Pinus bungeana (Pinaceae). Mitochondr. DNA Part A 2016, 27, 2971–2972. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Separating the wheat from the chaff: Mitigating the effects of noise in a plastome phylogenomic data set from Pinus L. (Pinaceae). BMC Evol. Biol. 2012, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.F.; Wang, Y.J.; Zu, Y.M.; Dong, W.L.; Wang, R.N.; Deng, T.T.; Li, Z.H. The complete chloroplast genome of the taiwan red pine Pinus taiwanensis (Pinaceae). Mitochondr. DNA Part A 2016, 27, 2732–2733. [Google Scholar] [CrossRef]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.-B.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhao, Z.; Xu, H.; Chen, S.; Dorje, G. Chloroplast genome structures in Gentiana (Gentianaceae), based on three medicinal alpine plants used in Tibetan herbal medicine. Curr. Genet. 2017, 63, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Iaffaldano, B.J.; Zhuang, X.; Cardina, J.; Cornish, K. Chloroplast genome resources and molecular markers differentiate rubber dandelion species from weedy relatives. BMC Plant Biol. 2017, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2016, 7, 1955. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dong, W.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Barkley, N.A.; Jenkins, T.M. Microsatellite markers in plants and insects. Part I: Applications of biotechnology. Genes Genomes Genom. 2009, 29, 623–627. [Google Scholar]
- Redwan, R.; Saidin, A.; Kumar, S. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15, 294. [Google Scholar] [CrossRef] [PubMed]
- Braukmann, T.W.A.; Kuzmina, M.; Stefanović, S. Loss of all plastid ndh genes in Gnetales and conifers: Extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 2009, 55, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Qian, Z.Q.; Liu, Z.L.; Deng, T.T.; Zu, Y.M.; Zhao, P.; Zhao, G.F. The complete chloroplast genome of armand pine Pinus armandii, an endemic conifer tree species to China. Mitochondr. DNA Part A 2016, 27, 2635–2636. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. Cap3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare H.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; Mclnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Goss, E.M.; Tabima, J.F.; Cooke, D.E.L.; Restrepo, S.; Fry, W.E.; Forbes, G.A.; Fieland, V.J.; Cardenas, M.; Grünwald, N.J. The Irish potato famine pathogen Phytophthora infestans originated in central Mexico rather than the Andes. Proc. Natl. Acad. Sci. USA 2014, 111, 8791–8796. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sequence data of Pinus massoniana are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Contents |
|---|---|
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ, psaMa |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Small subunit of ribosome | rps2, rps3, rps4, rps7, rps8, rps11, rps12b, rps14, rps15, rps18, rps19 |
| Large subunit of ribosome | rpl2b, rpl14, rpl16b, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 |
| Subunits of cytochrome b/f complex | petA, petBb, petDb, petG, petL, petN |
| Subunits of ATP synthase | atpA, atpB, atpE, atpFb, atpH, atpI |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1b, rpoC2 |
| ChlorophyII biosynthesis | chlB, chlL, chlN |
| Protease | clpP |
| Maturase | matK |
| Envelope membrane protein | cemA |
| Translation initiation factor | infA |
| Cytochrome c biogenesis | ccsA |
| Subunit Acetyl-CoA-Carboxylate | accD |
| Subunit of rubisco | rbcL |
| Ribosomal RNAs | rrn4.5, rrn5, rrn16, rrn23 |
| Conserved open reading frames | ycf1, ycf2, ycf3b, ycf4, ycf12, ycf68 |
| Transfer RNA | trnA-UGCb, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-UCC, trnG-GCCb, trnH-GUGa, trnI-GAUab, trnK-UUUb |
| trnL-CAA, trnL-UAAb, trnL-UAG, trnM-CAU, trnN-GUU, trnP-GGG, trnP-UGG, trnQ-UUG, trnR-ACG, trnR-CCG, trnR-UCU | |
| trnS-GCUa, trnS-GGA, trnS-UGA, trnT-GGUa, trnT-UGU, trnV-GAC, trnV-UACb, trnW-CCA, trnY-GUA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, Z.; Ye, Y.; Bai, T.; Xu, M.; Xu, L.-A. Complete Chloroplast Genome of Pinus massoniana (Pinaceae): Gene Rearrangements, Loss of ndh Genes, and Short Inverted Repeats Contraction, Expansion. Molecules 2017, 22, 1528. https://doi.org/10.3390/molecules22091528
Ni Z, Ye Y, Bai T, Xu M, Xu L-A. Complete Chloroplast Genome of Pinus massoniana (Pinaceae): Gene Rearrangements, Loss of ndh Genes, and Short Inverted Repeats Contraction, Expansion. Molecules. 2017; 22(9):1528. https://doi.org/10.3390/molecules22091528
Chicago/Turabian StyleNi, ZhouXian, YouJu Ye, Tiandao Bai, Meng Xu, and Li-An Xu. 2017. "Complete Chloroplast Genome of Pinus massoniana (Pinaceae): Gene Rearrangements, Loss of ndh Genes, and Short Inverted Repeats Contraction, Expansion" Molecules 22, no. 9: 1528. https://doi.org/10.3390/molecules22091528