The Small Glutathione Peroxidase Mimic 5P May Represent a New Strategy for the Treatment of Liver Cancer

 ,
, 
Abstract
:1. Introduction
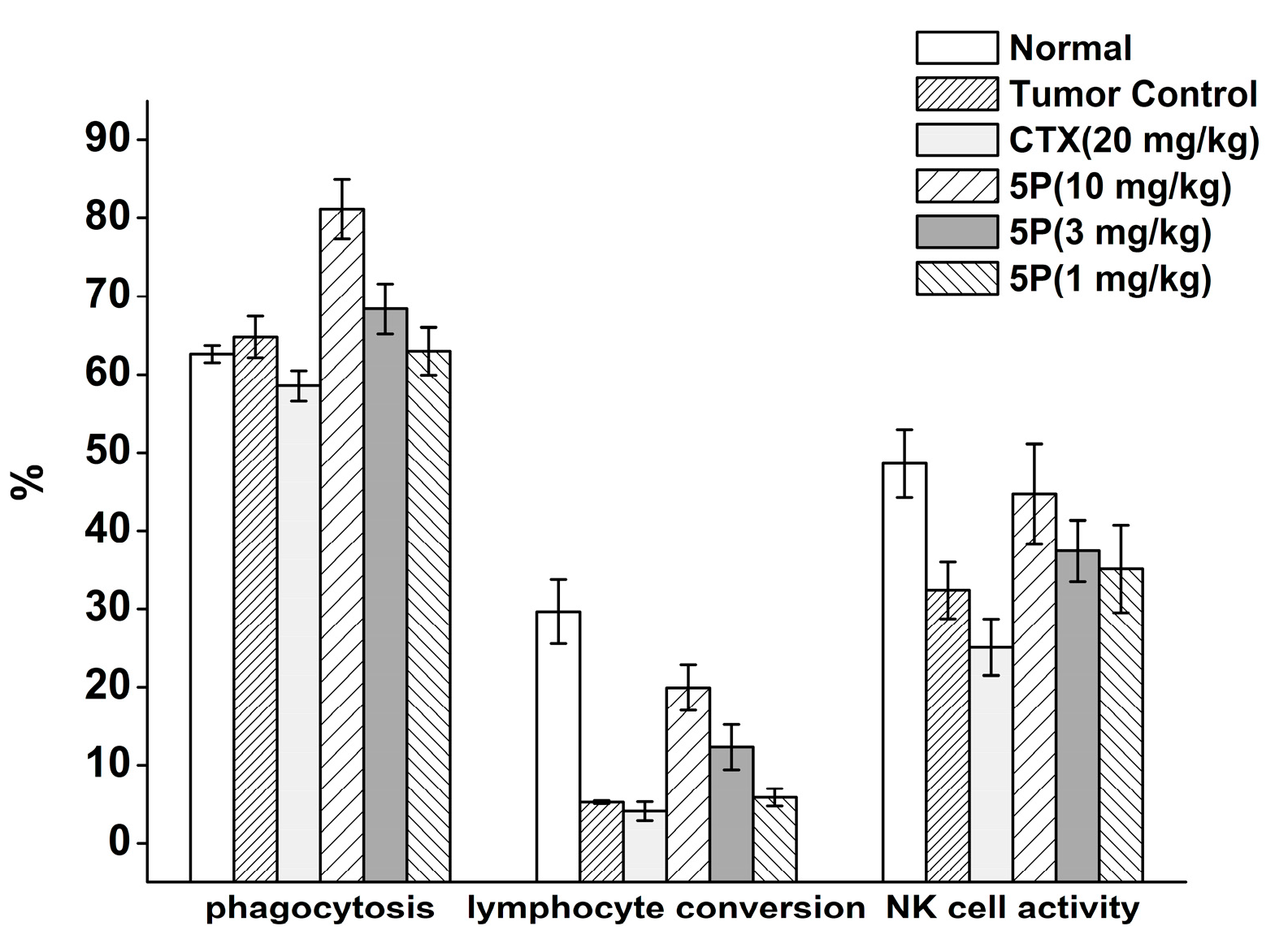
2. Results
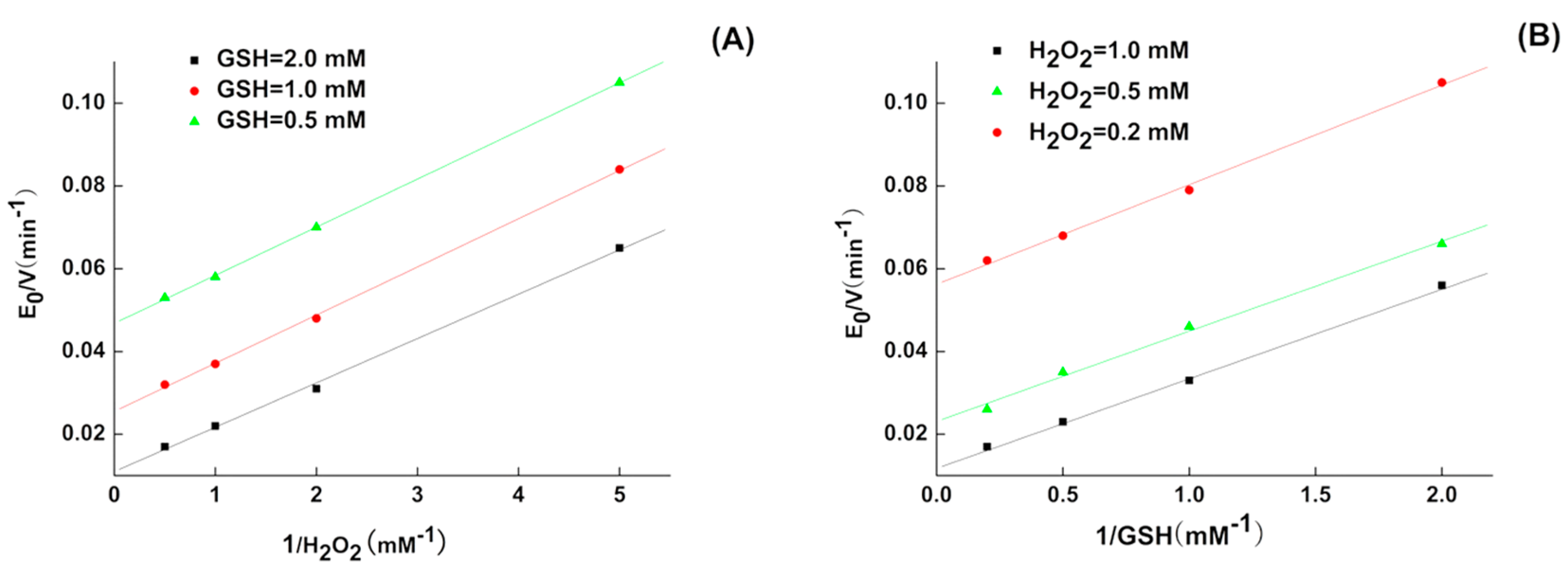
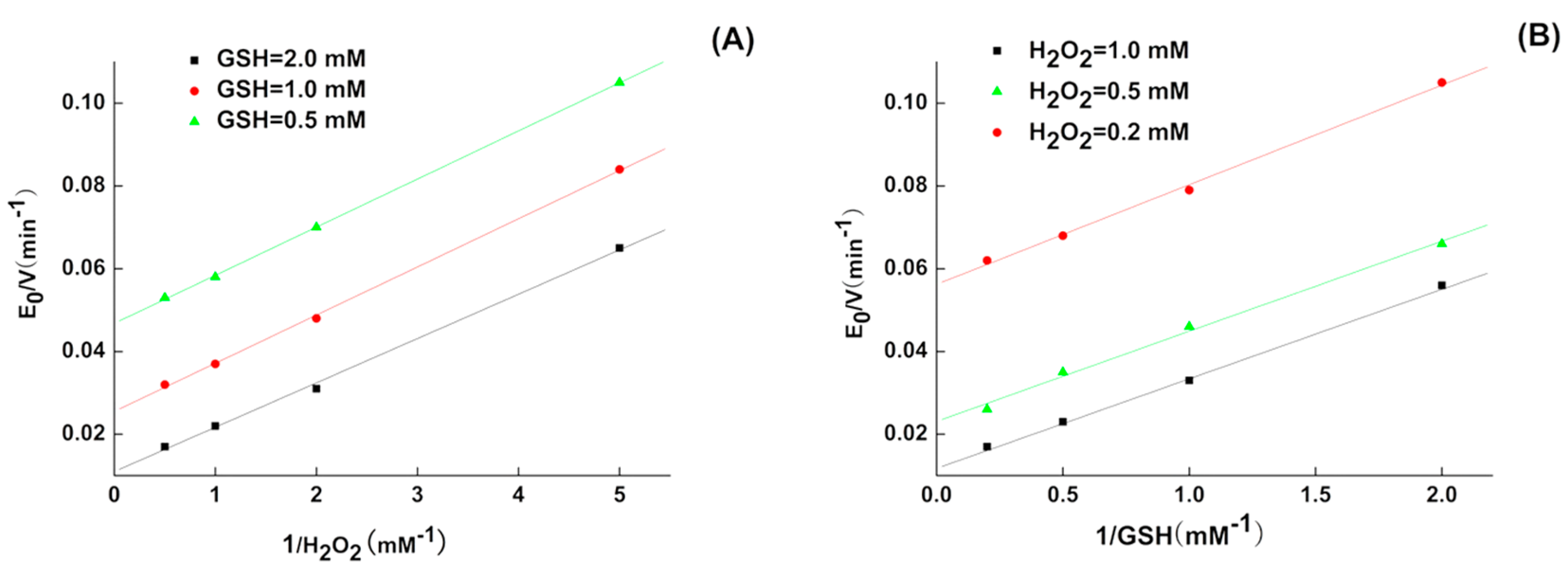
2.1. Kinetics of 5P
2.2. Inhibition of H22 Cells by 5P In Vitro
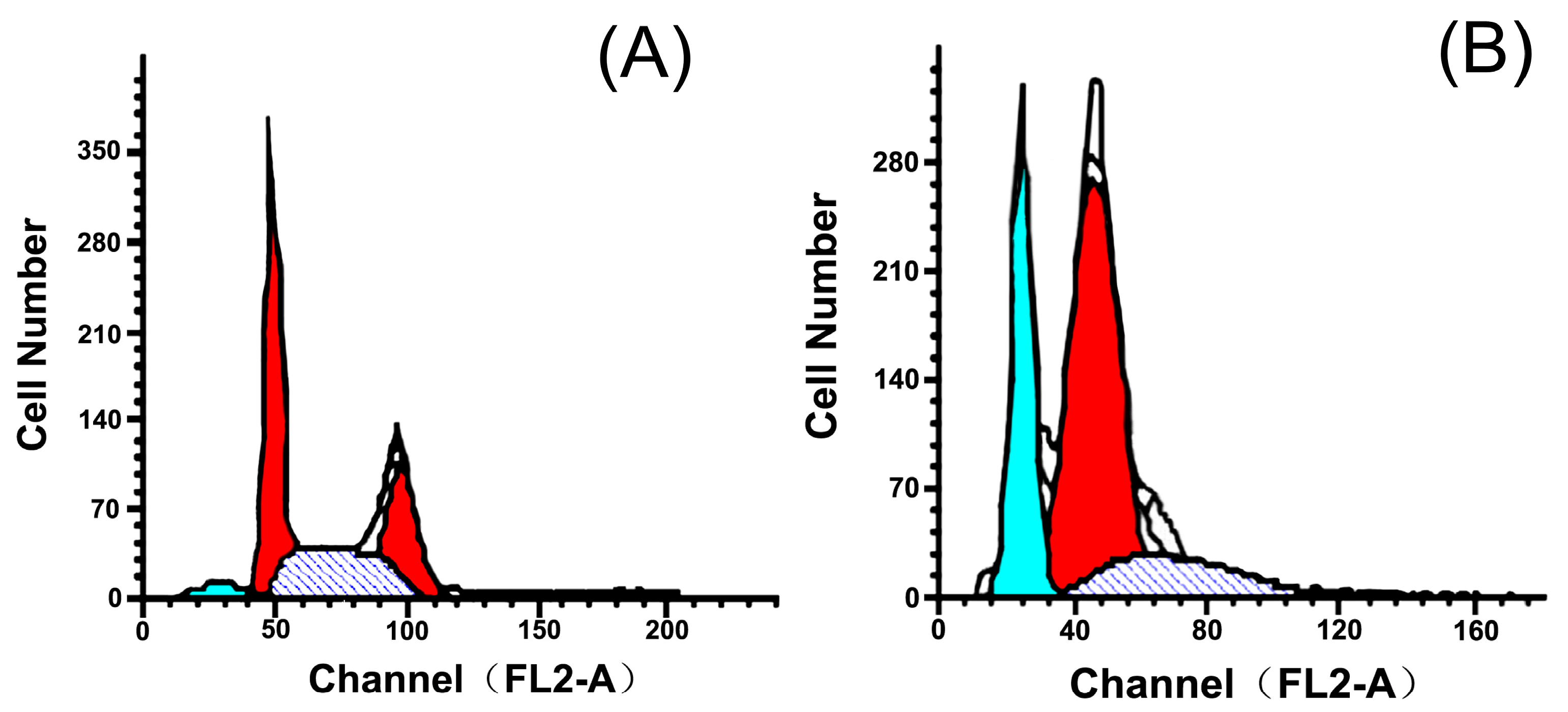
2.3. Effects of 5P on Apoptosis
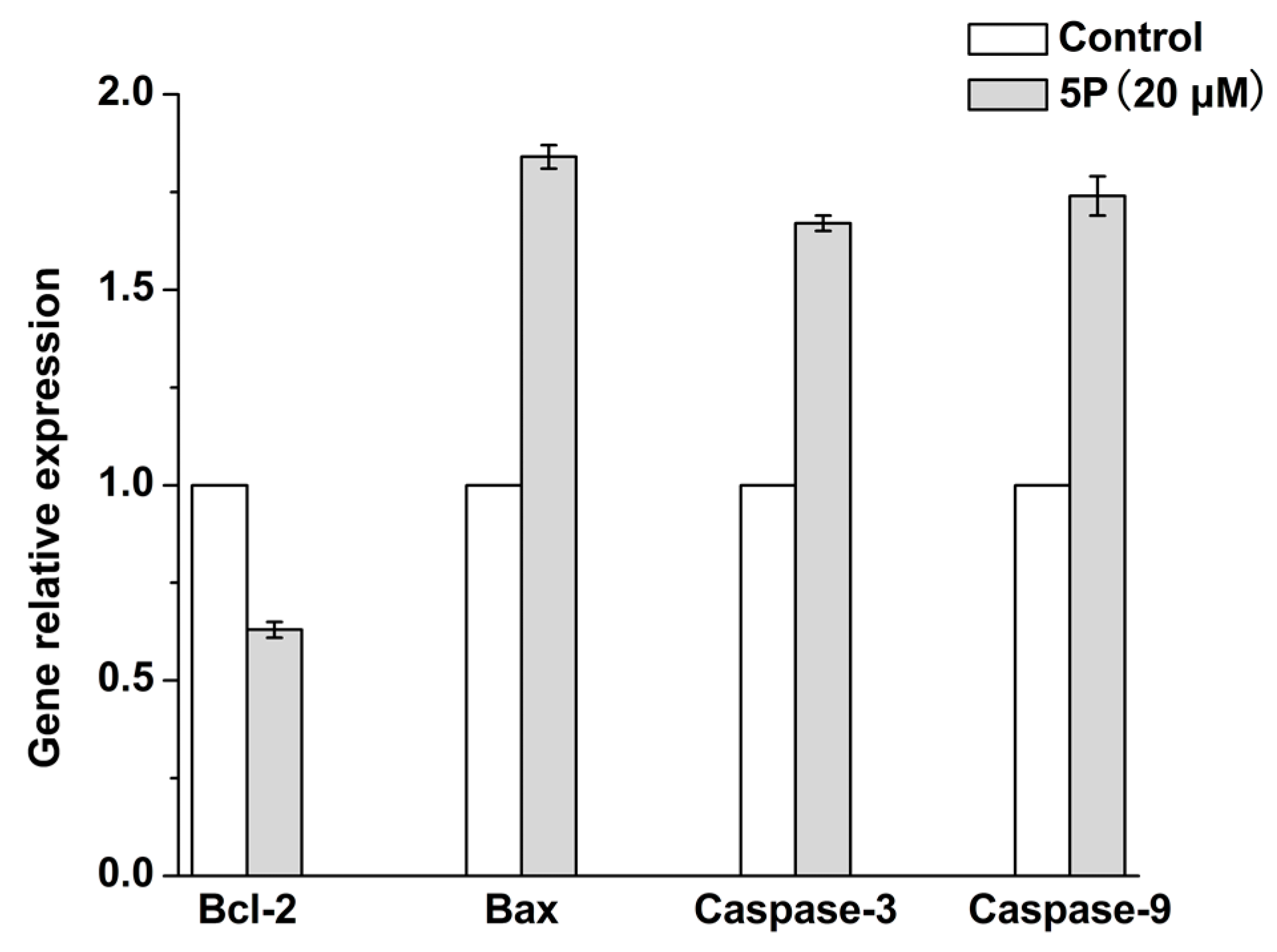
2.4. Effects of 5P on Apoptosis-Related Genes
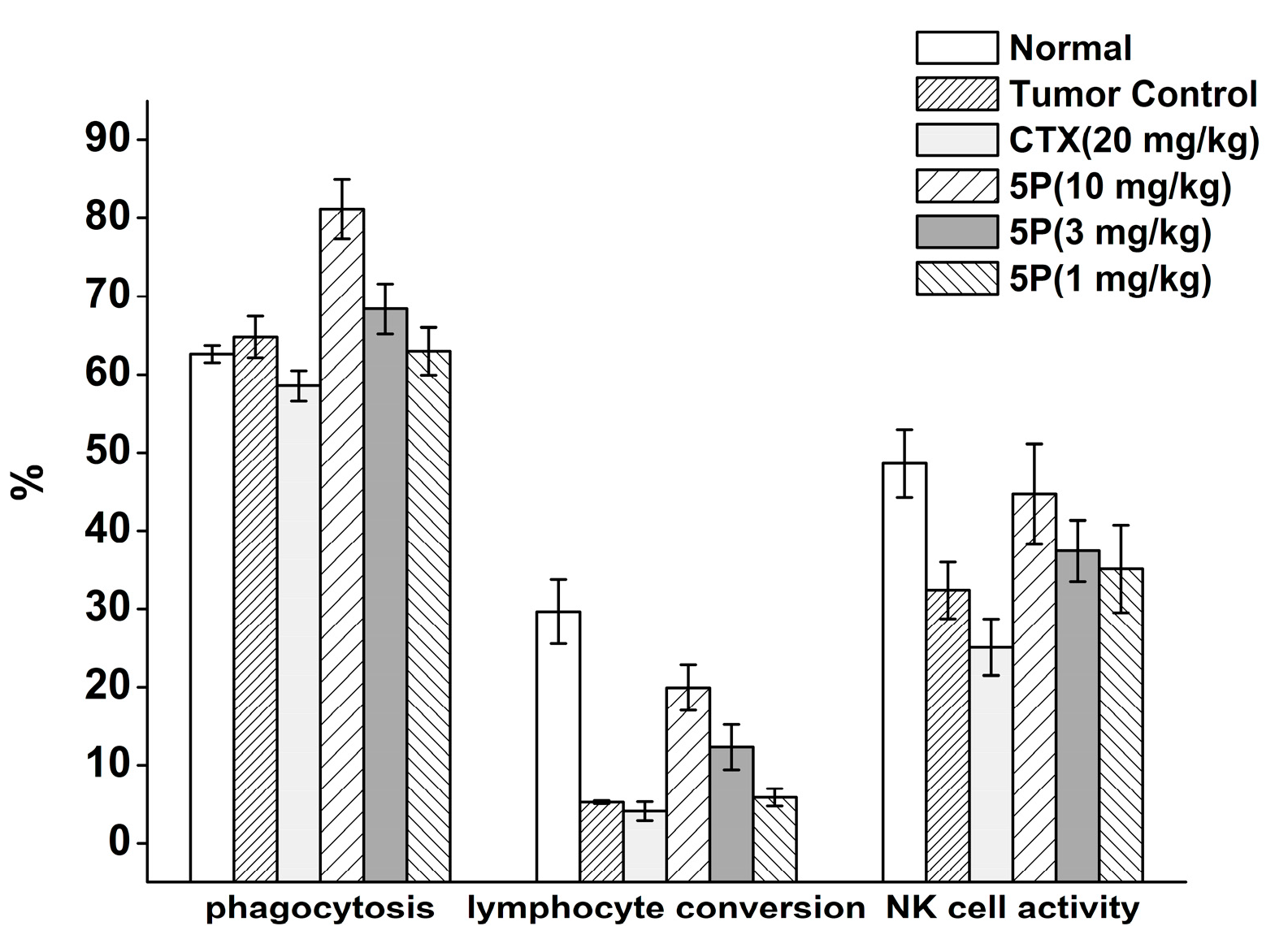
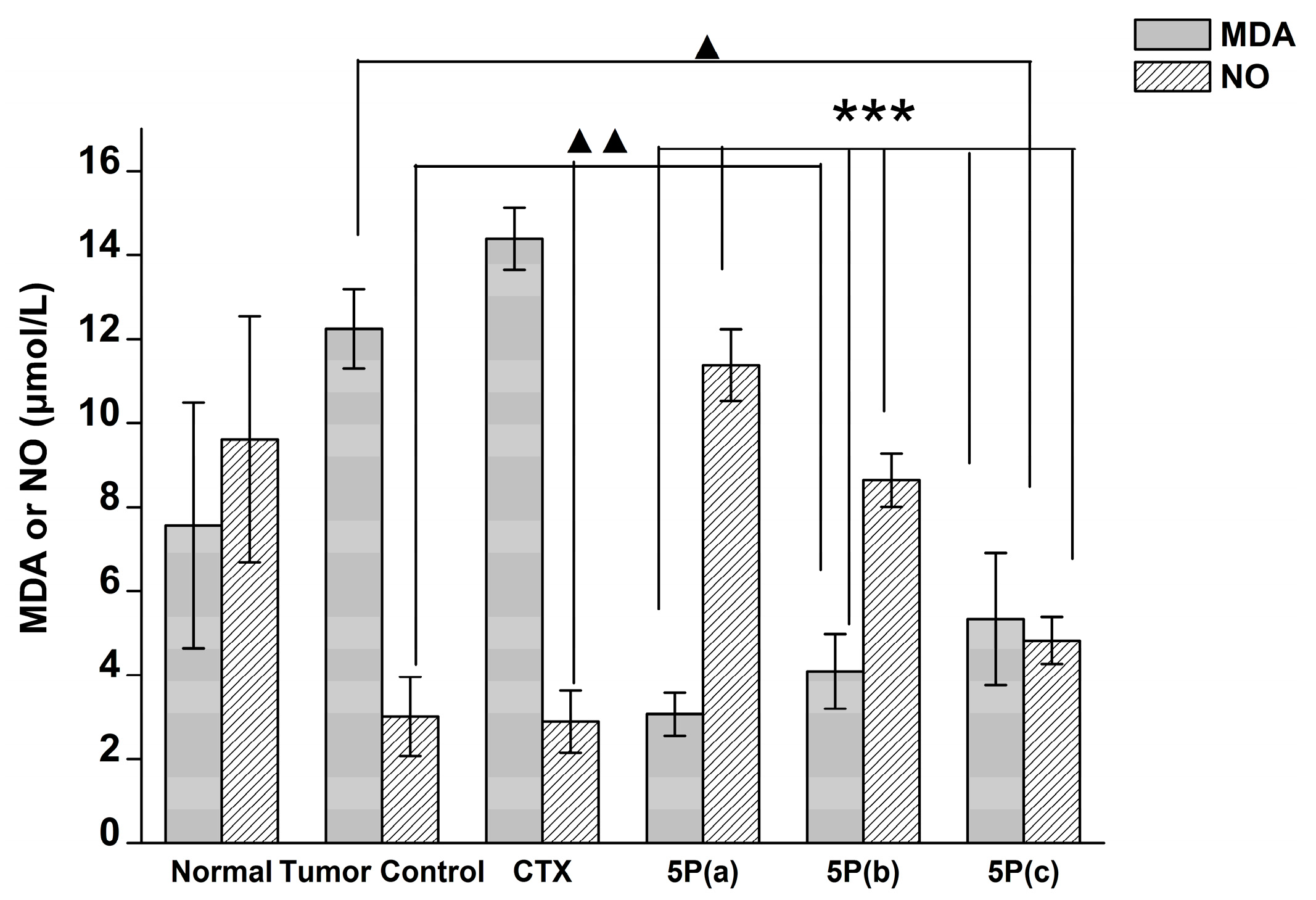
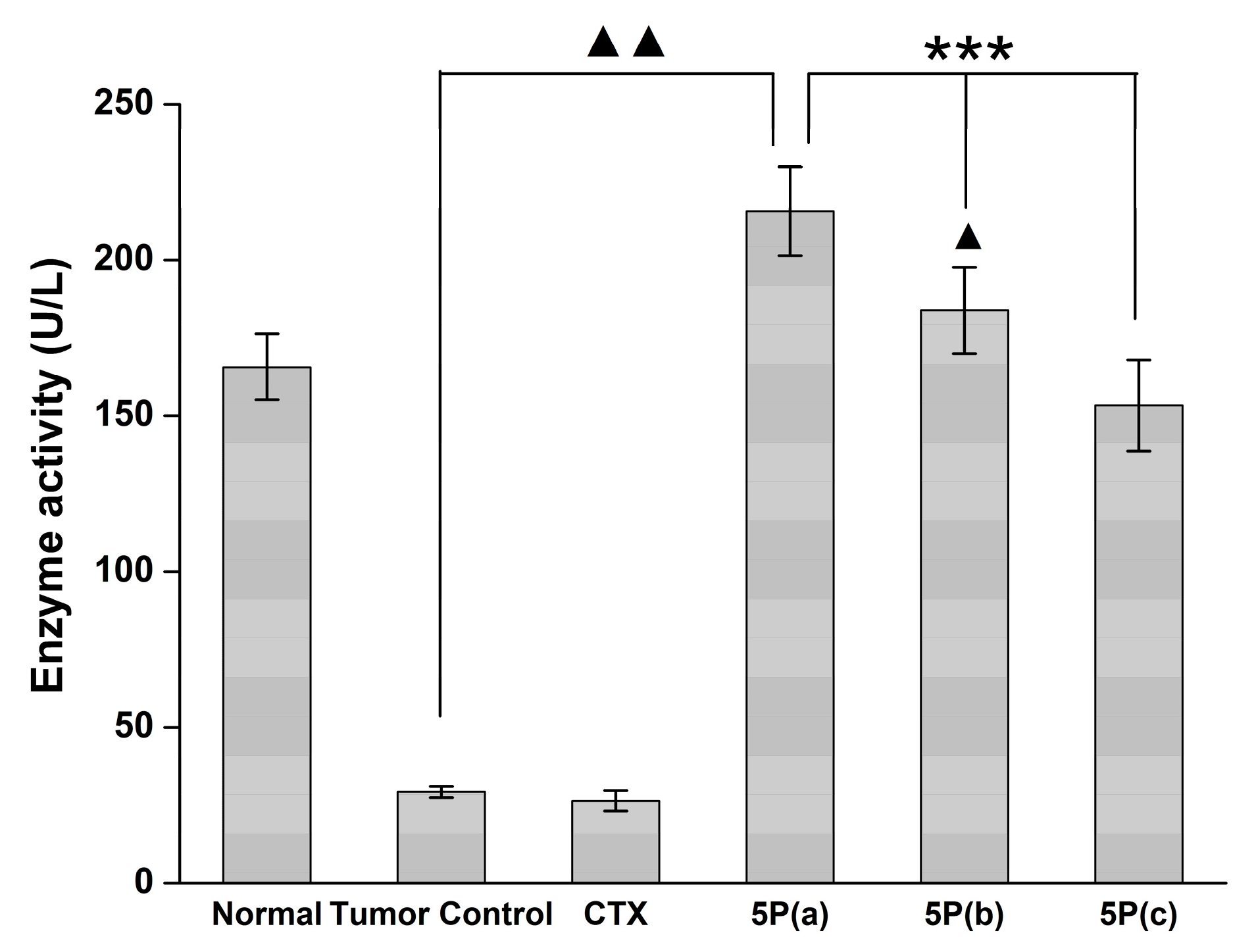
2.5. In Vivo Tumor Growth Model
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Materials
4.3. Synthesis of 5P
4.4. GPx-Like Activity and Kinetics Measurement
4.5. Cell Viability Assay
4.6. Apoptosis Assay
4.7. RT-PCR Analysis
4.8. In Vivo Tumor Growth Model
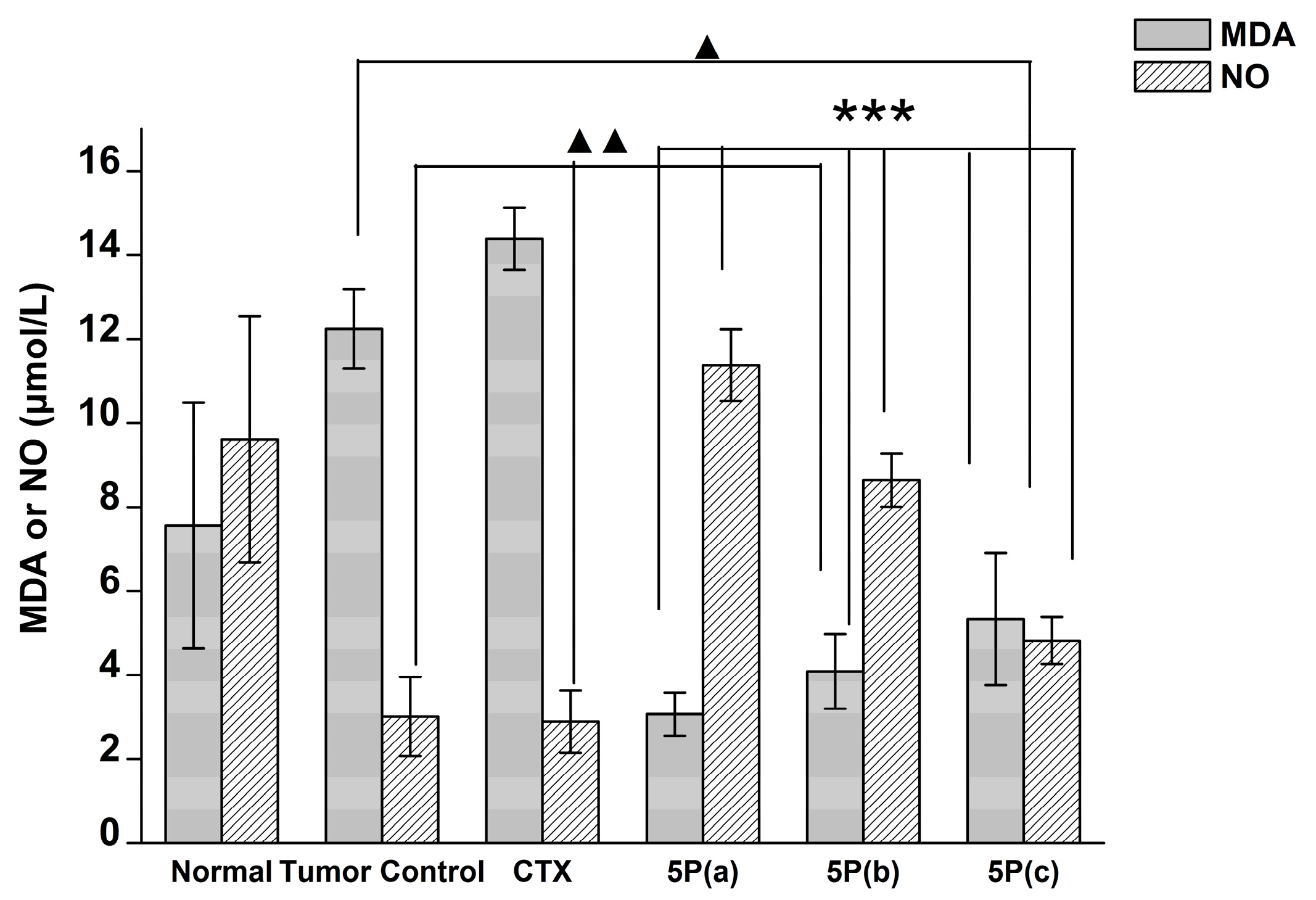
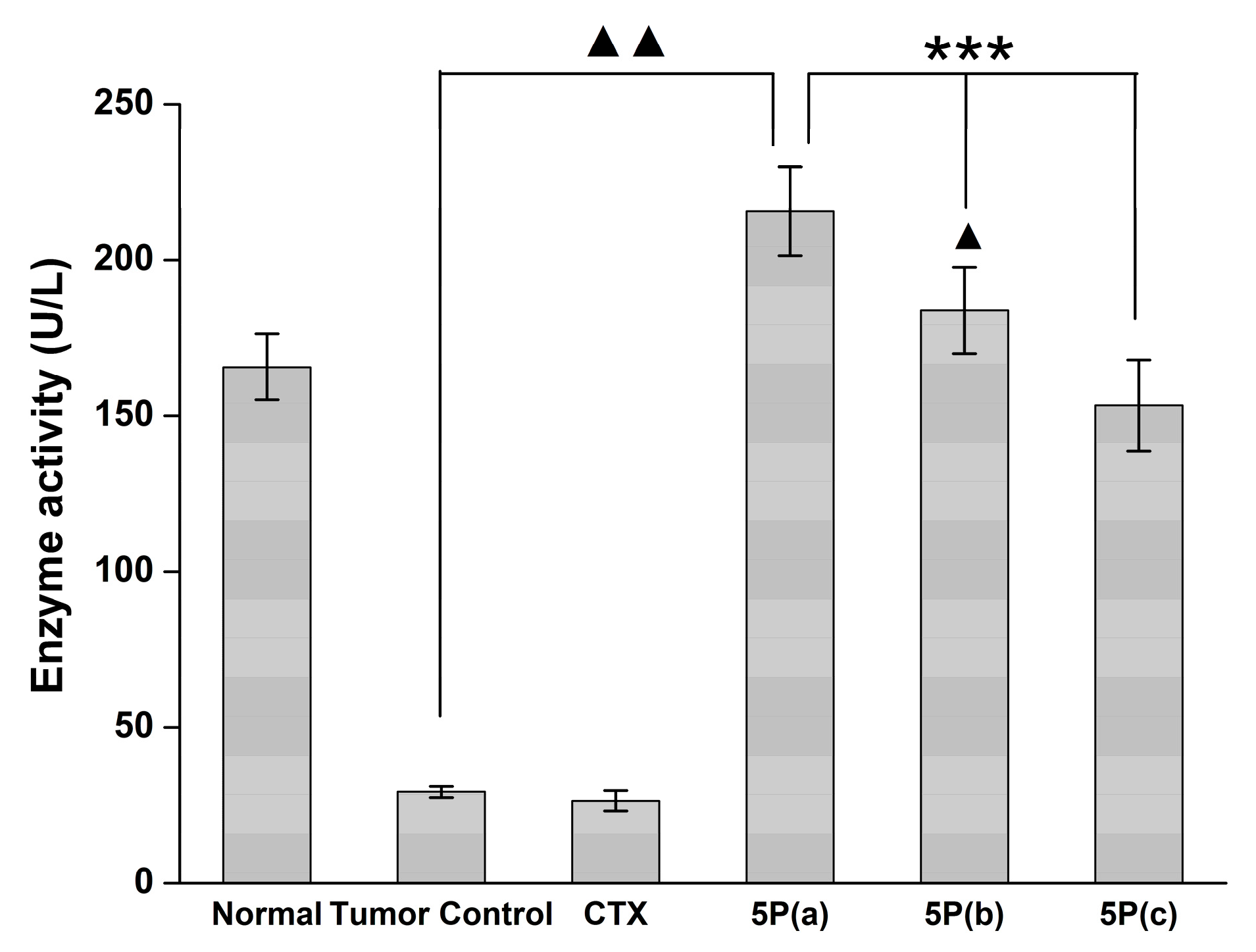
4.9. Blood MDA, NO, and GPX Activity
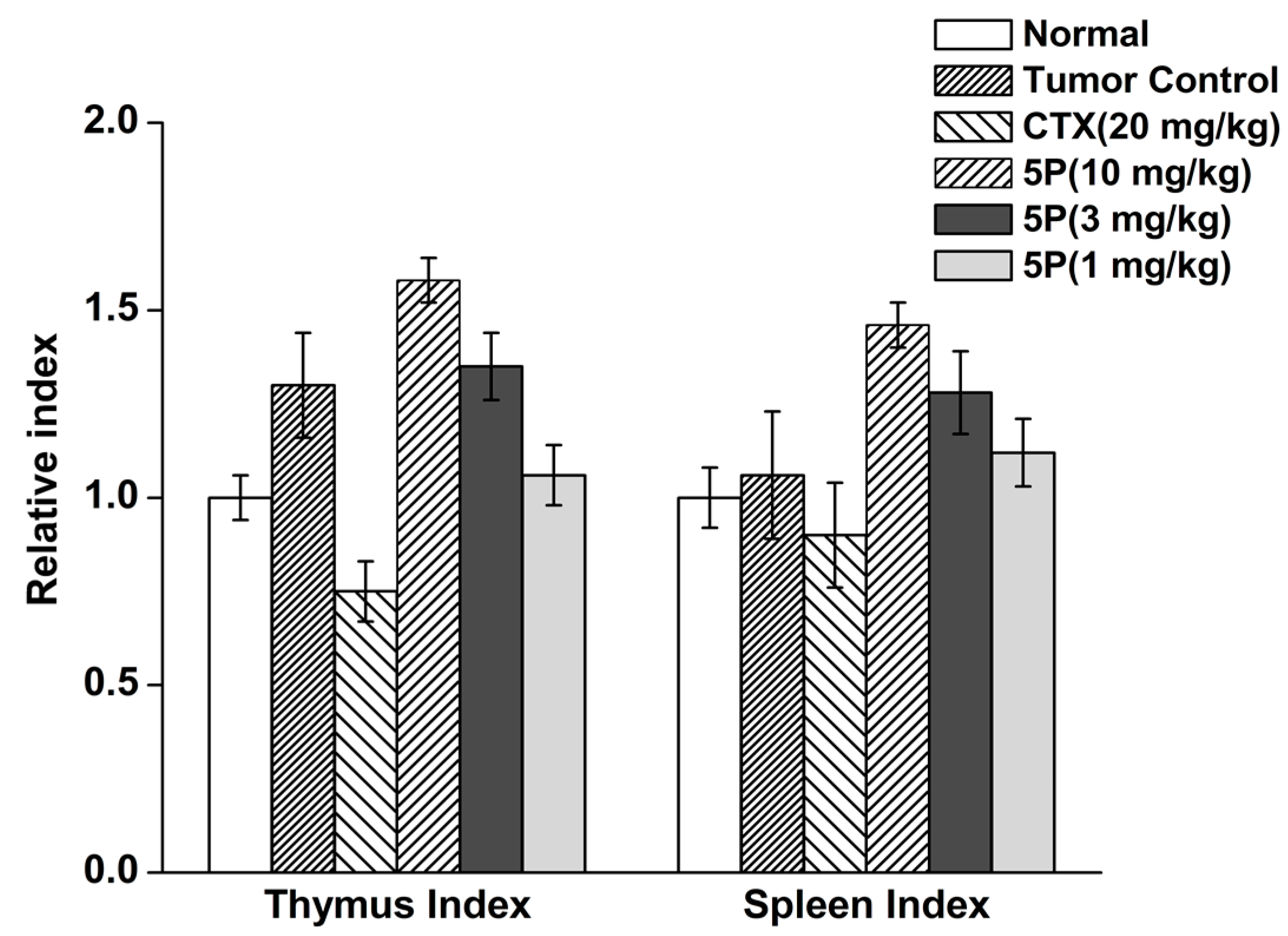
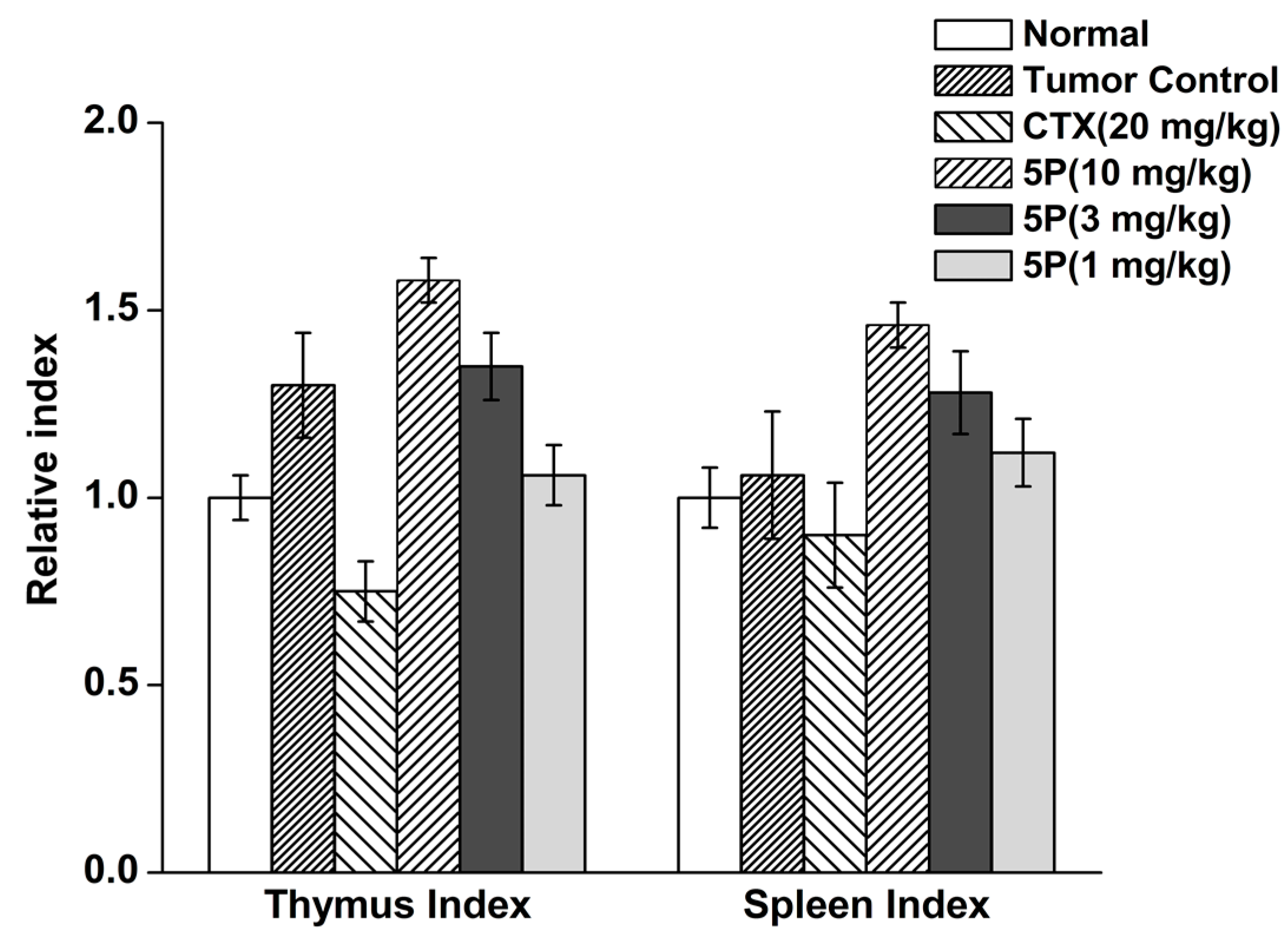
4.10. Tumor Weights, Thymus Index, and Spleen Index
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mates, J.M. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.C. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [PubMed]
- Flohe, L.; Gunzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef]
- Pan, T.; Liu, Y.; Si, C.; Bai, Y.; Qiao, S.; Zhao, L.; Xu, J.; Dong, Z.; Luo, Q.; Liu, J. Construction of ATP-switched allosteric antioxidant selenoenzyme. ACS Catal. 2017, 7, 1875–1879. [Google Scholar] [CrossRef]
- Jiang, Q.; Pan, Y.; Cheng, Y.; Li, H.; Li, H. Protection of rat liver against hepatic ischemia-reperfusion injury by a novel selenocysteine-containing 7-mer peptide. Mol. Med. Rep. 2016, 14, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Zucker, P.A.; Huang, R.R.C.; Spector, A. Development of synthetic compounds with glutathione peroxidase activity. J. Am. Chem. Soc. 1989, 111, 5936–5939. [Google Scholar] [CrossRef]
- Dinakarpandian, D.; Shenoy, B.C.; Hilvert, D.; McRee, D.E.; McTigue, M.; Carey, P.R. Electric fields in active sites: Substrate switching from null to strong fields in thiol-and selenol-subtilisins. Biochemistry 1999, 38, 6659–6667. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Molecular imprinting in cross-linked materials with the aid of molecular templates—A way towards artificial antibodies. Angew. Chem. Int. Ed. 1995, 34, 1812–1832. [Google Scholar] [CrossRef]
- Iwaoka, M.; Tomoda, S. A model study on the effect of an amino group on the antioxidant activity of glutathione peroxidase. J. Am. Chem. Soc. 1994, 116, 2557–2561. [Google Scholar] [CrossRef]
- Meyer, E.; Cole, G.; Radhakrishnan, R.; Epp, O. Structure of native porcine pancreatic elastase at 1.65 resolution. Acta Crystallogr. Sect. B Struct. Sci. 1988, 44, 26–38. [Google Scholar] [CrossRef]
- Sun, Y.; Li, T.; Chen, H.; Zhang, K.; Zheng, K.; Mu, Y.; Yan, G.; Li, W.; Shen, J.; Luo, G. Selenium-containing 15-mer peptides with high glutathione peroxidase-like activity. J. Biol. Chem. 2004, 279, 37235–37240. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.G.; Bag, P.P.; Kumakura, F.; Iwaoka, M.; Priyadarsini, K.I. Role of substrate reactivity in the glutathione peroxidase (GPx) activity of selenocystine. Bull. Chem. Soc. Jpn. 2010, 83, 703–708. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, X.Y.; Si, C.Y.; Gao, Y.Z.; Zhao, L.L.; Hou, C.X.; Shoseyov, O.; Luo, Q.; Liu, J.Q. Construction of a highly stable artificial glutathione peroxidase on a protein nanoring. Org. Biomol. Chem. 2014, 12, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Ren, X.; Xue, Y.; Zhang, K.; Liu, J.; Luo, G.; Zheng, J.; Mu, Y.; Shen, J. A novel dicyclodextrinyl ditelluride compound with antioxidant activity. FEBS Lett. 2001, 507, 377–380. [Google Scholar] [CrossRef]
- Abd El-Wahab, A.E.; Ghareeb, D.A.; Sarhan, E.E.; Abu-Serie, M.M.; El Demellawy, M.A. In vitro biological assessment of berberis vulgaris and its active constituent, berberine: Antioxidants, anti-acetylcholinesterase, anti-diabetic and anticancer effects. BMC Complement. Altern. Med. 2013, 13, 218. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xia, C.; Wang, B.; Chen, H.; Wang, T.; He, Q.; Cao, H.; Wang, Y. Effects of quantum dots on the ROS amount of liver cancer stem cells. Colloids Surf. B Biointerfaces 2017, 155, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Waziri, P.M.; Abdullah, R.; Yeap, S.K.; Omar, A.R.; Abdul, A.B.; Kassim, N.K.; Malami, I.; Karunakaran, T.; Imam, M.U. Clausenidin from Clausena excavata induces apoptosis in hepG2 cells via the mitochondrial pathway. J. Ethnopharmacol. 2016, 194, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Yin, Y.; Xing, J.; Li, C.; Kou, L.; Hu, B.; Wu, Z.W.; Wang, J.J.; Xu, G.X. Therapeutic efficacy of bifidobacterium longum-mediated human granulocyte colony-stimulating factor and/or endostatin combined with cyclophosphamide in mouse-transplanted tumors. Cancer Sci. 2009, 100, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C.; Fu, X.-C. Enhancement of bleomycin—Iron free radical damage to DNA by antioxidants and their inhibition of lipid peroxidation. FEBS Lett. 1981, 123, 71–74. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Tokuyama, Y.; Igarashi, M.; Miyazawa, T. Tumor growth suppression by α-eleostearic acid, a linolenic acid isomer with a conjugated triene system, via lipid peroxidation. Carcinogenesis 2004, 25, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Yang, G.H.; Zheng, R.B.; Yu, X.T.; Peng, S.Z.; Xie, J.H.; Chen, J.N.; Wang, X.F.; Su, Z.R.; Zhang, X.J. The immune-regulating effect of Xiao’er Qixingcha in constipated mice induced by high-heat and high-protein diet. BMC Complement. Altern. Med. 2017, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, H.; Zou, D.; He, X.; Yu, X.; Li, Y. Experimental study of chemotherapy related leukocytopenia treated by various peroal leucocyte increasing drugs. Afr. J. Tradit. Complement. Altern. Med. AJTCAM 2017, 14, 155–164. [Google Scholar]
- Lok, H.C.; Sahni, S.; Jansson, P.J.; Kovacevic, Z.; Hawkins, C.L.; Richardson, D.R. A nitric oxide storage and transport system that protects activated macrophages from endogenous nitric oxide cytotoxicity. J. Biol. Chem. 2016, 291, 27042–27061. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, E.S.; Antinarelli, L.M.R.; Silva, N.P.; Souza, I.O.; Meinel, R.S.; Rocha, M.N.; Soares, R.P.P.; da Silva, A.D. Quin line derivatives: Synthesis, leishmanicidal activity and involvement of mitochondrial oxidative stress as mechanism of action. Chem. Biol. Interact. 2016, 260, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Pita-Lopez, M.L.; Pera, A.; Solana, R. Adaptive memory of human NK-like CD8+ T-cells to aging, and viral and tumor antigens. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Tabynov, K.; Yespembetov, B.; Matikhan, N.; Ryskeldinova, S.; Zinina, N.; Kydyrbayev, Z.; Assanzhanova, N.; Tabynov, K.; Renukaradhya, G.J.; Mukhitdinova, G.; et al. First evaluation of an influenza viral vector based brucella abortus vaccine in sheep and goats: Assessment of safety, immunogenicity and protective efficacy against brucella melitensis infection. Vet. Microbiol. 2016, 197, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Koide, T.; Itoh, H.; Otaka, A.; Yasui, H.; Kuroda, M.; Esaki, N.; Soda, K.; Fujii, N. Synthetic study on selenocystine-containing peptides. Chem. Pharm. Bull. 1993, 41, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Takei, T.; Urabe, Y.; Asahina, Y.; Hojo, H.; Nomura, T.; Dedachi, K.; Arai, K.; Iwaoka, M. Model study using designed selenopeptides on the importance of the catalytic triad for the antioxidative functions of glutathione peroxidase. J. Phys. Chem. B 2013, 118, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Pascual, P.; Martinez-Lara, E.; Bárcena, J.A.; López-Barea, J.; Toribio, F. Direct assay of glutathione peroxidase activity using high-performance capillary electrophoresis. J. Chromatogr. B Biomed. Sci. Appl. 1992, 581, 49–56. [Google Scholar] [CrossRef]
- Flohé, L.; Loschen, G.; Günzler, W.A.; Eichele, E.; Glutathione peroxidase, V. The kinetic mechanism. Hoppe-Seylers Z. Physiol. Chem. 1972, 353, 987–1000. [Google Scholar]
- Scheving, L.E. Circadian rhythms in cell proliferation: Their importance when investigating the basic mechanism of normal versus abnormal growth. Prog. Clin. Biol. Res. 1981, 59, 39. [Google Scholar]
- Nagata, S. Fas-mediated apoptosis. In Mechanisms of Lymphocyte Activation and Immune Regulation VI; Springer: Berlin/Heidelberg, Germany, 1996; pp. 119–124. [Google Scholar]
- Brüne, B.; Sandau, K.; Von Knethen, A. Apoptotic cell death and nitric oxide: Activating and antagonistic transducing pathways. Biochemistry 1998, 63, 817–825. [Google Scholar]
Sample Availability: Samples of the compounds 5P are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPX Mimic | kmax (min−1) | Km,GSH (mM) | Km,H2O2 (mM) | kmax/Km,GSH (M−1 min−1) | kmax/Km,H2O2 (M−1 min−1) |
|---|---|---|---|---|---|
| 5P | 333.2 ± 8 | 9.4 ± 0.28 | 3.9 ± 0.12 | 3.5 × 104 | 8.54 × 104 |
| Group | Dose (μM) | Absorption Value (OD 570 nm) | Inhibition Ratio (%) |
|---|---|---|---|
| control | - | 0.336 ± 0.026 | - |
| 5P | 5 | 0.353 ± 0.019 | - |
| 5P | 10 | 0.266 ± 0.025 ** | 20.84 |
| 5P | 15 | 0.172 ± 0.022 *** | 48.81 |
| 5P | 20 | 0.150 ± 0.008 *** | 55.36 |
| 5P | 25 | 0.139 ± 0.007 *** | 58.63 |
| Group | Dose | G0/G1 | G2/M | S | Apoptosis (%) |
|---|---|---|---|---|---|
| Control | - | 41.33 | 27.30 | 31.37 | 4.52 |
| 5P | 20 | 79.55 | 0.15 | 20.30 | 27.51 |
| Group | Dose mg/kg | Body Weight (g) Start | Body Weight (g) End |
|---|---|---|---|
| Normal | - | 34.86 ± 2.06 | 40.24 ± 1.67 |
| Tumor Control | - | 34.94 ± 2.39 | 44.19 ± 2.92 ▲▲ |
| CTX | 20 | 34.91 ± 2.56 | 31.99 ± 2.27 *** ▲▲▲ |
| 5P | 10 | 34.53 ± 1.98 | 43.34 ± 3.54 ▲ |
| 5P | 3 | 34.34 ± 2.55 | 43.69 ± 3.10 ▲ |
| 5P | 1 | 34.78 ± 3.66 | 43.78 ± 3.90 ▲ |
| Group | Dose (mg/kg) | Tumor Weight (g) | Tumor Inhibition Ratio (%) |
|---|---|---|---|
| Normal | - | - | - |
| Tumor Control | - | 1.09 ± 0.30 | - |
| CTX | 20 | 0.43 ± 0.16 *** | 60.94 |
| 5P | 10 | 0.59 ± 0.14 ** | 46 |
| 5P | 3 | 0.71 ± 0.14 * | 34.86 |
| 5P | 1 | 0.91 ± 0.30 | 16.91 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, J.; Wang, B.; Zhu, X.; Qu, X.; Huang, Y.; Lv, S.; Mu, Y.; Luo, G. The Small Glutathione Peroxidase Mimic 5P May Represent a New Strategy for the Treatment of Liver Cancer. Molecules 2017, 22, 1495. https://doi.org/10.3390/molecules22091495
Yin J, Wang B, Zhu X, Qu X, Huang Y, Lv S, Mu Y, Luo G. The Small Glutathione Peroxidase Mimic 5P May Represent a New Strategy for the Treatment of Liver Cancer. Molecules. 2017; 22(9):1495. https://doi.org/10.3390/molecules22091495
Chicago/Turabian StyleYin, Juxin, Bingmei Wang, Xuejun Zhu, Xiaonan Qu, Yi Huang, Shaowu Lv, Ying Mu, and Guimin Luo. 2017. "The Small Glutathione Peroxidase Mimic 5P May Represent a New Strategy for the Treatment of Liver Cancer" Molecules 22, no. 9: 1495. https://doi.org/10.3390/molecules22091495