Forbidden Coherence Transfer of 19F Nuclei to Quantitatively Measure the Dynamics of a CF3-Containing Ligand in Receptor-Bound States

Abstract
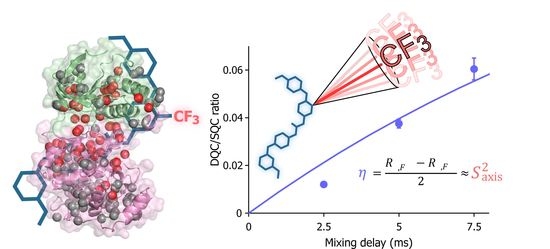
:
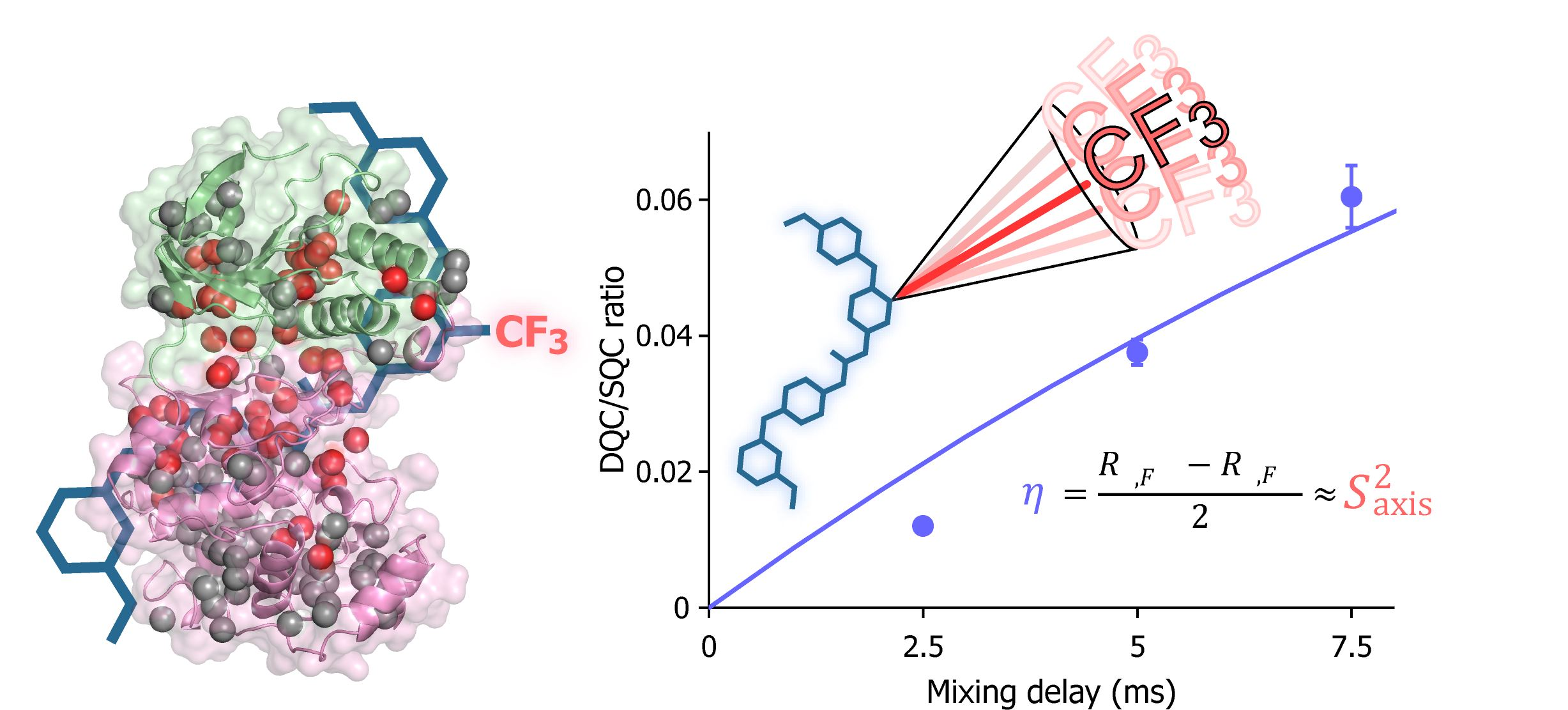{kind=link}
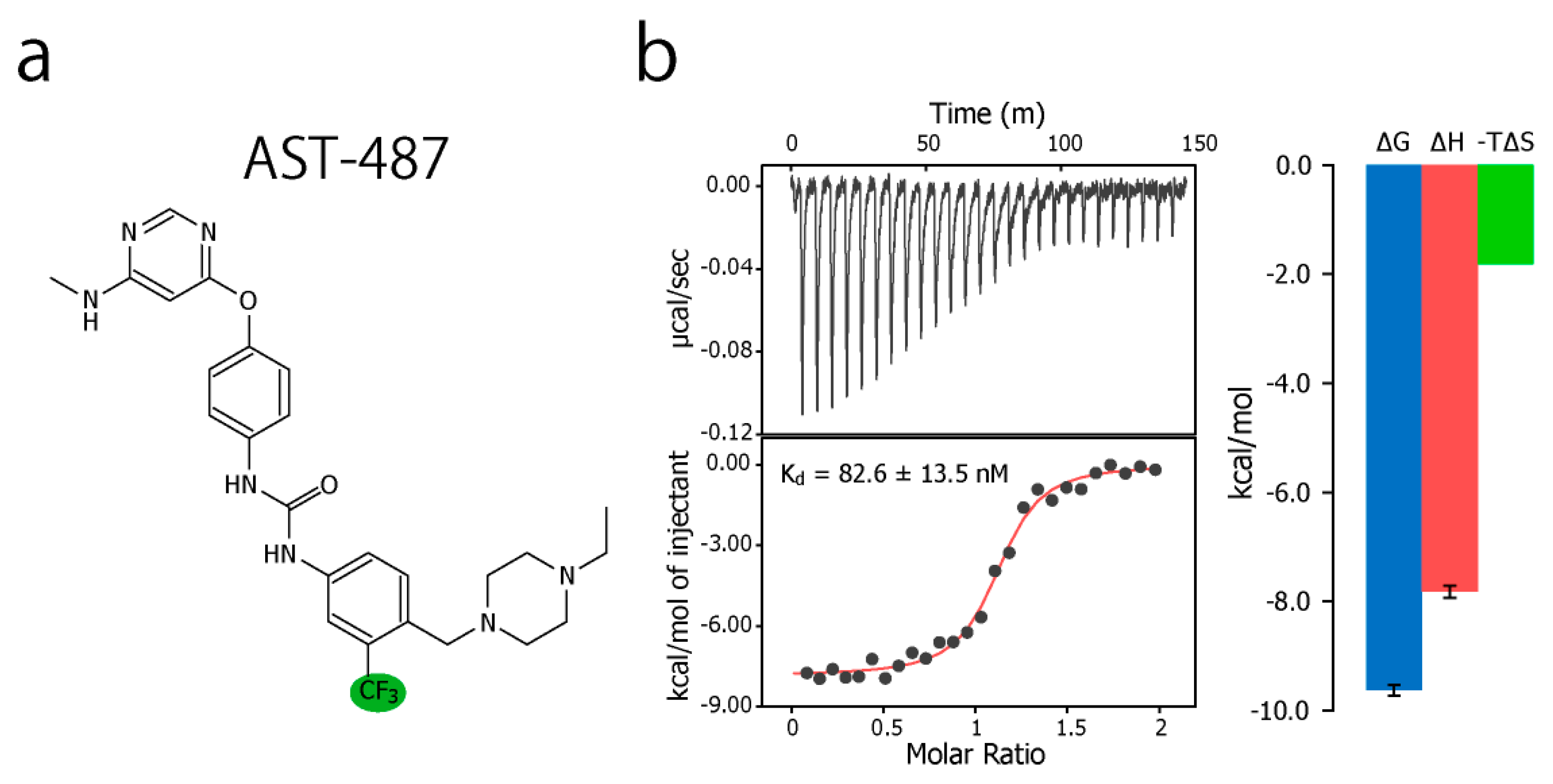{kind=link}
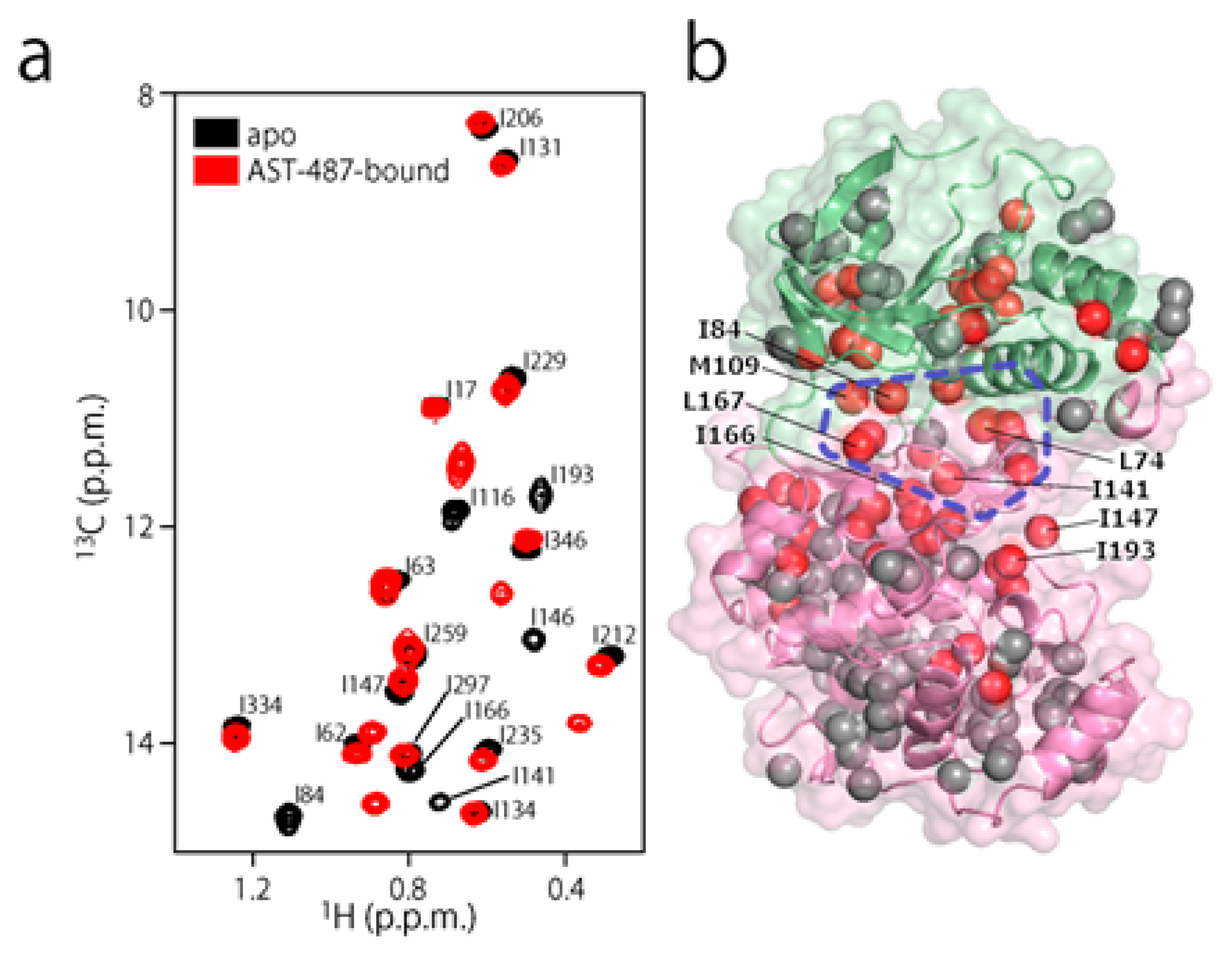{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
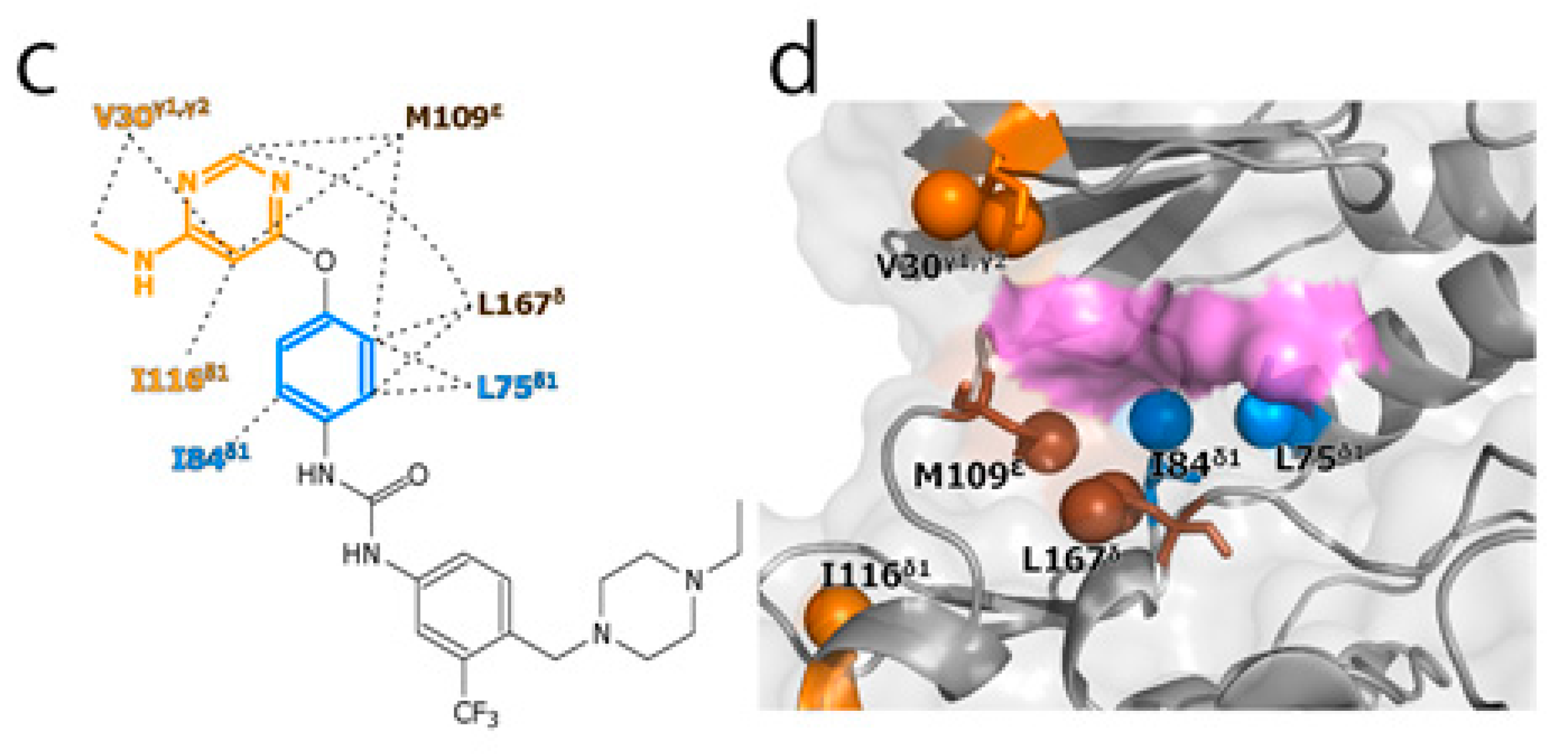
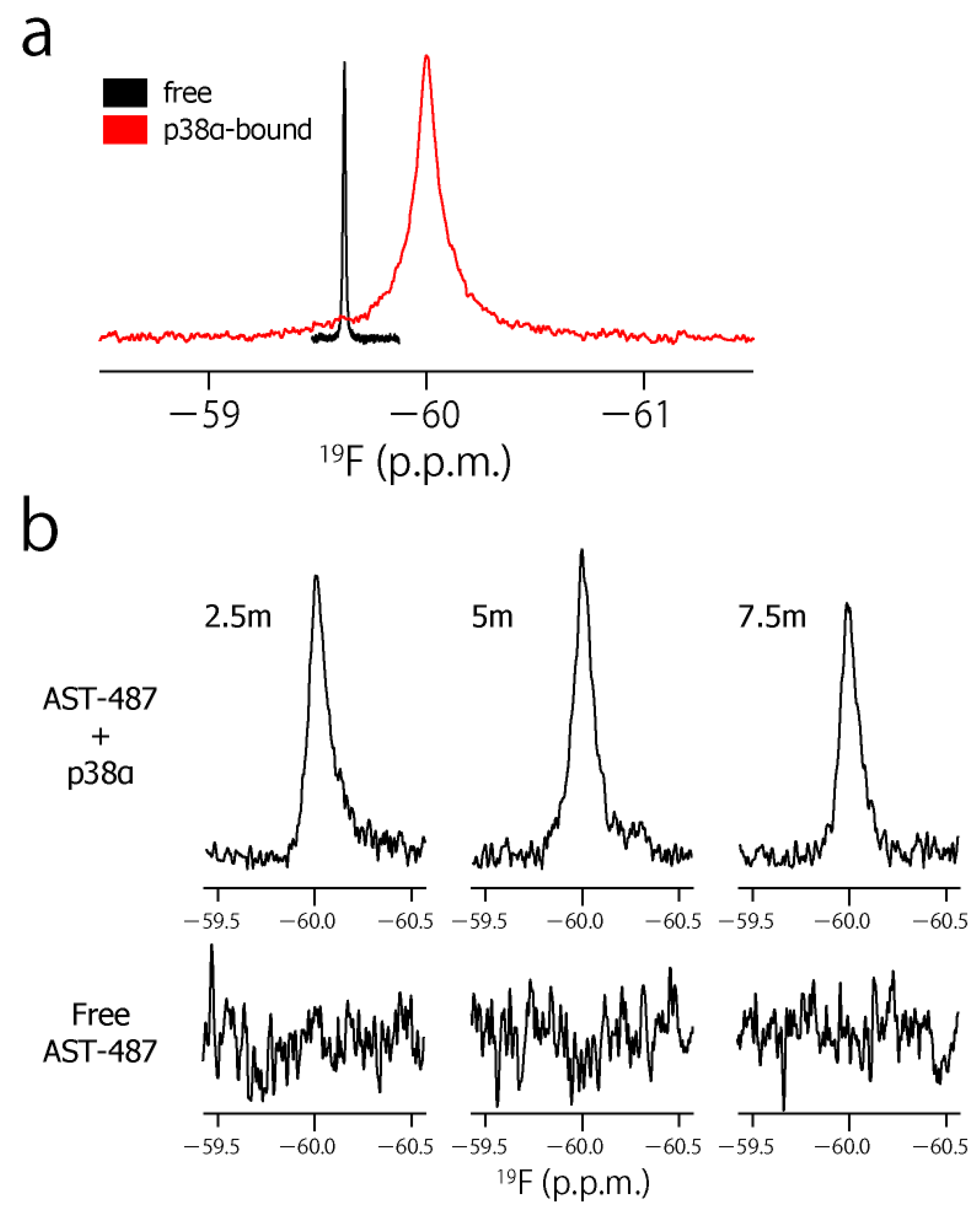
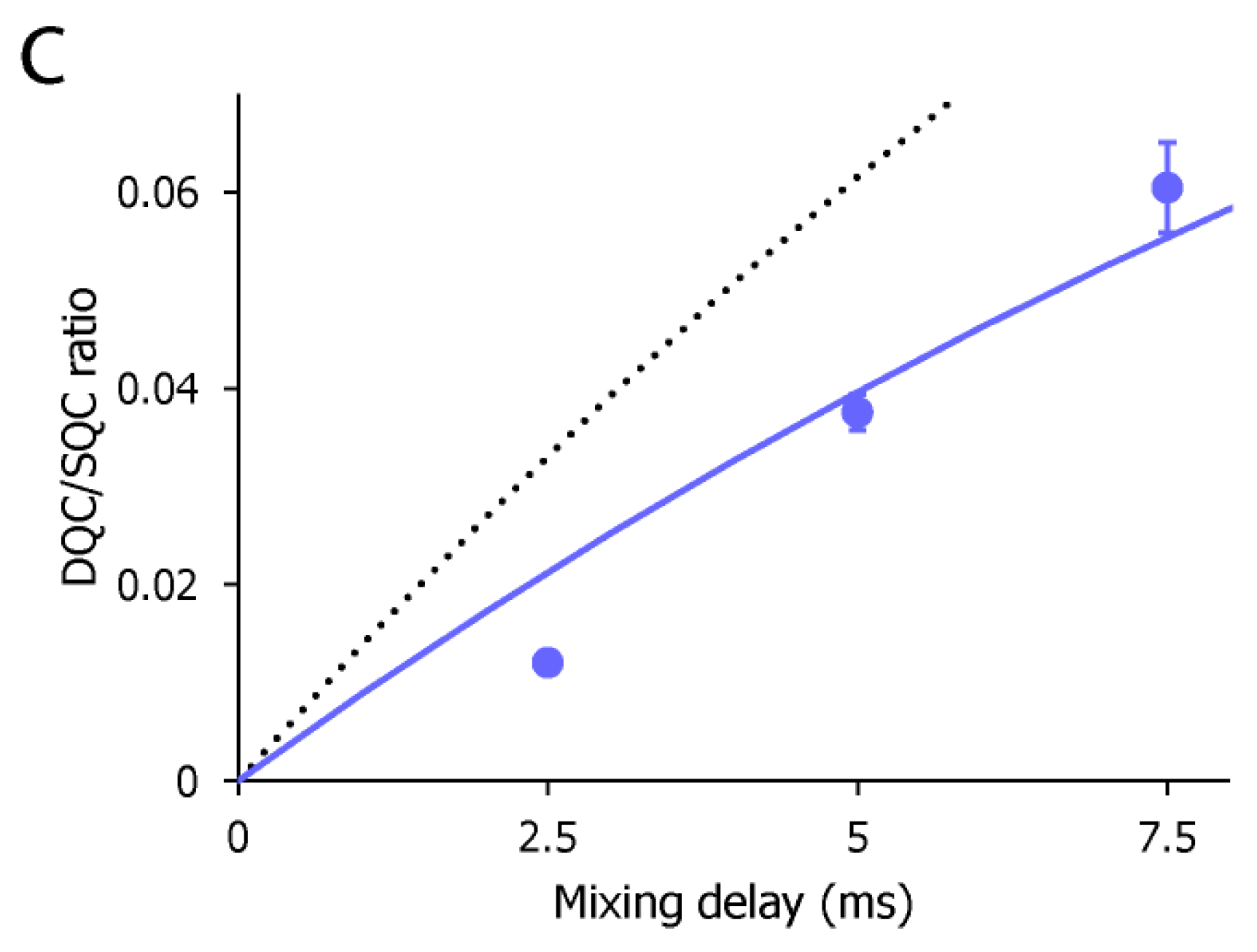
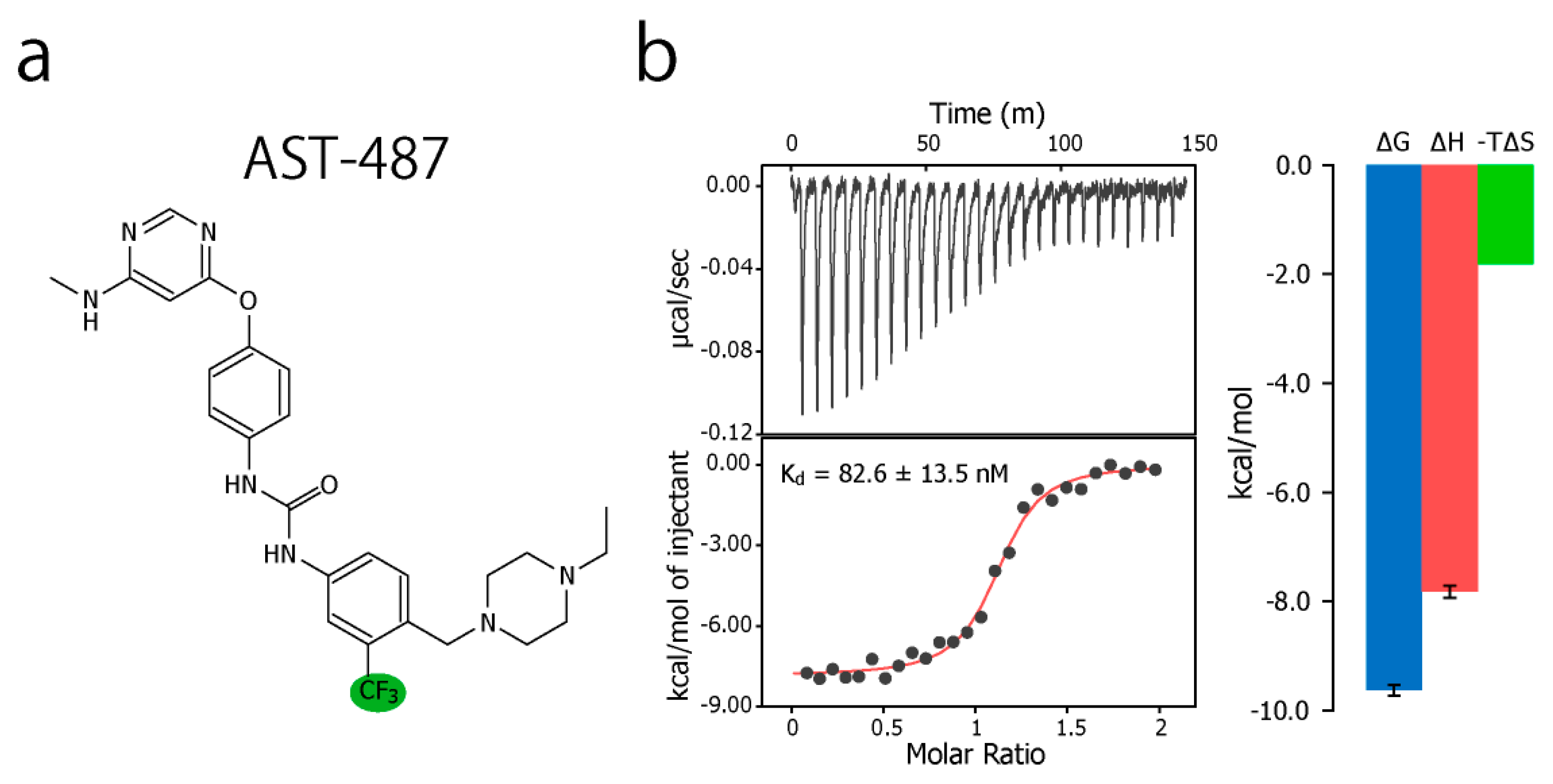
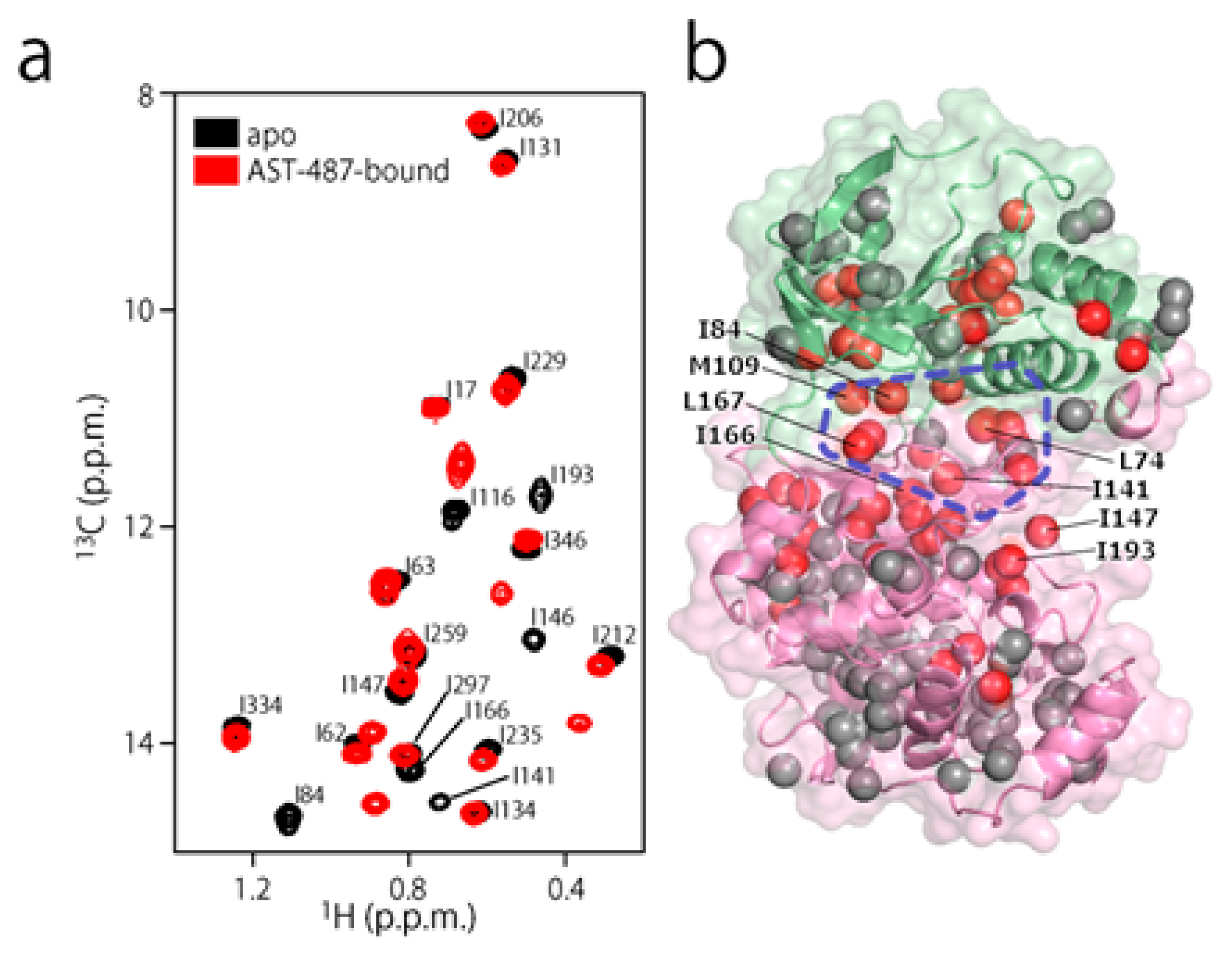
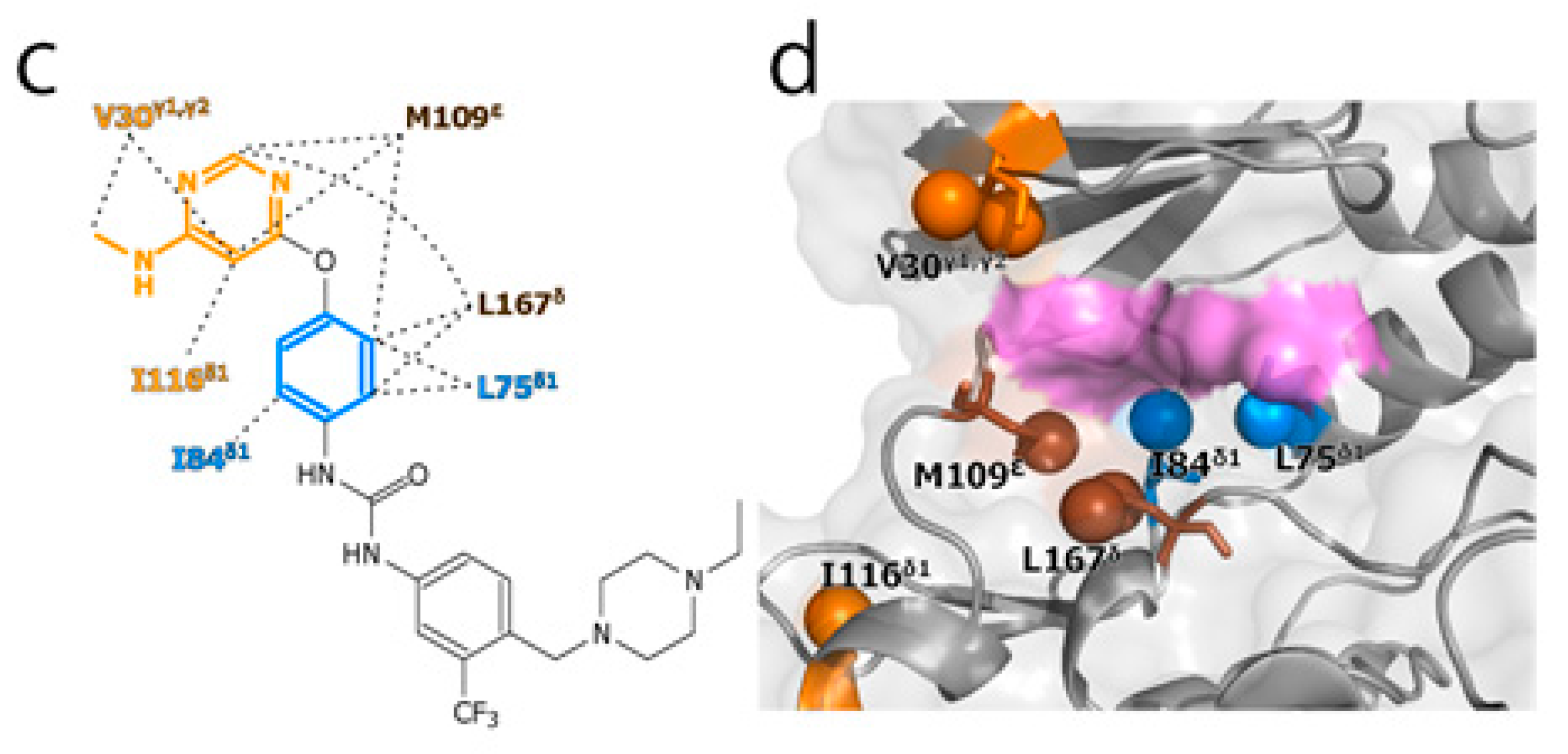
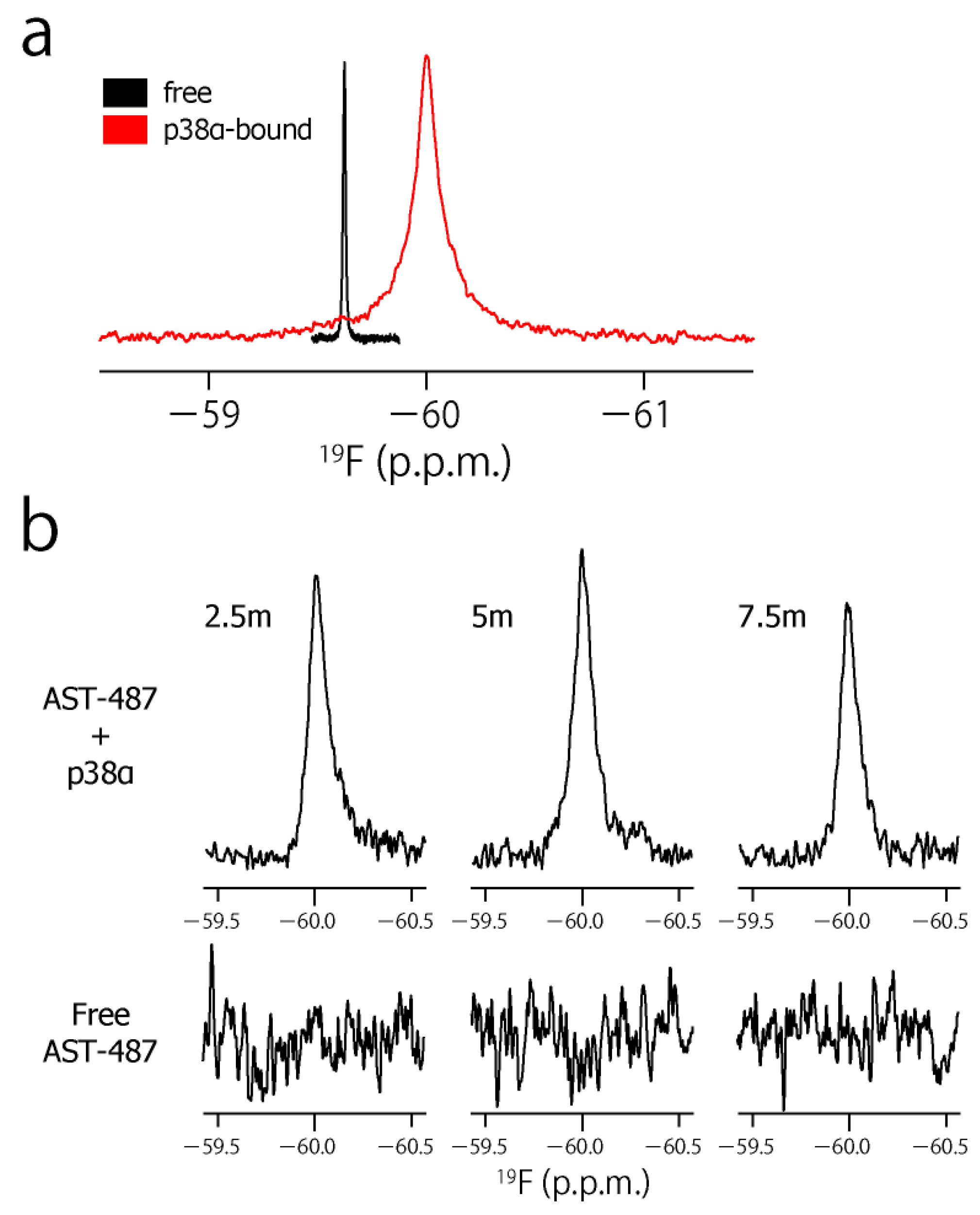
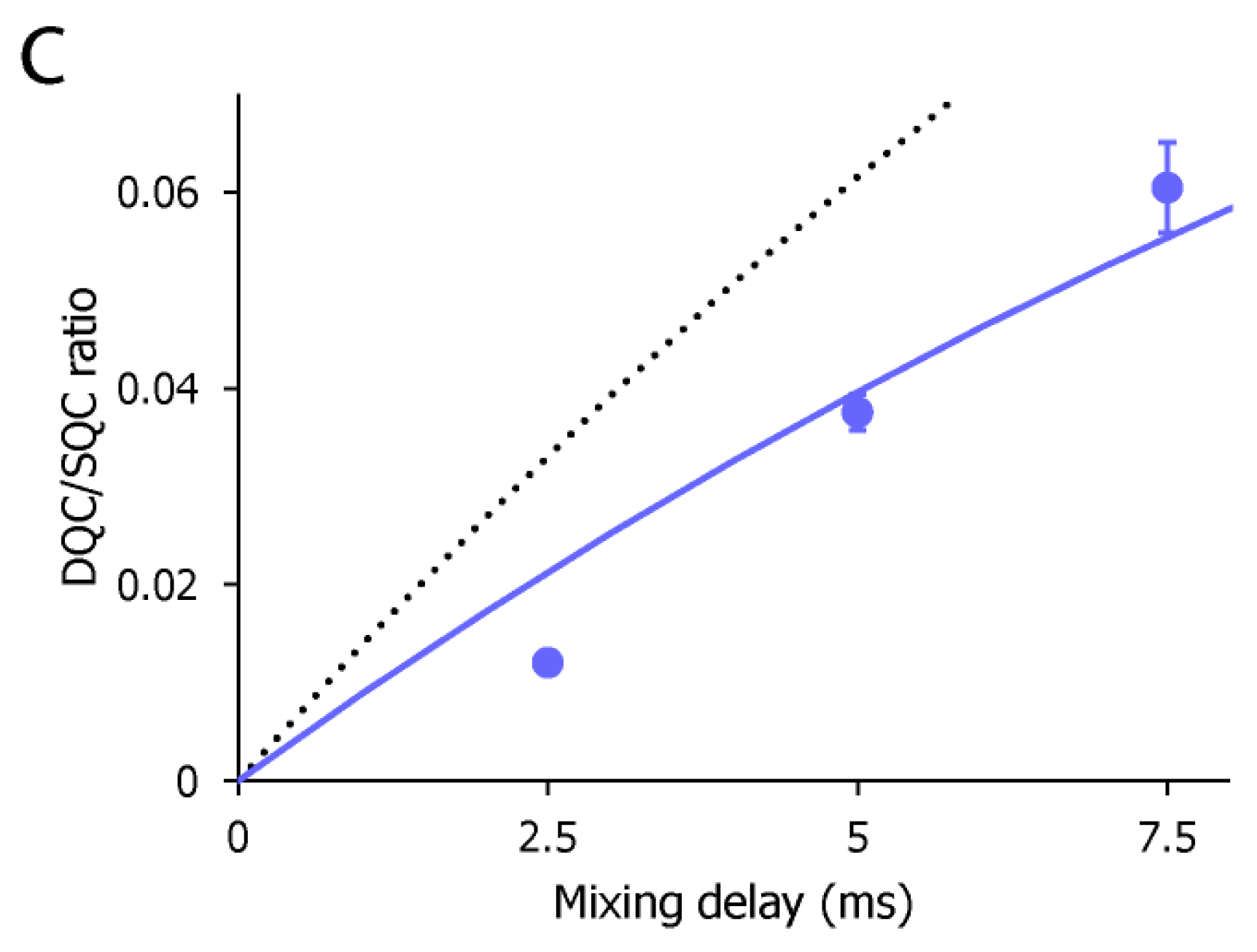
2. Results and Discussion
3. Materials and Methods
3.1. Preparation of the Recombinant p38α
3.2. NMR Sample Preparation
3.3. NMR Experiments
3.4. ITC Experiments
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klebe, G. Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discov. 2015, 14, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Tarcsay, Á.; Keserű, G.M. Is there a link between selectivity and binding thermodynamics profiles? Drug Discov. Today 2015, 20, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.; Nalivaika, E.A.; Ozen, A.; et al. Molecular basis for drug resistance in HIV-1 protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka, H.; Schön, A.; Freire, E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry 2003, 42, 13659–13666. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.-Y.; Yang, D.-Y.; Selzle, H.L.; Schlag, E.W. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. USA 2003, 100, 12683–12687. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.-Y.; Schlag, E.W.; Selzle, H.L.; Yang, D.-Y. Molecular dynamics of hydrogen bonds in protein—D2O: The solvent isotope effect. J. Phys. Chem. A 2008, 112, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Rajeshwar, T.R.; Krishnan, M. Direct determination of site-specific noncovalent interaction strengths of proteins from NMR-derived fast side chain motional parameters. J. Phys. Chem. B 2017, 121, 5174–5186. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Bertini, I.; Cowburn, D.; Dalvit, C.; Giralt, E.; Jahnke, W.; James, T.L.; Homans, S.W.; Kessler, H.; Luchinat, C.; et al. Perspectives on nmr in drug discovery: A technique comes of age. Nat. Rev. Drug Discov. 2008, 7, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef] [PubMed]
- Lepre, C.A.; Moore, J.M.; Peng, J.W. Theory and applications of NMR-based screening in pharmaceutical research. Chem. Rev. 2004, 104, 3641–3676. [Google Scholar] [CrossRef] [PubMed]
- Wand, A. Dynamic activation of protein function: A view emerging from NMR spectroscopy. Nat. Struct. Biol. 2001, 8, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E. New views of functionally dynamic proteins by solution nmr spectroscopy. J. Mol. Biol. 2016, 428, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. Exploring sparsely populated states of macromolecules by diamagnetic and paramagnetic NMR relaxation. Protein Sci. 2011, 20, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Giovanni, L.; Attila, S. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar]
- Ban, D.; Sabo, T.M.; Griesinger, C.; Lee, D. Measuring dynamic and kinetic information in the previously inaccessible supra-τ(c) window of nanoseconds to microseconds by solution nmr spectroscopy. Molecules 2013, 18, 11904–11937. [Google Scholar] [CrossRef] [PubMed]
- Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Tugarinov, V.; Sprangers, R.; Kay, L.E. Probing side-chain dynamics in the proteasome by relaxation violated coherence transfer nmr spectroscopy. J. Am. Chem. Soc. 2007, 129, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, Y.; Takeuchi, K.; Arutaki, M.; Tokunaga, Y.; Takizawa, T.; Hanzawa, H.; Shimada, I. Improvement of ligand affinity and thermodynamic properties by NMR-based evaluation of local dynamics and surface complementarity in the receptor-bound state. Angew. Chem. Int. Ed. Engl. 2016, 55, 14606–14609. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Kamlet, A.S.; Ritter, T. Catalysis for fluorination and trifluoromethylation. Nature 2011, 473, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Tomashenko, O.A.; Grushin, V.V. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 2011, 111, 4475–4521. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Dalvit, C. Fluorine local environment: From screening to drug design. Drug Discov. Today 2012, 17, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Ardini, E.; Fogliatto, G.P.; Mongelli, N.; Veronesi, M. Reliable high-throughput functional screening with 3-fabs. Drug Discov. Today 2004, 9, 595–602. [Google Scholar] [CrossRef]
- Norton, R.S.; Leung, E.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of (19)F-NMR in fragment-based drug discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef] [PubMed]
- Schindler, J.F.; Monahan, J.B.; Smith, W.G. P38 pathway kinases as anti-inflammatory drug targets. J. Dent. Res. 2007, 86, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Dodeller, F.; Schulze-Koops, H. The p38 mitogen-activated protein kinase signaling cascade in cd4 t cells. Arthritis Res. Ther. 2006, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The chembl bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Akeno-Stuart, N.; Croyle, M.; Knauf, J.A.; Malaguarnera, R.; Vitagliano, D.; Santoro, M.; Stephan, C.; Grosios, K.; Wartmann, M.; Cozens, R.; et al. The ret kinase inhibitor NVP-ast487 blocks growth and calcitonin gene expression through distinct mechanisms in medullary thyroid cancer cells. Cancer Res. 2007, 67, 6956–6964. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Deibler, K.K.; Mishra, R.K.; Clutter, M.R.; Antanasijevic, A.; Bergan, R.; Caffrey, M.; Scheidt, K.A. A chemical probe strategy for interrogating inhibitor selectivity across the mek kinase family. ACS Chem. Biol. 2017, 12, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Prosser, R.S. Activation of the a2a adenosine g-protein-coupled receptor by conformational selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Heidenreich, D.; Ono, Y.; Sugiki, T.; Yokoyama, K.I.; Suzuki, E.I.; Fujiwara, T.; Kojima, C. Protein 19F-labeling using transglutaminase for the nmr study of intermolecular interactions. J. Biomol. NMR 2017. [Google Scholar] [CrossRef] [PubMed]
- Prosser, R.S.; Kim, T.H. Nuts and bolts of CF3 and CH3 NMR toward the understanding of conformational exchange of GPCRs. Methods Mol. Biol. 2015, 1335, 39–51. [Google Scholar] [PubMed]
- Didenko, T.; Liu, J.J.; Horst, R.; Stevens, R.C.; Wüthrich, K. Fluorine-19 NMR of integral membrane proteins illustrated with studies of gpcrs. Curr. Opin. Struct. Biol. 2013, 23, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Harkins, P.C.; Ulevitch, R.J.; Han, J.; Cobb, M.H.; Goldsmith, E.J. The structure of mitogen-activated protein kinase p38 at 2.1-a resolution. Proc. Natl. Acad. Sci. USA 1997, 94, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Canagarajah, B.J.; Boehm, J.C.; Kassisà, S.; Cobb, M.H.; Young, P.R.; Abdel-Meguid, S.; Adams, J.L.; Goldsmith, E.J. Structural basis of inhibitor selectivity in map kinases. Structure 1998, 6, 1117–1128. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Takeuchi, K.; Takahashi, H.; Shimada, I. Allosteric enhancement of map kinase p38α’s activity and substrate selectivity by docking interactions. Nat. Struct. Mol. Biol. 2014, 21, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.K.; Gardner, K.H.; Mueller, G.A.; Willis, R.C.; Kay, L.E. A robust and cost-effective method for the production of val, leu, ile (δ1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR 1999, 13, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Schanda, P.; Kupce, E.; Brutscher, B. Sofast-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 2005, 33, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Amero, C.; Schanda, P.; Durá, M.A.; Ayala, I.; Marion, D.; Franzetti, B.; Brutscher, B.; Boisbouvier, J. Fast two-dimensional NMR spectroscopy of high molecular weight protein assemblies. J. Am. Chem. Soc. 2009, 131, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Keller, D.G. Sparky 3, University of California, San Francisco. Available online: http://www.cgl.ucsf.edu/home/sparky/ (ver 3.114 distributed on 12th July 2007).
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts. Iupac recommendations 2001. Solid State Nucl. Magn. Reson. 2002, 22, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Hilty, C.; Wider, G.; Wüthrich, K. Effective rotational correlation times of proteins from NMR relaxation interference. J. Magn. Reson. 2006, 178, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E.; Prestegard, J.H. Methyl group dynamics from relaxation of double quantum filtered NMR signals: Application to deoxycholate. J. Am. Chem. Soc. 1987, 109, 3829–3835. [Google Scholar] [CrossRef]
- Shaka, A.J.; Keeler, J.; Frenkiel, T.; Freeman, R.A.Y. An improved sequence for broadband decoupling: Waltz-16. J. Magn. Reson. 1983, 52, 335–338. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound AST-487 (Purity 99.90%, Catalog No. SYN1210) is not available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokunaga, Y.; Takeuchi, K.; Shimada, I. Forbidden Coherence Transfer of 19F Nuclei to Quantitatively Measure the Dynamics of a CF3-Containing Ligand in Receptor-Bound States. Molecules 2017, 22, 1492. https://doi.org/10.3390/molecules22091492
Tokunaga Y, Takeuchi K, Shimada I. Forbidden Coherence Transfer of 19F Nuclei to Quantitatively Measure the Dynamics of a CF3-Containing Ligand in Receptor-Bound States. Molecules. 2017; 22(9):1492. https://doi.org/10.3390/molecules22091492
Chicago/Turabian StyleTokunaga, Yuji, Koh Takeuchi, and Ichio Shimada. 2017. "Forbidden Coherence Transfer of 19F Nuclei to Quantitatively Measure the Dynamics of a CF3-Containing Ligand in Receptor-Bound States" Molecules 22, no. 9: 1492. https://doi.org/10.3390/molecules22091492