The Role of Kinase Modulators in Cellular Senescence for Use in Cancer Treatment


Abstract
:1. Introduction
2. Cancer and Senescence
3. Kinase Modulators and Senescence
3.1. BCR-ABL Inhibitors: Imatinib and Dasatinib
3.2. EGFR Family Modulators: Gefitinib, Erlotinib, Lapatinib, and Cetuximab
3.3. Angiogenesis Inhibitors: Sunitinib, Axitinib, and Bevacizumab
3.4. B-RAF Inhibitor: Vemurafenib
3.5. MEK Inhibitor: Trametinib
3.6. CDK Inhibitor: Palbociclib
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [Google Scholar]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Rudge, S.A.; Wakelam, M.J. SnapShot: Lipid kinases and phosphatases. Cell 2013, 155, 1654. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Noble, M.E.; Johnson, L.N. The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 2012, 81, 587–613. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A. Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 2001, 101, 2271–2290. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Zhang, J.; Fabbro, D.; Schioth, H.B. Advances in kinase targeting: Current clinical use and clinical trials. Trends Pharmacol. Sci. 2014, 35, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: Relevance to cancer therapies. Trends Biochem. Sci. 2014, 39, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.D.; Flanagan, M.E.; Telliez, J.B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023–5038. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat. Rev. Drug Discov. 2013, 12, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Nakamura, K.; MacLauchlan, S.; Ngo, D.T.; Shimizu, I.; Fuster, J.J.; Katanasaka, Y.; Yoshida, S.; Qiu, Y.; Yamaguchi, T.P.; et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2014, 20, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.A.; Toader, D.; Ferguson, A.D. Structural approaches to obtain kinase selectivity. Trends Pharmacol. Sci. 2012, 33, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 2017, 16, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A. Tyrosine kinase inhibitors: Views of selectivity, sensitivity, and clinical performance. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 161–185. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Ott, P.A.; Shapiro, G.I. The biology and clinical development of MEK inhibitors for cancer. Drugs 2014, 74, 2111–2128. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, M.C.; Velasco-Velazquez, M.; Aguirre-Alvarado, C.; Pestell, R.G. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: Past and present. Expert Opin. Investig. Drugs 2014, 23, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Papadopoulos, N. The safety of therapeutic monoclonal antibodies: Implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis 2013, 228, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; LoRusso, P.M.; Purdom, M.; Ward, D. Dermatologic side effects associated with gefitinib therapy: Clinical experience and management. Clin. Lung Cancer 2003, 4, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Loaiza, N.; Demaria, M. Cellular senescence and tumor promotion: Is aging the key? Biochim. Biophys. Acta 2016, 1865, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Wolstein, J.M.; Lee, D.H.; Michaud, J.; Buot, V.; Stefanchik, B.; Plotkin, M.D. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2010, 299, F1486–F1495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.H.; Du, J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Murphy, A.J.; Sayers, S.; Li, R.; Yvan-Charvet, L.; Davis, J.Z.; Krishnamurthy, J.; Liu, Y.; Puig, O.; Sharpless, N.E.; et al. Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Sone, H.; Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005, 48, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Lieu, C.A.; Demaria, M.; Laberge, R.M.; Campisi, J.; Andersen, J.K. Environmental stress, ageing and glial cell senescence: A novel mechanistic link to Parkinson’s disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Niederlander, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Brown, T.D.; Heiner, A.D.; Buckwalter, J.A. Chondrocyte senescence, joint loading and osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S96–S103. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J. Biol. Chem. 2009, 284, 9529–9539. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998, 16, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Yoshizuka, N.; New, L.; Moser, B.A.; Li, Y.; Liao, R.; Xie, C.; Chen, J.; Deng, Q.; Yamout, M.; et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell 2007, 128, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr. Opin. Cell Biol. 2010, 22, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Serrano, M. p53: Guardian of the genome and policeman of the oncogenes. Cell Cycle 2007, 6, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; d’Adda di Fagagna, F. Cellular senescence: Hot or what? Curr. Opin. Genet. Dev. 2009, 19, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kahlem, P.; Dorken, B.; Schmitt, C.A. Cellular senescence in cancer treatment: Friend or foe? J. Clin. Investig. 2004, 113, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Helton, E.S.; Chen, X. p53 modulation of the DNA damage response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Chen, X. Tumor suppression by p53: Making cells senescent. Histol. Histopathol. 2010, 25, 515–526. [Google Scholar] [PubMed]
- Klump, K.E.; McGinnis, J.F. The role of reactive oxygen species in ocular malignancy. Adv. Exp. Med. Biol. 2014, 801, 655–659. [Google Scholar] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Cahu, J.; Bustany, S.; Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012, 3, e446. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Golomb, L.; Sagiv, A.; Pateras, I.S.; Maly, A.; Krizhanovsky, V.; Gorgoulis, V.G.; Oren, M.; Ben-Yehuda, A. Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ. 2015, 22, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Drullion, C.; Tregoat, C.; Lagarde, V.; Tan, S.; Gioia, R.; Priault, M.; Djavaheri-Mergny, M.; Brisson, A.; Auberger, P.; Mahon, F.X.; et al. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012, 3, e373. [Google Scholar] [CrossRef] [PubMed]
- DiPiro, J.T.; Talbert, R.L.; Yee, G.C.; Wells, B.G.; Posey, L.M. Pharmacotherapy a Pathophysiologic Approach 9/E; McGraw-Hill Education: New York, NY, USA, 2014. [Google Scholar]
- Sen, B.; Peng, S.; Tang, X.; Erickson, H.S.; Galindo, H.; Mazumdar, T.; Stewart, D.J.; Wistuba, I.; Johnson, F.M. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci. Transl. Med. 2012, 4, 136ra70. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Sen, B.; Mazumdar, T.; Byers, L.A.; Diao, L.; Wang, J.; Tong, P.; Giri, U.; Heymach, J.V.; Kadara, H.N.; et al. Dasatinib induces DNA damage and activates DNA repair pathways leading to senescence in non-small cell lung cancer cell lines with kinase-inactivating BRAF mutations. Oncotarget 2016, 7, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Biology of Cancer, 2nd ed.; Taylor & Francis Group: Abingdon, UK, 2013. [Google Scholar]
- Hotta, K.; Tabata, M.; Kiura, K.; Kozuki, T.; Hisamoto, A.; Katayama, H.; Takigawa, N.; Fujimoto, N.; Fujiwara, K.; Ueoka, H.; et al. Gefitinib induces premature senescence in non-small cell lung cancer cells with or without EGFR gene mutation. Oncol. Rep. 2007, 17, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, C.D.; Diefendorf, L.P.; Jette, D.F.; Mohammed, A.; Moses, M.A.; Searleman, S.A.; Stevens, D.A.; Wilton, K.M.; Mondal, S. Inhibition of the epidermal growth factor receptor by erlotinib prevents immortalization of human cervical cells by Human Papillomavirus type 16. Virology 2011, 421, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.B.; Yuan, L.; Yang, P.; Sun, T.; Chen, R.; Xiang, H.; Chen, J.; Wu, H.; Radiloff, D.R.; Wang, X.F. EGF promotes mammalian cell growth by suppressing cellular senescence. Cell Res. 2015, 25, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Zacarias-Fluck, M.F.; Morancho, B.; Vicario, R.; Luque Garcia, A.; Escorihuela, M.; Villanueva, J.; Rubio, I.T.; Arribas, J. Effect of cellular senescence on the growth of HER2-positive breast cancers. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Cho, B.J.; Choi, E.J.; Park, J.M.; Kim, D.H.; Kim, I.A. Radiosensitizing effect of lapatinib in human epidermal growth factor receptor 2-positive breast cancer cells. Oncotarget 2016, 7, 79089–79100. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Browne, B.; Crown, J.; O’Brien, N.; Slamon, D.J.; O’Donovan, N. Lapatinib-induced senescent-like phenotype in HER2-positive breast cancer cells. J. Clin. Oncol. 2011, 29, 583. [Google Scholar] [CrossRef]
- McDermott, M.; Browne, B.; Szabo, A.; Crown, J.; O’Donovan, N. Irreversible panHER tyrosine kinase inhibitors (TKIs) to induce irreversible senescence in HER2 positive breast cancer cells. J. Clin. Oncol. 2016, 34, e12092. [Google Scholar]
- Brunton, L.; Chabner, B.; Knollman, B. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, Twelfth ed.; McGraw-Hill Education: New York, NY, USA, 2011. [Google Scholar]
- Liu, X.; Guo, W.J.; Zhang, X.W.; Cai, X.; Tian, S.; Li, J. Cetuximab enhances the activities of irinotecan on gastric cancer cell lines through downregulating the EGFR pathway upregulated by irinotecan. Cancer Chemother. Pharmacol. 2011, 68, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Moses, M.A. Molecular regulation of tumor angiogenesis: Mechanisms and therapeutic implications. In Cancer: Cell Structures, Carcinogens and Genomic Instability; Birkhäuser: Basel, Switzerland, 2006; pp. 223–268. [Google Scholar]
- Zhu, Y.; Xu, L.; Zhang, J.; Hu, X.; Liu, Y.; Yin, H.; Lv, T.; Zhang, H.; Liu, L.; An, H.; et al. Sunitinib induces cellular senescence via p53/Dec1 activation in renal cell carcinoma cells. Cancer Sci. 2013, 104, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Amantini, C.; Santoni, M.; Soriani, A.; Nabissi, M.; Cardinali, C.; Santoni, A.; Santoni, G. Axitinib induces DNA damage response leading to senescence, mitotic catastrophe, and increased NK cell recognition in human renal carcinoma cells. Oncotarget 2015, 6, 36245–36259. [Google Scholar] [PubMed]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Cardinali, C.; Santoni, M.; Bernardini, G.; Santoni, A.; Santoni, G. Axitinib induces senescence-associated cell death and necrosis in glioma cell lines: The proteasome inhibitor, bortezomib, potentiates axitinib-induced cytotoxicity in a p21(Waf/Cip1) dependent manner. Oncotarget 2017, 8, 3380–3395. [Google Scholar] [PubMed]
- Hasan, M.R.; Ho, S.H.; Owen, D.A.; Tai, I.T. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int. J. Cancer 2011, 129, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Borst, A.; Adam, C.; Becker, T.M.; Motschenbacher, S.; Windhovel, S.; Hufnagel, A.L.; Houben, R.; Meierjohann, S. Vemurafenib induces senescence features in melanoma cells. J. Investig. Dermatol. 2013, 133, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Koseła-Paterczyk, H.; Kozak, K.; Rutkowski, P. Trametinib: A MEK inhibitor for management of metastatic melanoma. Oncotargets Ther. 2015, 8, 2251–2259. [Google Scholar]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Schick, U.; Kyula, J.; Barker, H.; Patel, R.; Zaidi, S.; Gregory, C.; Hafsi, H.; Roulstone, V.; Deutsch, E.; McLaughlin, M.; et al. Trametinib radiosensitises RAS- and BRAF-mutated melanoma by perturbing cell cycle and inducing senescence. Radiother. Oncol. 2015, 117, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sung, T.; Jessen, B.A.; Thibault, S.; Finkelstein, M.B.; Khan, N.K.; Sacaan, A.I. Mechanistic investigation of bone marrow suppression associated with palbociclib and its differentiation from cytotoxic chemotherapies. Clin. Cancer Res. 2016, 22, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M. Cancer: Final act of senescence. Nature 2011, 479, 481–482. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Worth, A.J.; Snyder, N.W.; Lee, J.V.; Sivanand, S.; Liu, Q.; Blair, I.A.; Wellen, K.E.; Zhang, R. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015, 11, 893–901. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
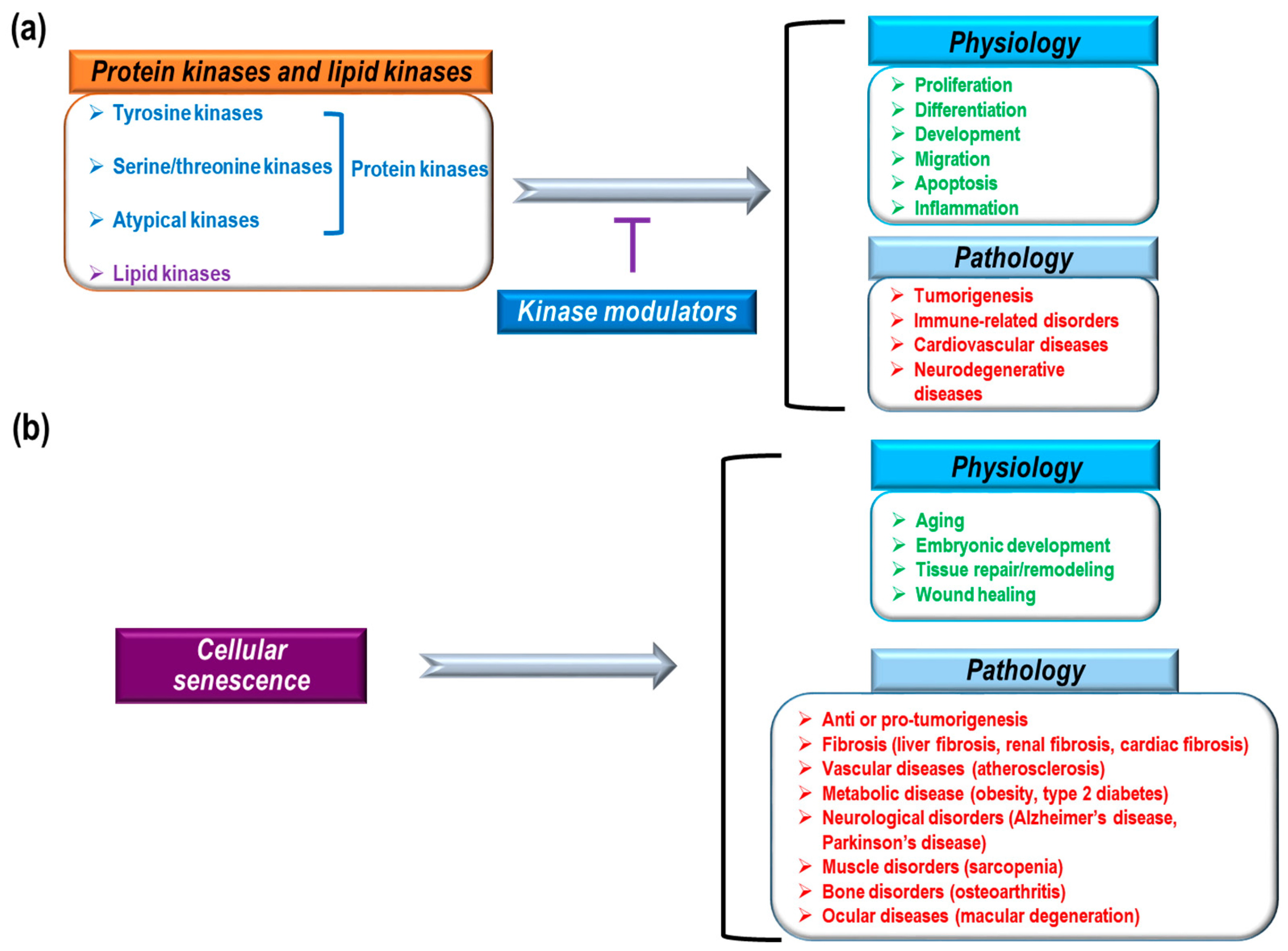

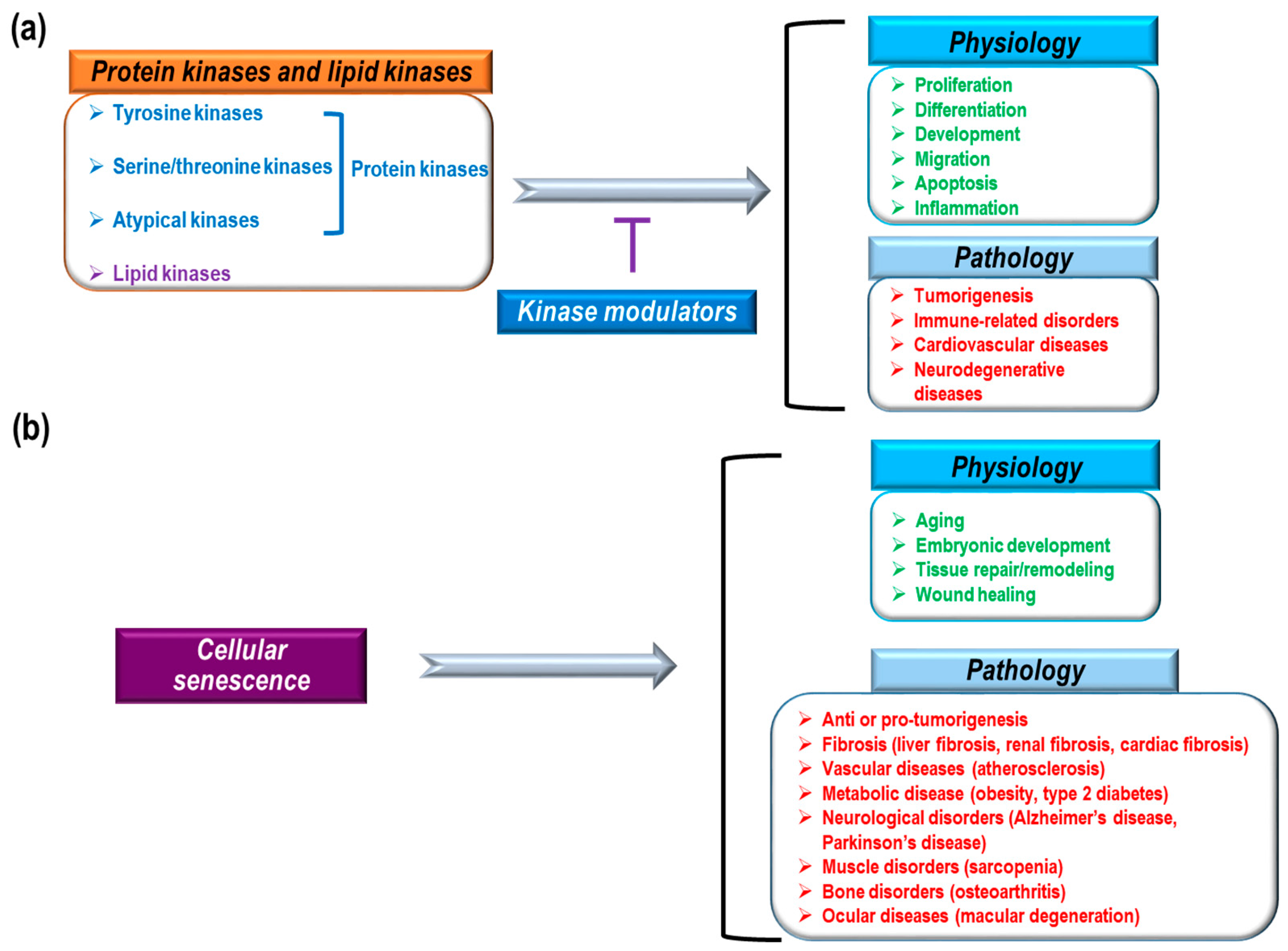{kind=link}
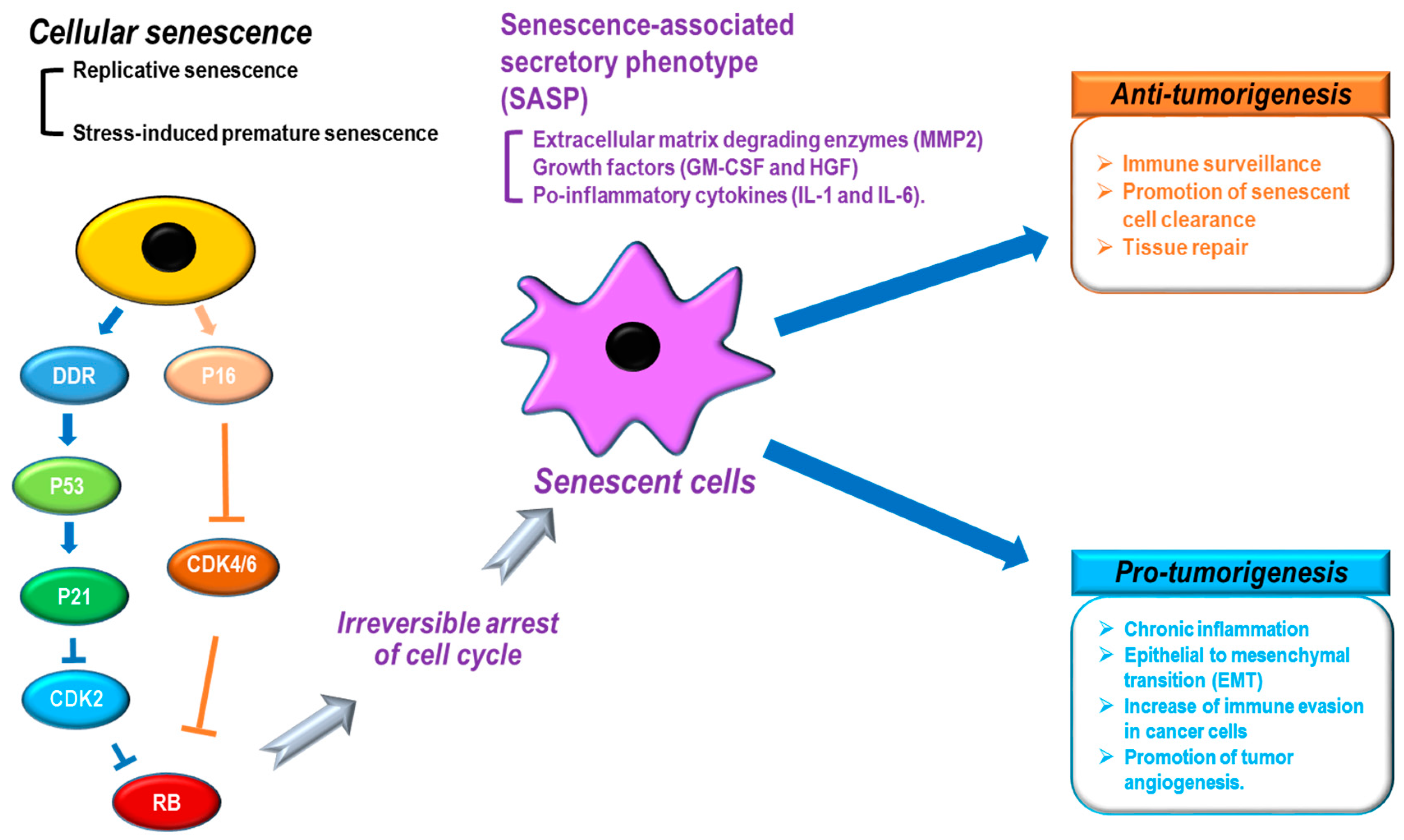
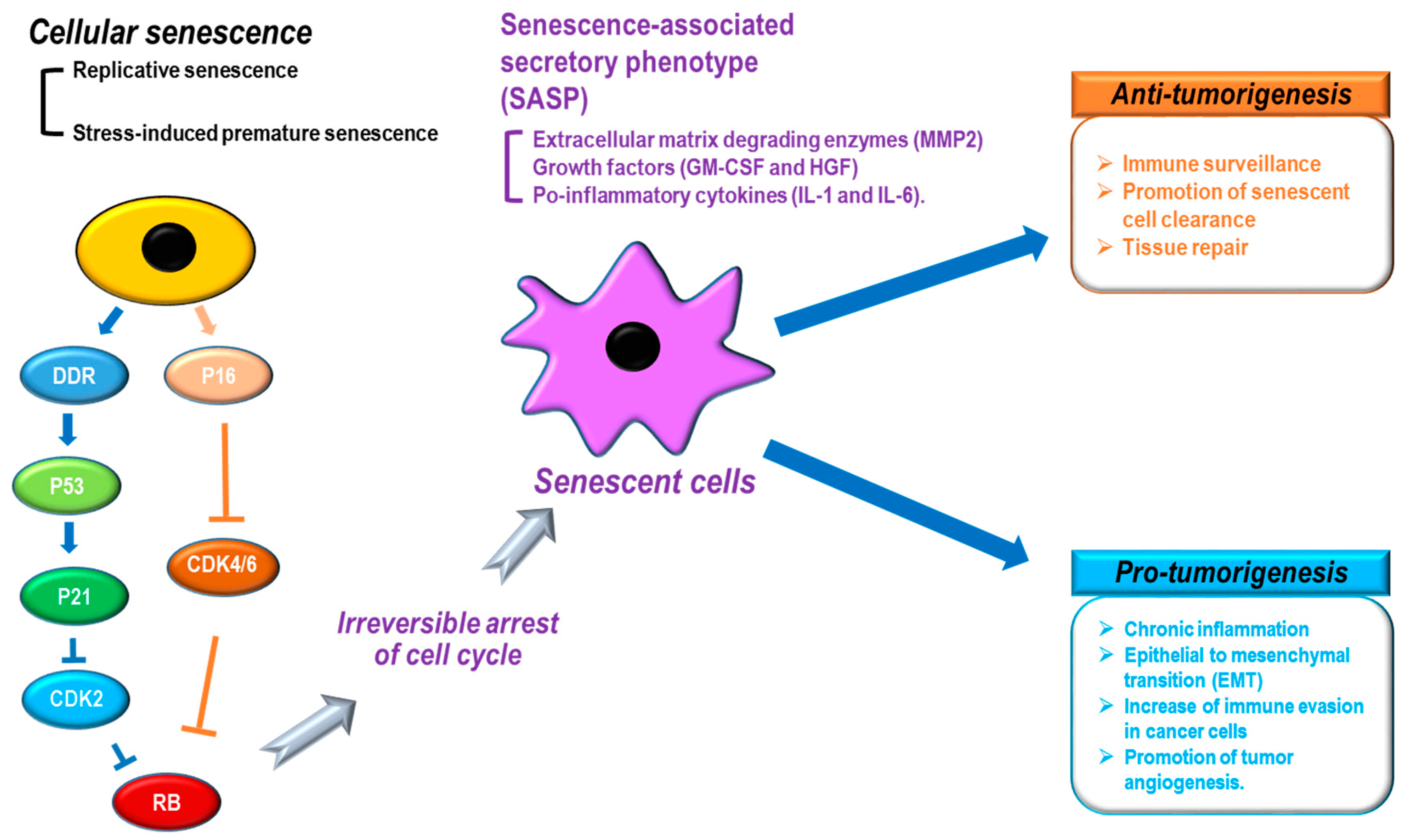{kind=link}
| Drugs | Target | Effect on Senescence | References |
|---|---|---|---|
| Imatinib | BCR-ABL | Induction | [82] |
| Dasatinib | BCR-ABL | Induction | [84,85] |
| Gefitinib | EGFR | Induction | [87] |
| Erlotinib | EGFR | Induction | [88,89] |
| Lapatinib | EGFR, HER2 | Suppression, Induction | [90,93,94,95] |
| Cetuximab | EGFR | No change | [97] |
| Sunitinib | VEGFR, multikinases | Induction | [99] |
| Axitinib | VEGFR, multikinases | Induction | [100,101] |
| Bevacizumab | VEGF | Induction | [102] |
| Vemurafenib | B-RAF | Induction | [104] |
| Trametinib | MEK1/2 | Induction | [107] |
| Palbociclib | CDK4/6 | Induction | [109] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.S.; Baek, J.; Han, S.-Y. The Role of Kinase Modulators in Cellular Senescence for Use in Cancer Treatment. Molecules 2017, 22, 1411. https://doi.org/10.3390/molecules22091411
Lee CS, Baek J, Han S-Y. The Role of Kinase Modulators in Cellular Senescence for Use in Cancer Treatment. Molecules. 2017; 22(9):1411. https://doi.org/10.3390/molecules22091411
Chicago/Turabian StyleLee, Chang Sup, Juhwa Baek, and Sun-Young Han. 2017. "The Role of Kinase Modulators in Cellular Senescence for Use in Cancer Treatment" Molecules 22, no. 9: 1411. https://doi.org/10.3390/molecules22091411