Determination of Structural Requirements of N-Substituted Tetrahydro-β-Carboline Imidazolium Salt Derivatives Using in Silico Approaches for Designing MEK-1 Inhibitors


Abstract
:1. Introduction
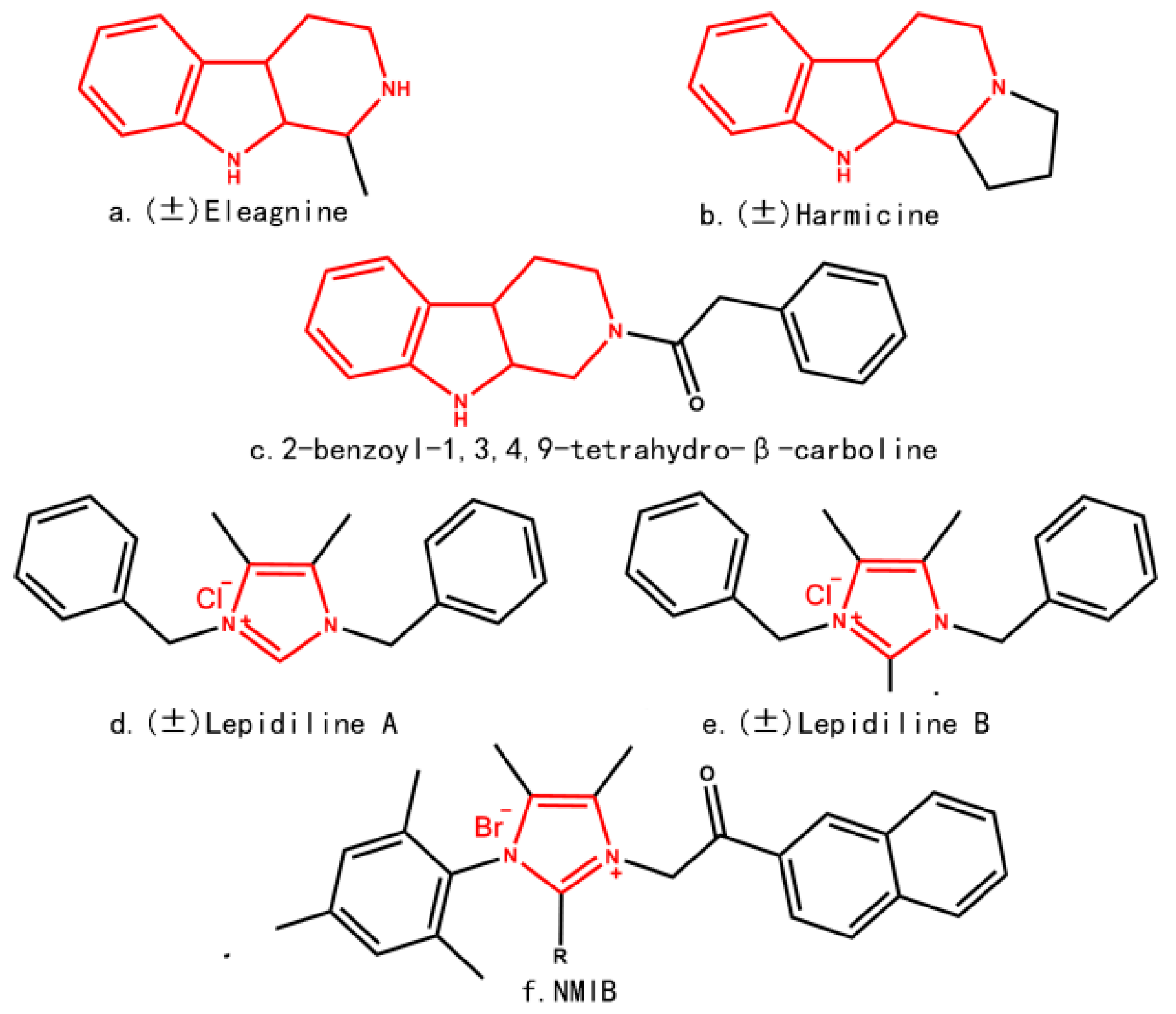
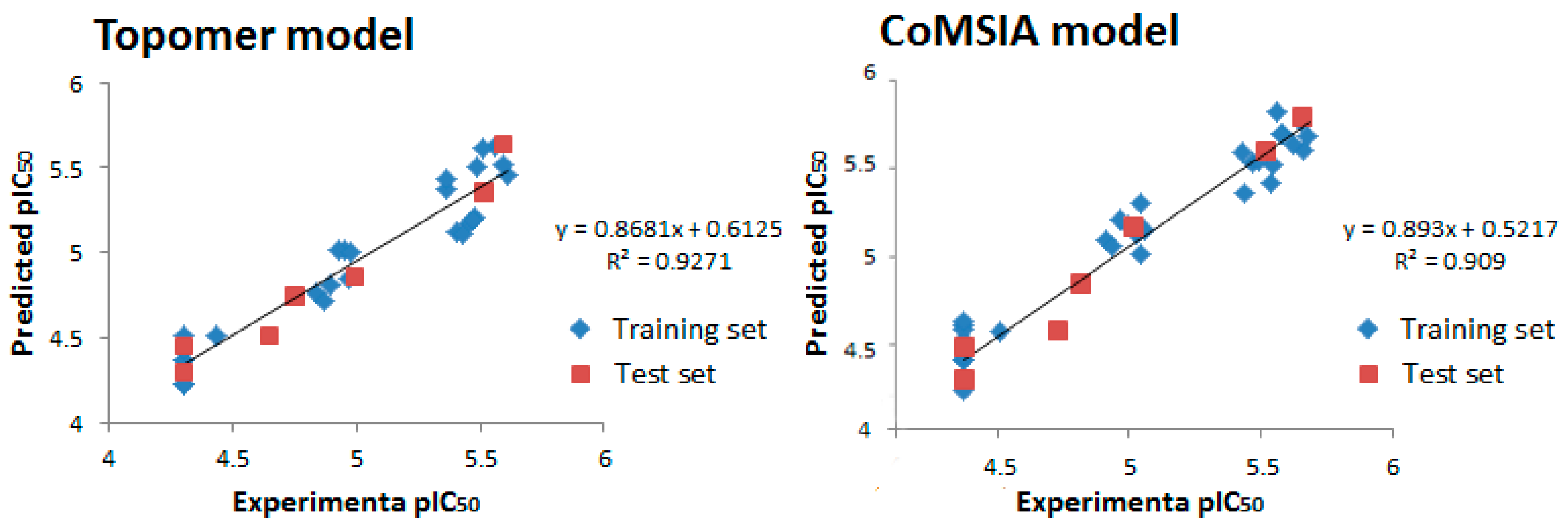
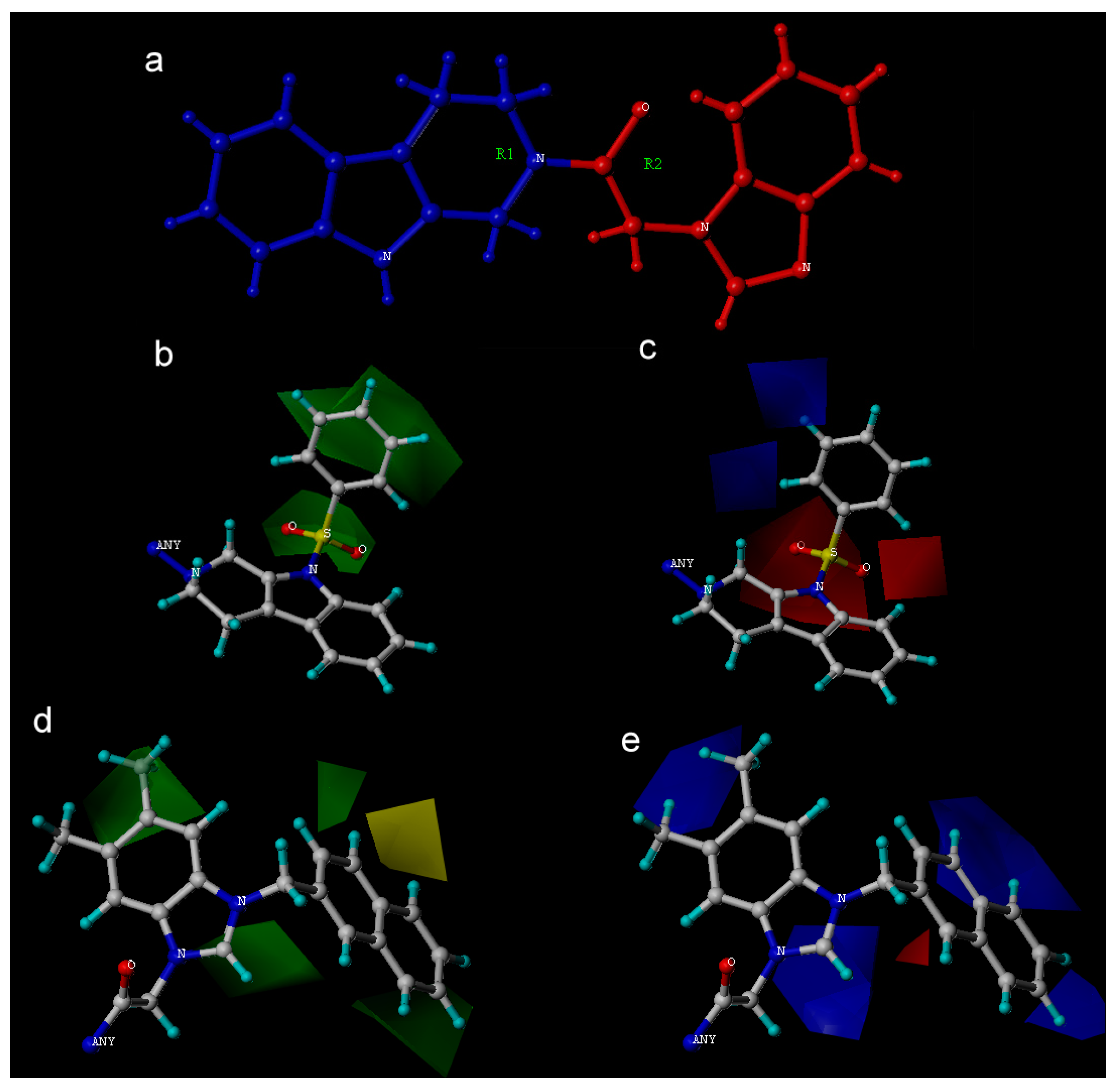
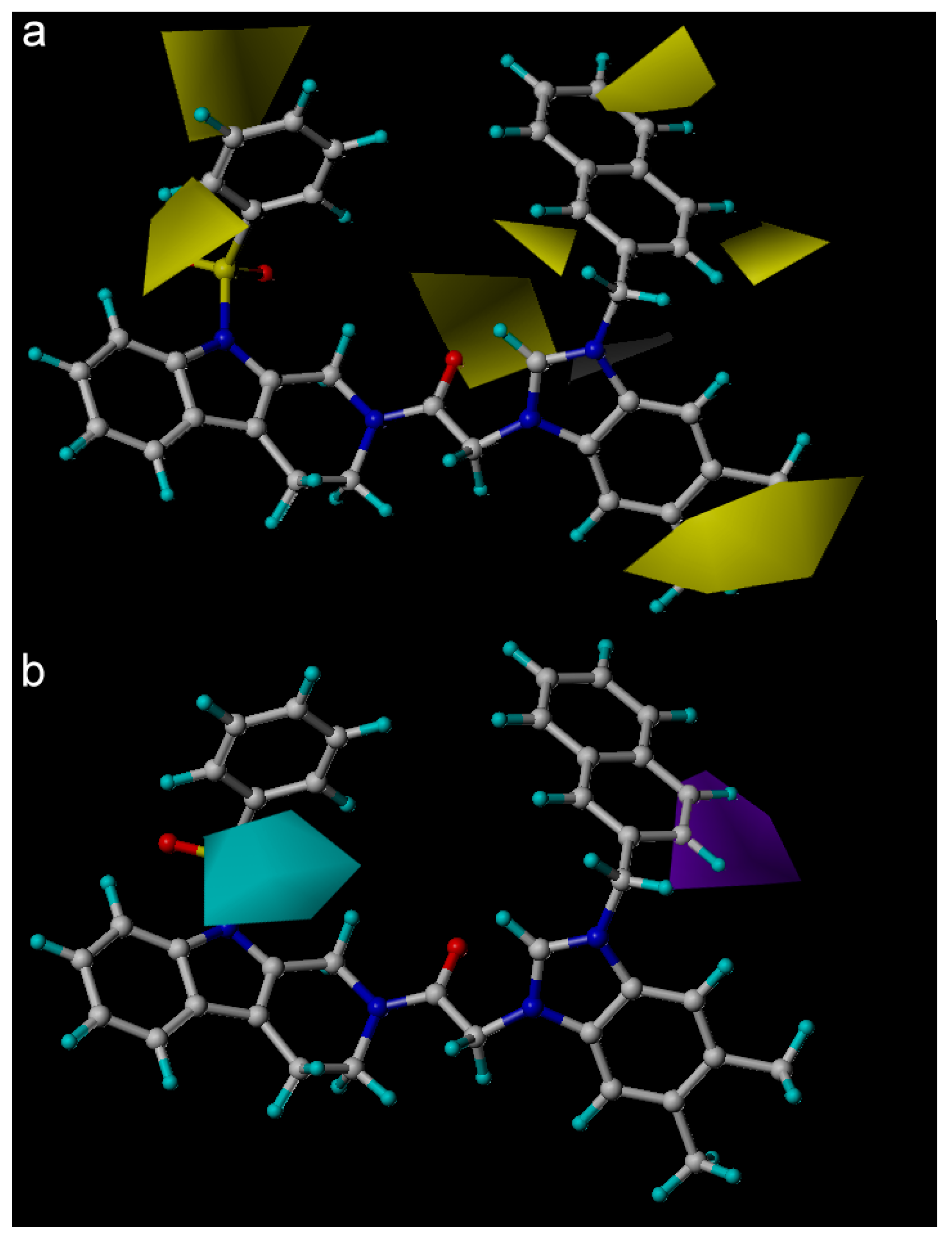
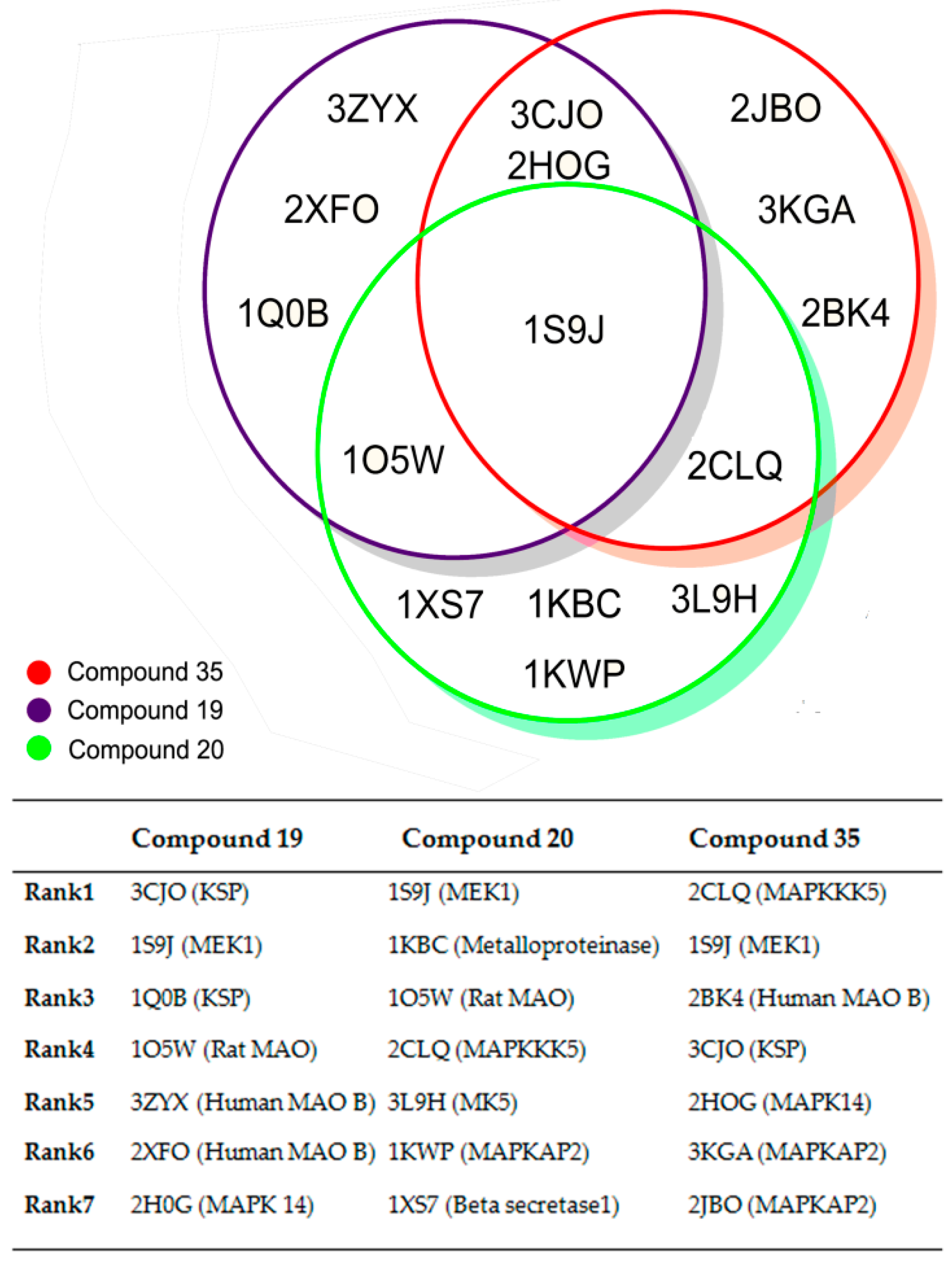
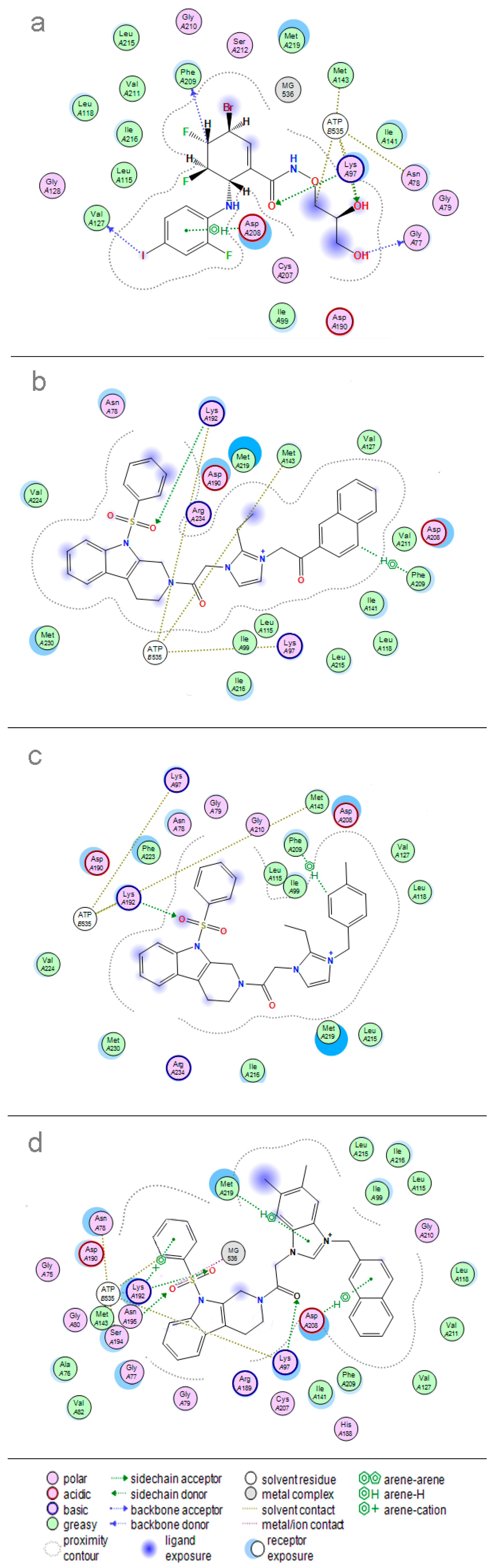
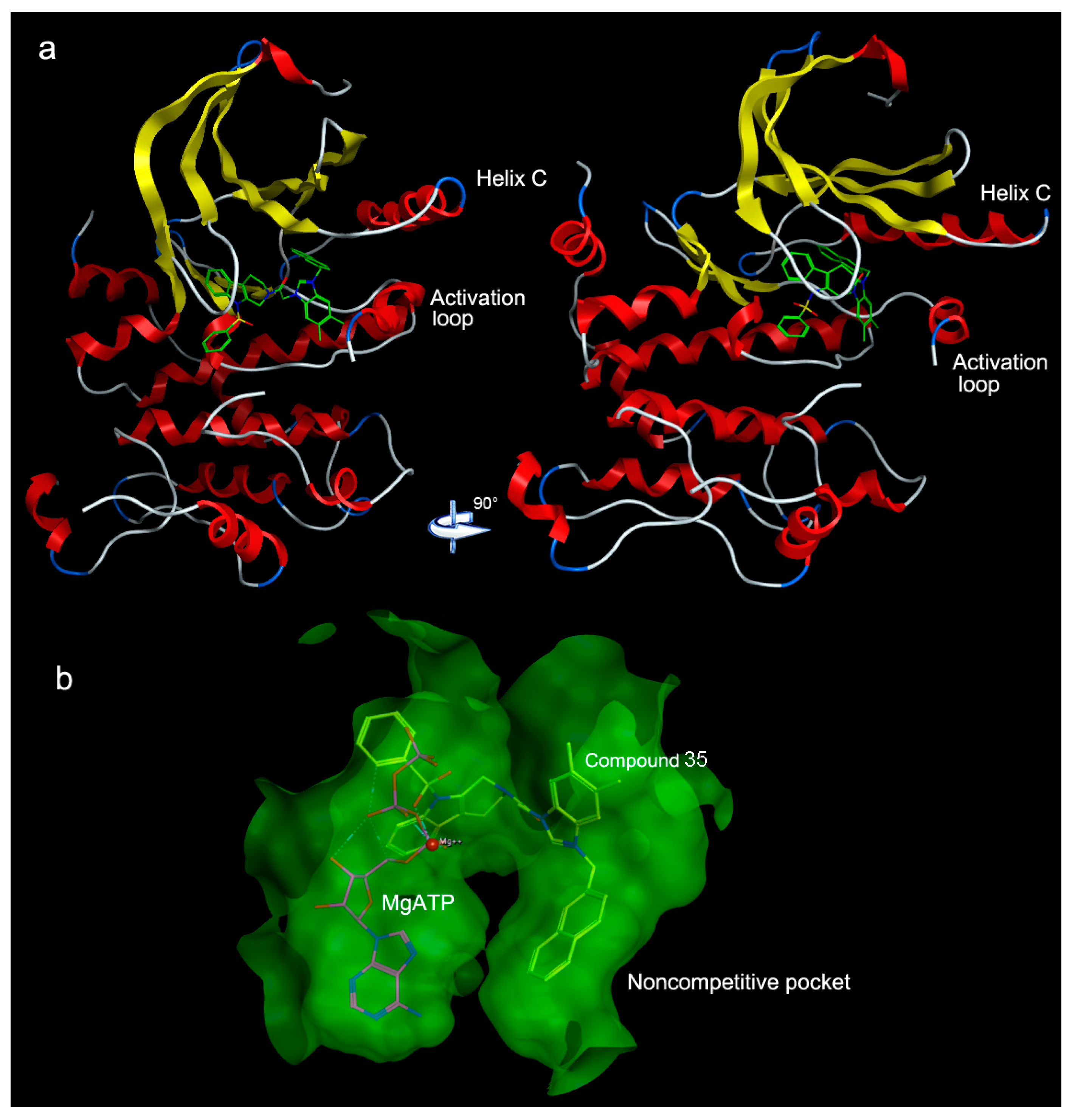
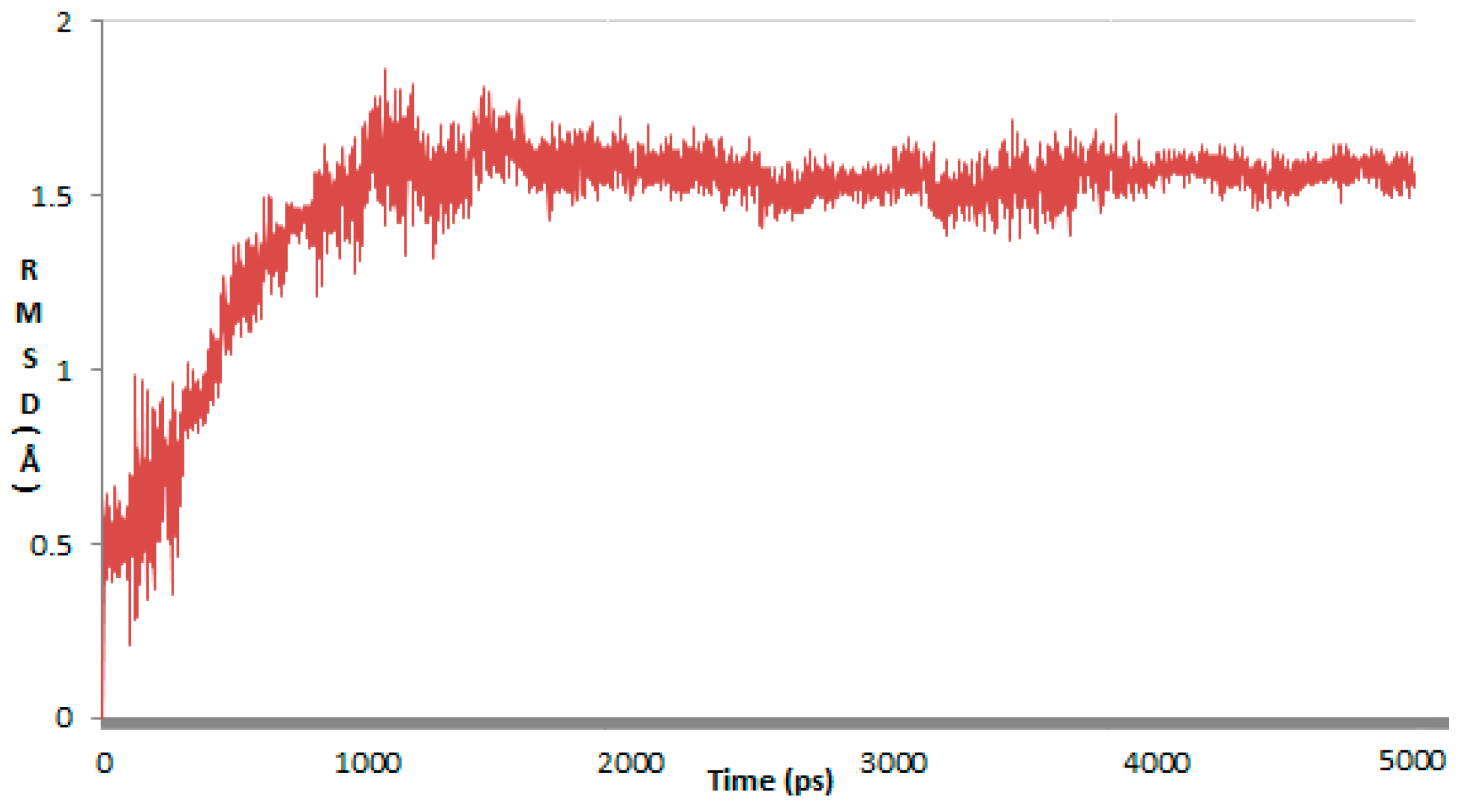
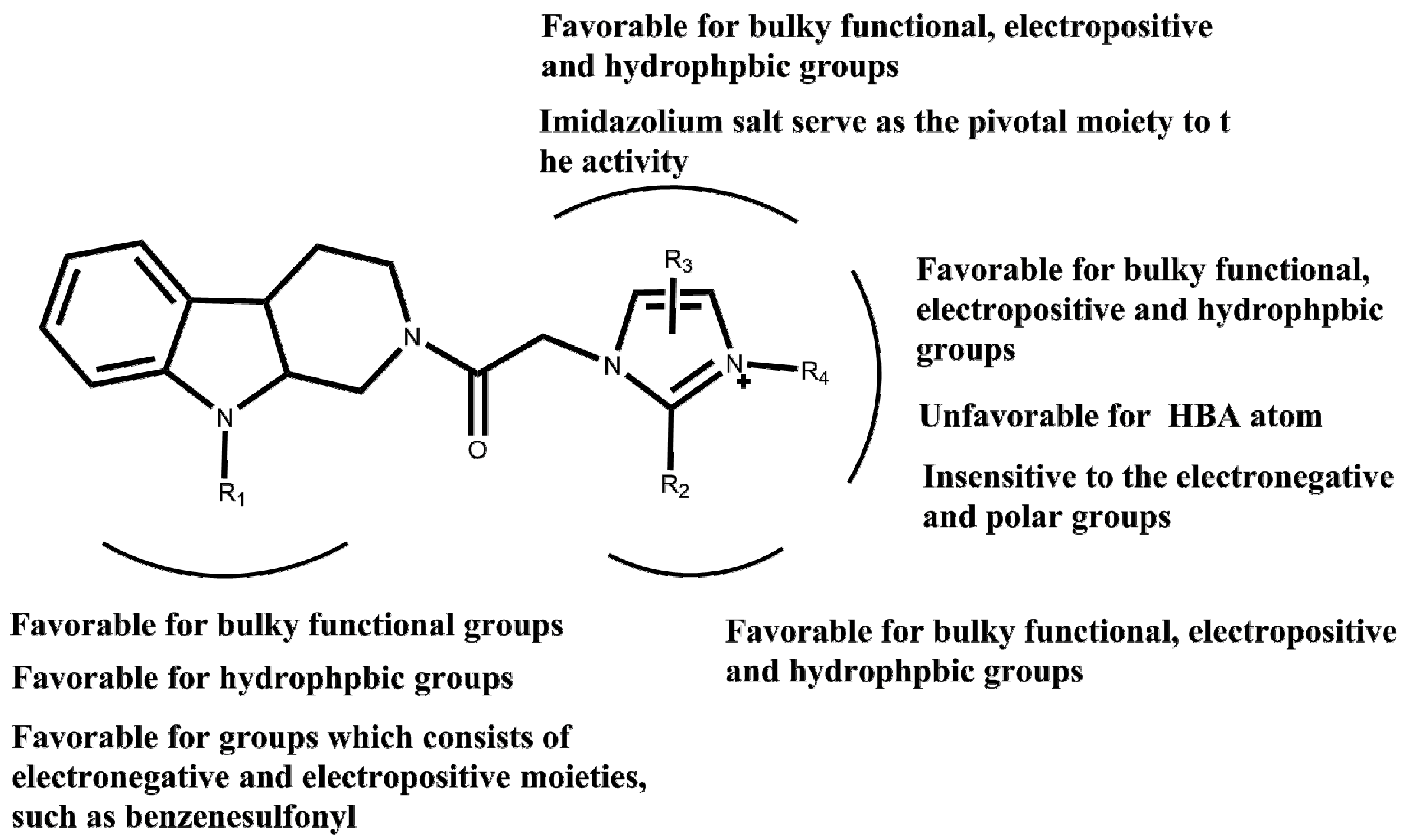
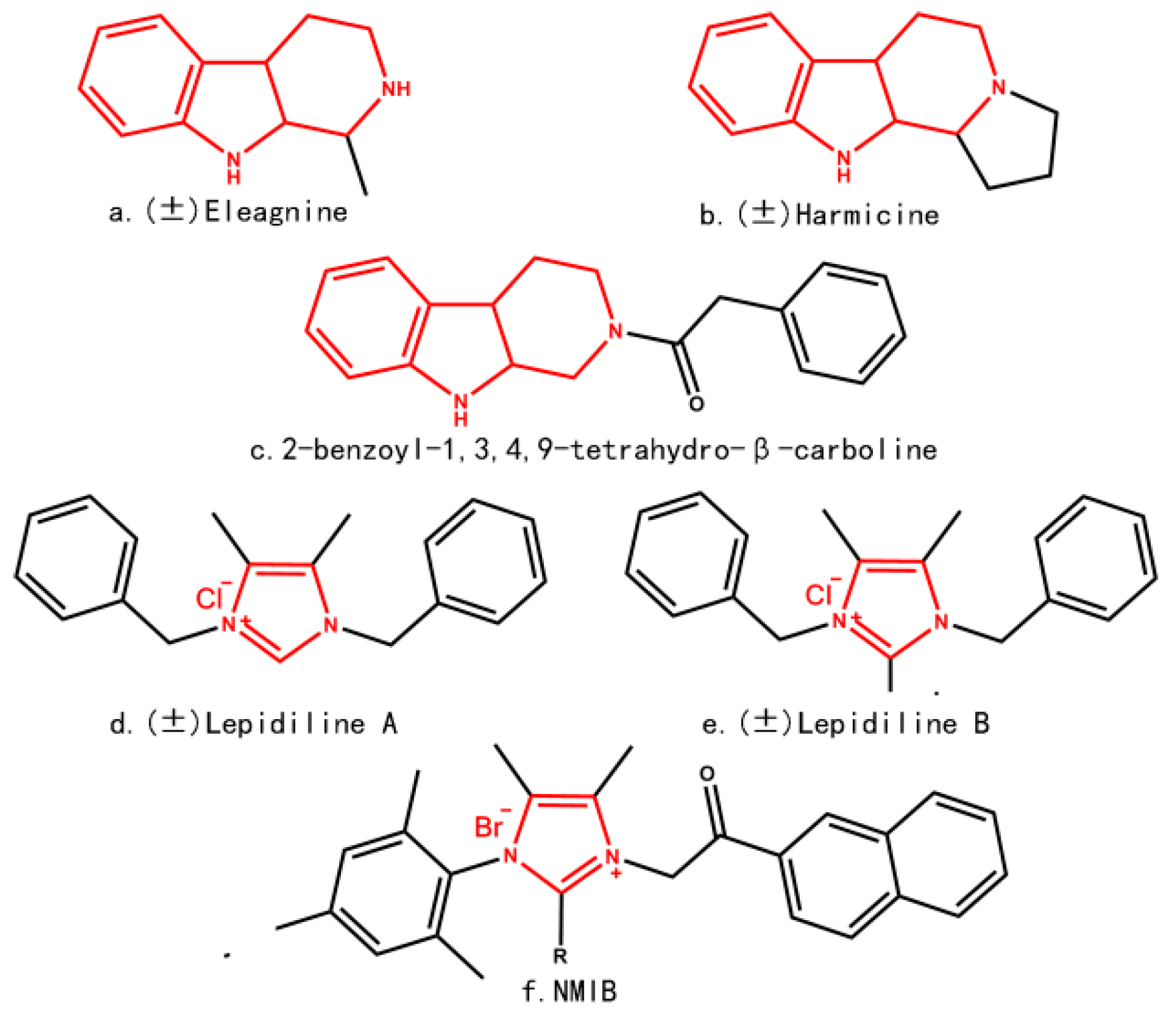
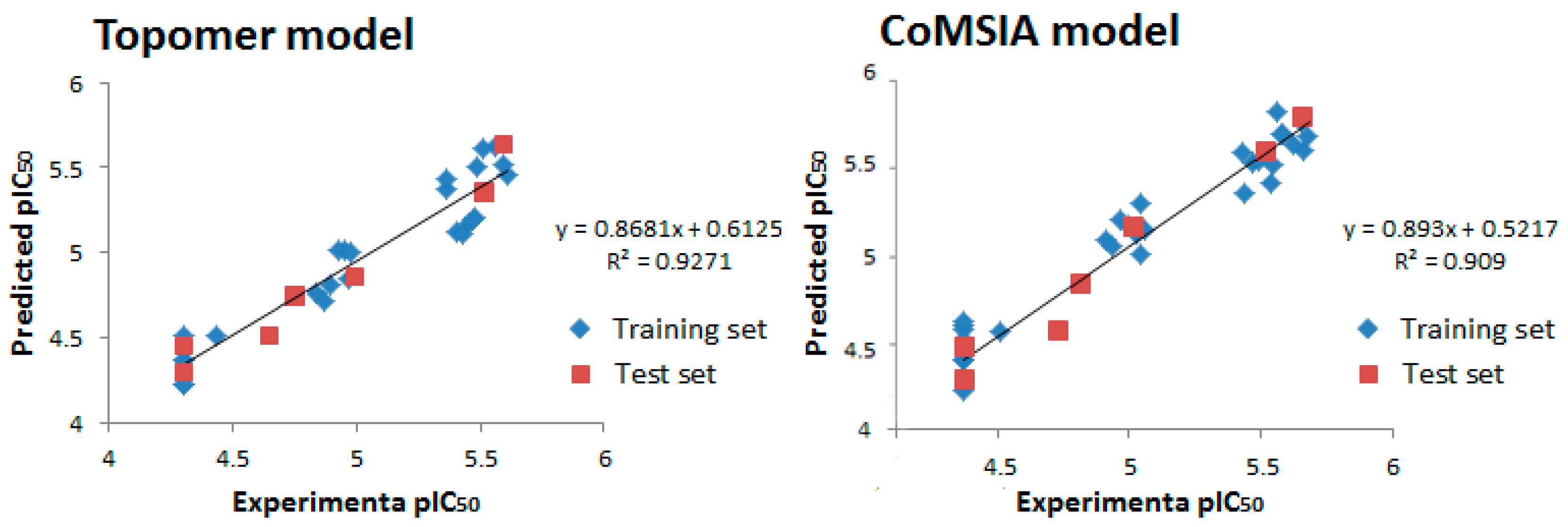
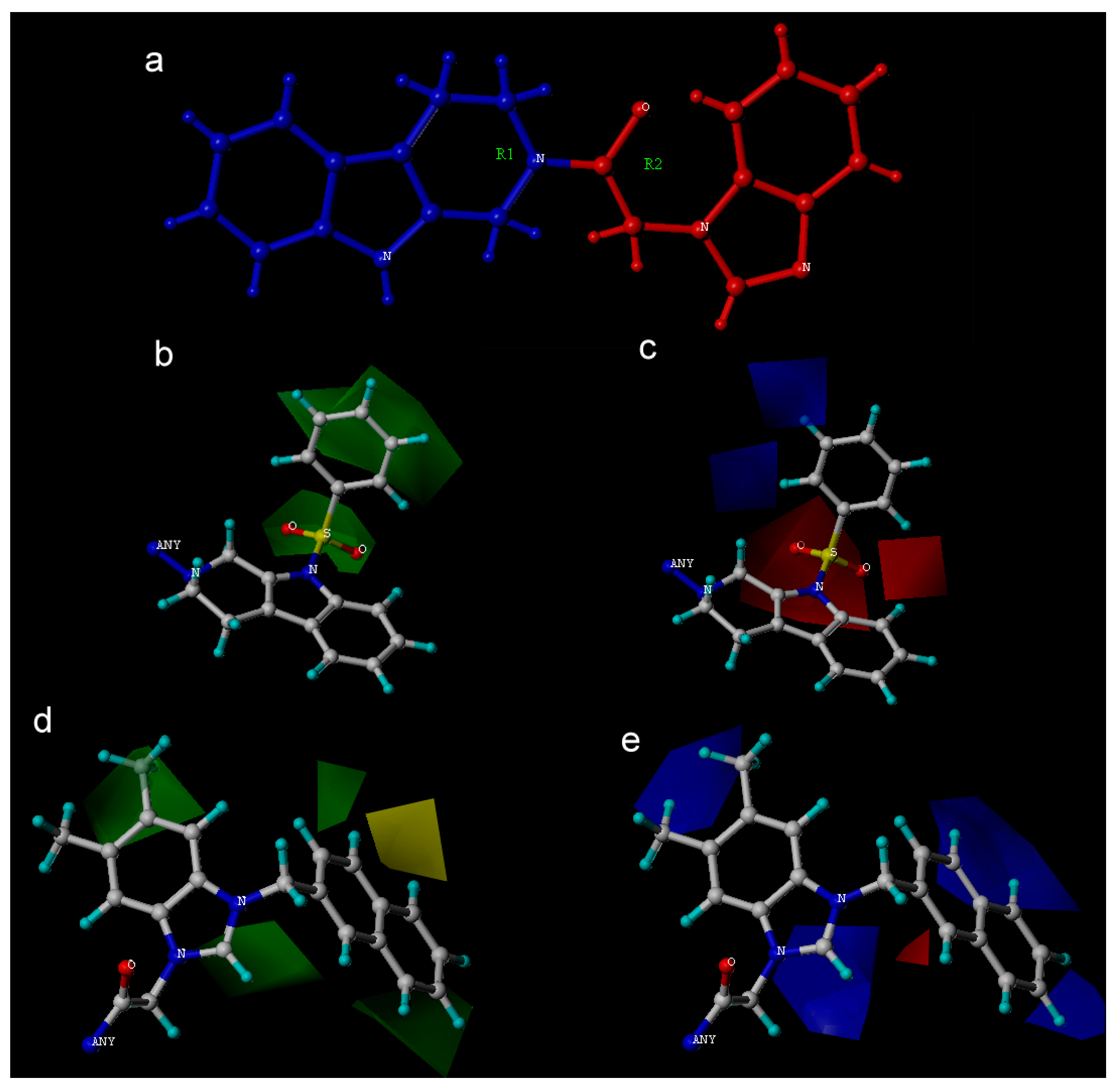
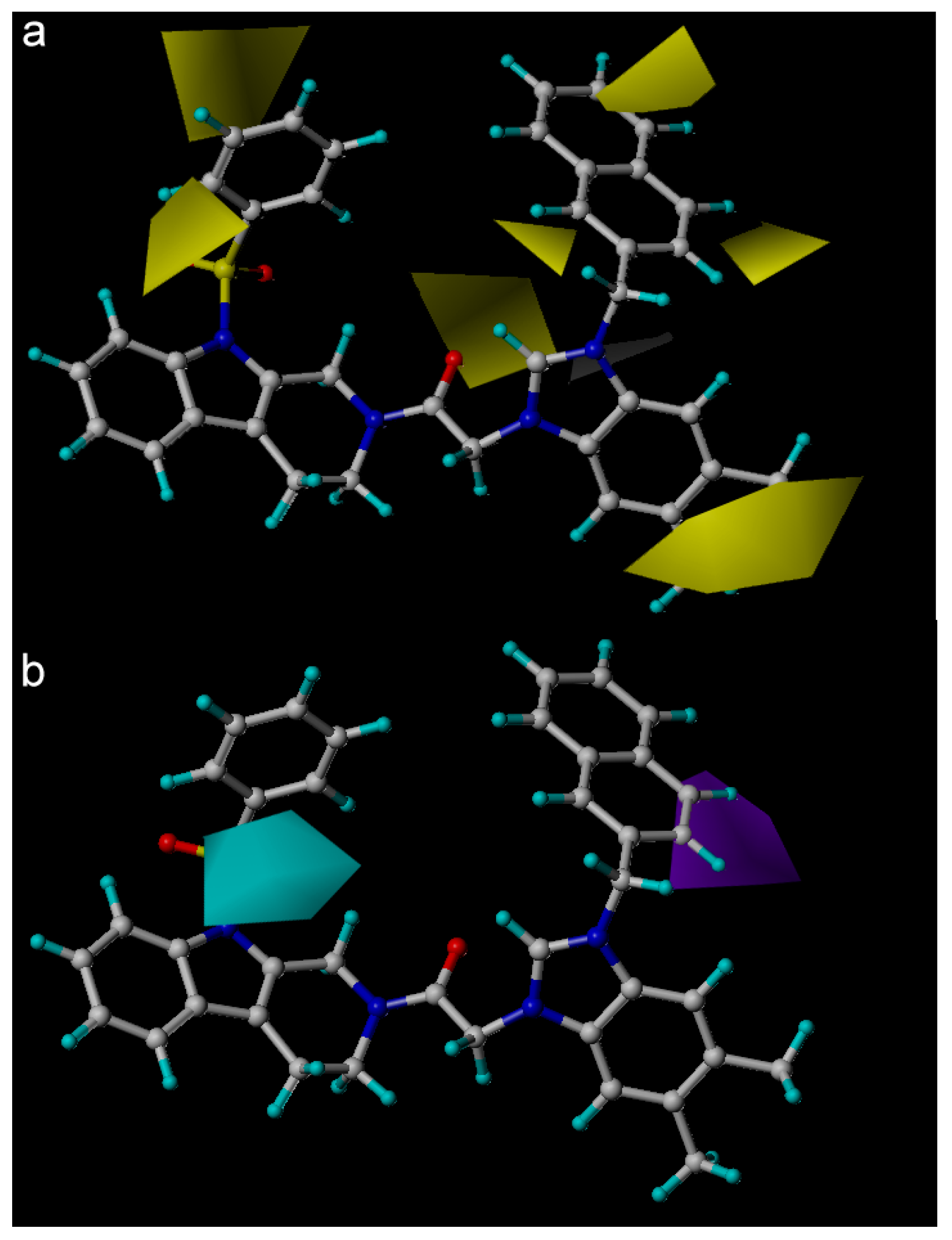
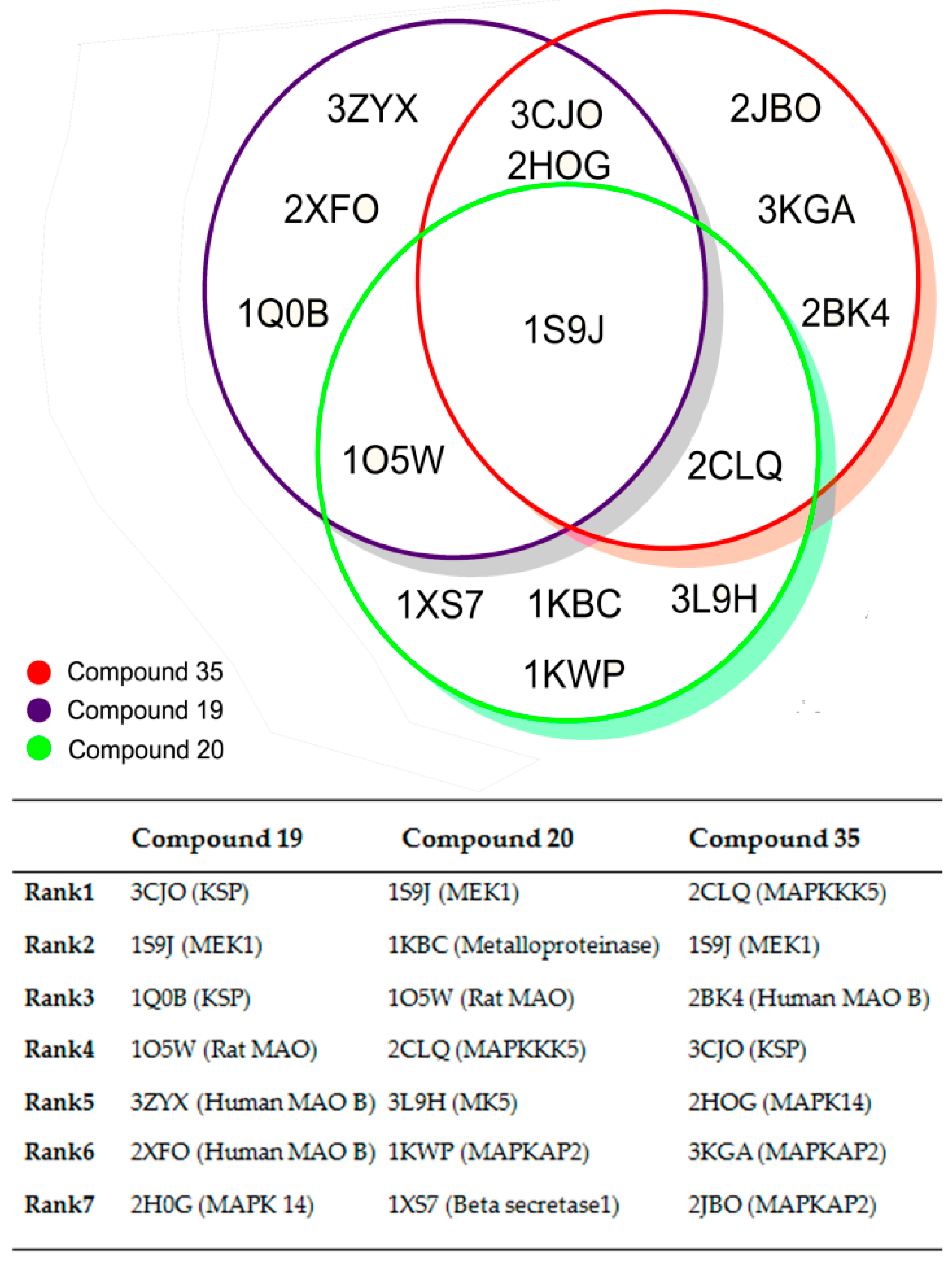
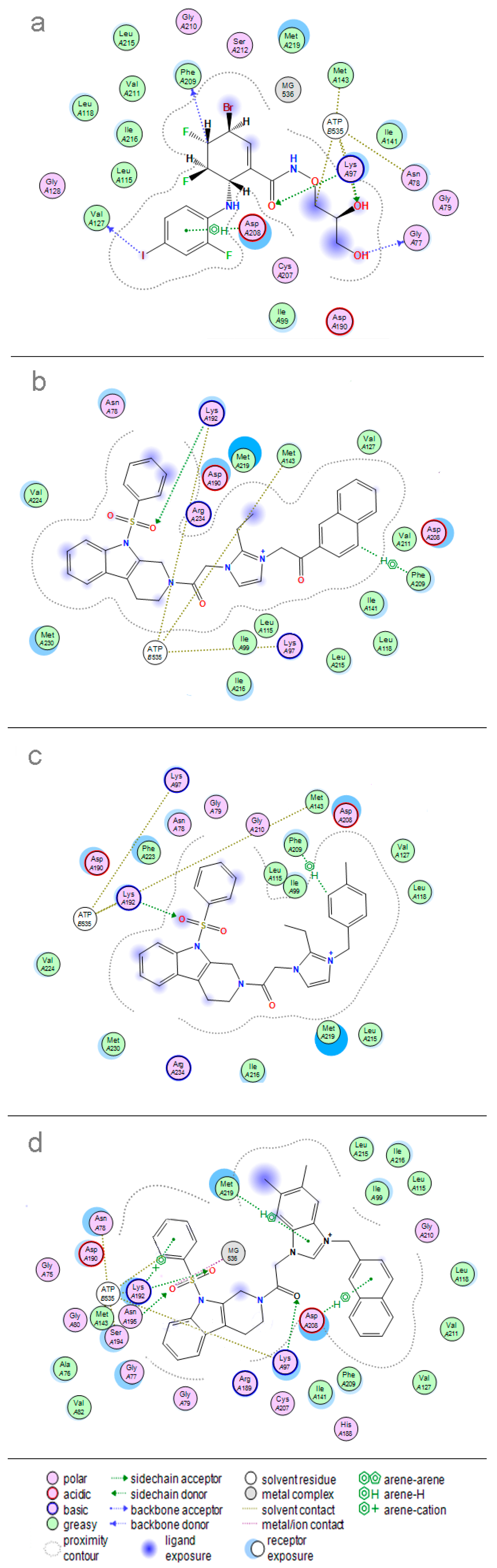
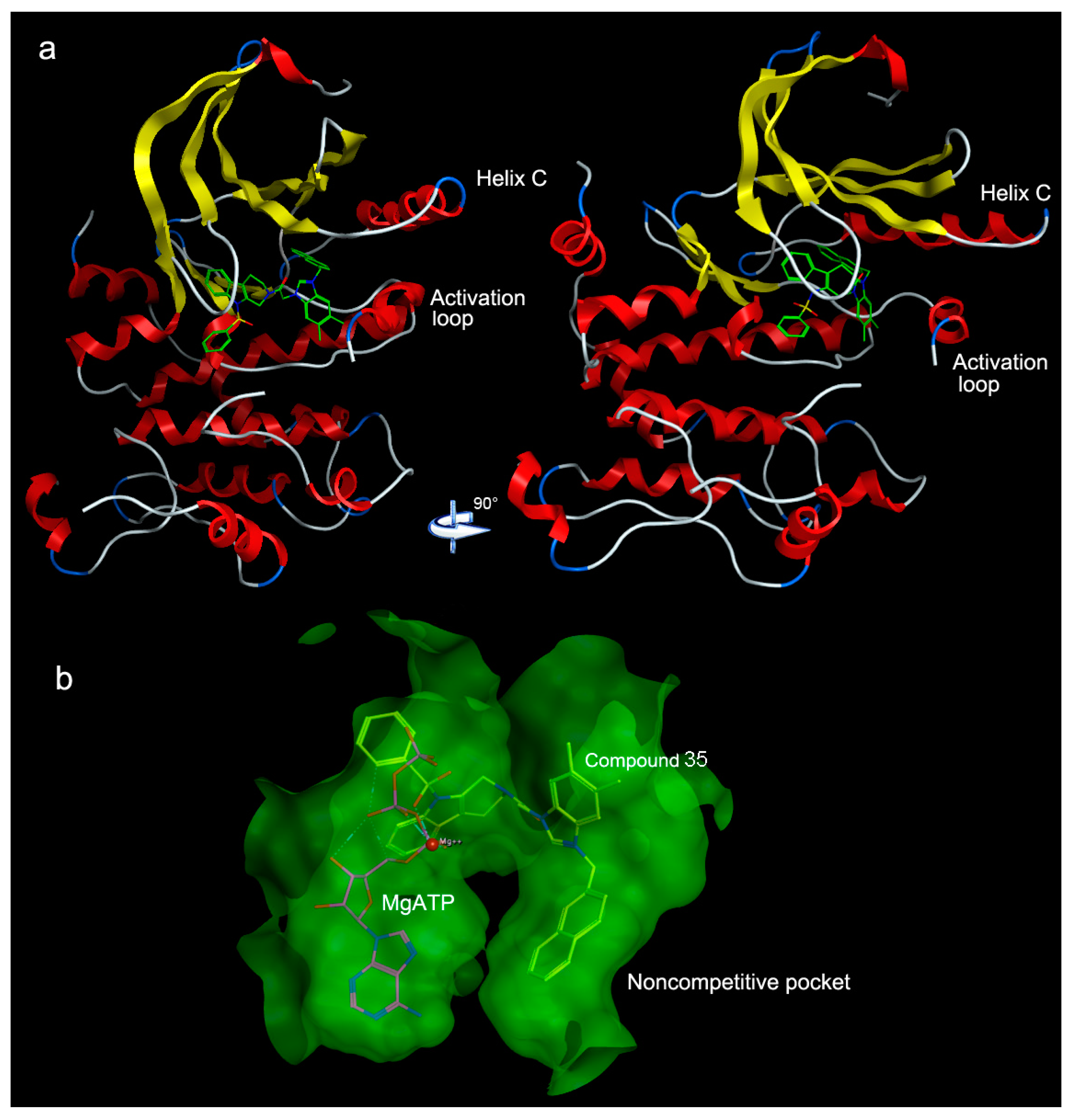
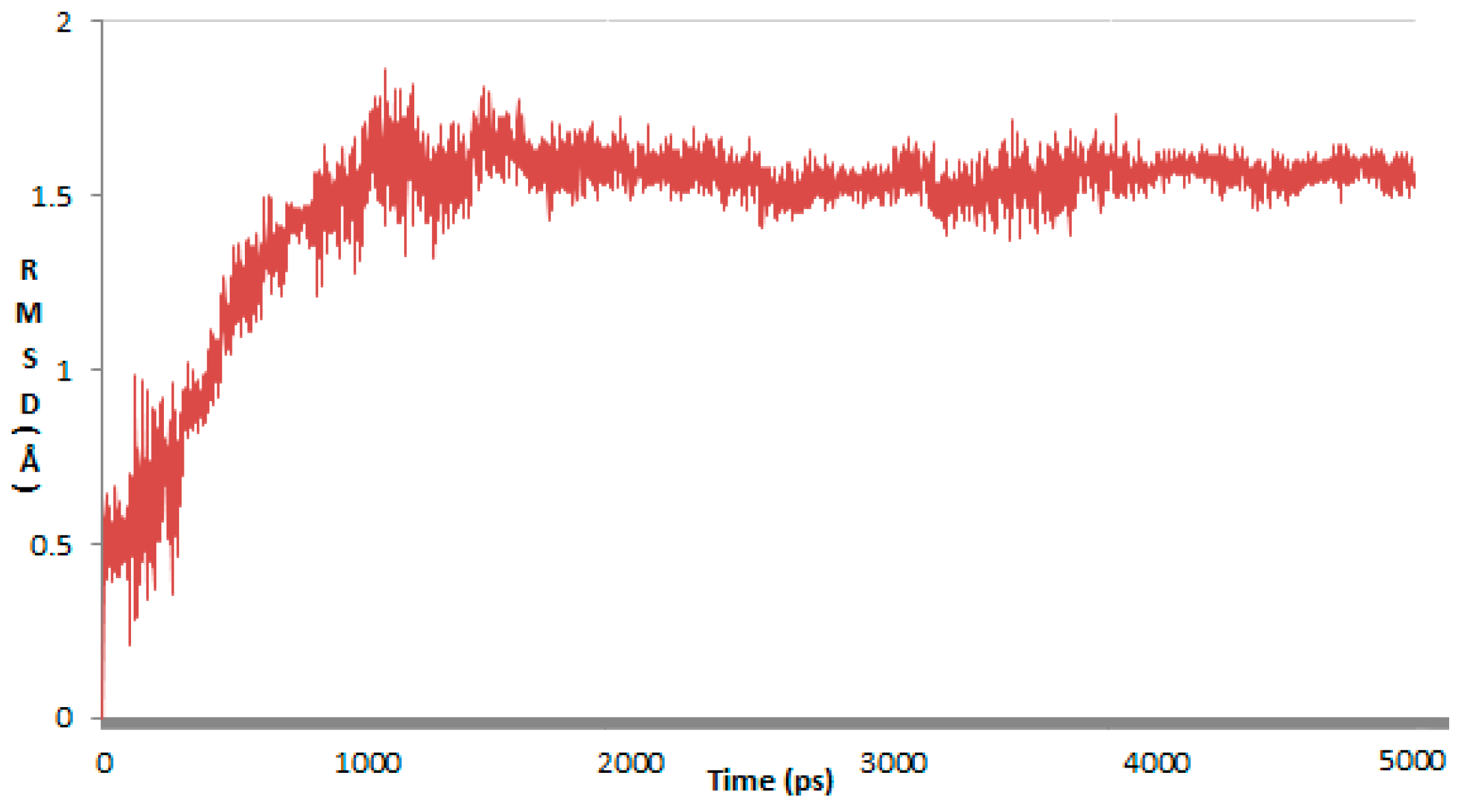
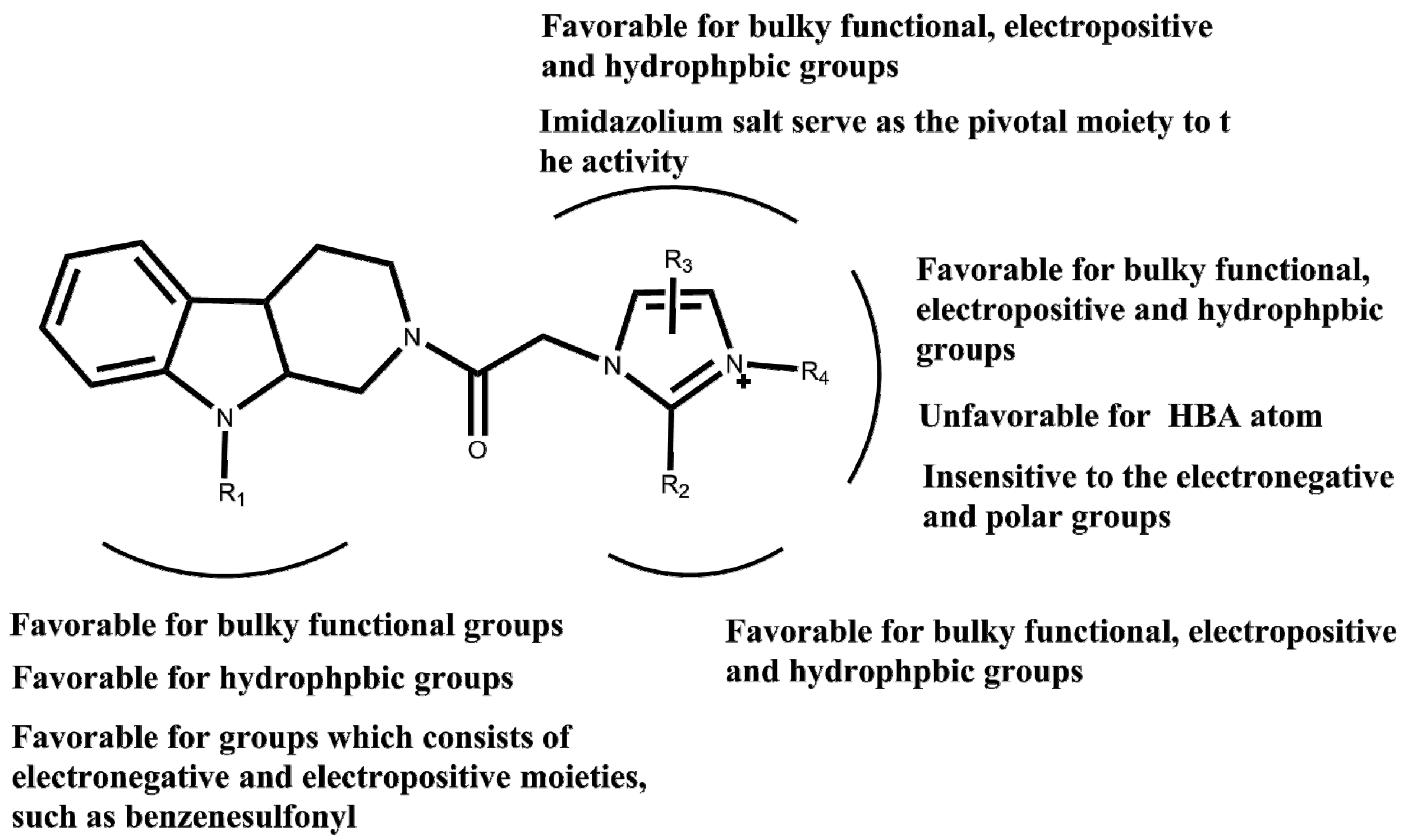
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO Website. Available online: http://www.who.int (assessed on 16 February 2017).
- Yan, Y.; Caruso, F. Particle carriers for combating multidrug resistant cancer. ACS Nano 2013, 7, 9512–9517. [Google Scholar] [CrossRef] [PubMed]
- Cholewinski, G.; Dzierzbicka, K.; Koodziejczyk, M.M. Natural and syntheticacridines/acridones as antitumor agents: Their biological activities and methods of synthesis. Pharmacol. Rep. 2011, 63, 305–335. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Z.F.; Deng, G.G.; Chen, W.; Li, M.; Yang, L.J. Synthesis and antitumor activity of novel n-substituted tetrahydro-β-carboline imidazolium salt derivatives. Org. Biomol. Chem. 2016, 14, 9423–9430. [Google Scholar] [CrossRef] [PubMed]
- Rook, Y.; Schmidtke, K.U.; Gaube, F.; Schepmann, D.; Wünsch, B.; Heilmann, J. Bivalent β-carbolines as potential multitarget anti-alzheimer agents. J. Med. Chem. 2010, 53, 3611. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.S.; Koskinen, A.M.P. ChemInform Abstract: Harmicine, a Tetracyclic Tetrahydro-β-carboline: From the First Synthetic Precedent to Isolation from Natural Sources to Target-Oriented Synthesis (Review). Chem. Heterocycl. Compd. 2015, 50, 1367–1387. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Lazar, C.; Crandall, I.E.; Szarek, W.A. Anti-plasmodium activity of imidazolium and triazolium salts. Bioorg. Med. Chem. 2010, 18, 6184–6196. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, C.; Barresi, V.; Berellini, G.; Musumarra, G. Design and synthesis of trans, 2-(furan-2-yl)vinyl heteroaromatic iodides with antitumour activity. Bioorg. Med. Chem. 2008, 16, 4150. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yu, L.Q.; Jiang, C.; Yang, L.; Wu, W.T.; You, Q.D. Discovery of tetrahydro-β-carbolines as inhibitors of the mitotic kinesin ksp. Bioorg. Med. Chem. 2010, 18, 4167. [Google Scholar] [CrossRef] [PubMed]
- Radulescu, M.C.; Bucur, M.P.; Bucur, B.; Radu, G.L. Biosensor based on inhibition of monoamine oxidases A and B for detection of β-carbolines. Talanta 2015, 137, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.I.; Meyers, M.J.; Anderson, D.R.; Hegde, S.; Mahoney, M.W.; Vernier, W.F. Novel tetrahydro-β-carboline-1-carboxylic acids as inhibitors of mitogen activated protein kinase-activated protein kinase 2 (mk-2). Bioorg. Med. Chem. Lett. 2007, 17, 4657–4663. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Zheng, B.L.; He, K.; Zheng, Q.Y. Imidazole alkaloids from lepidium meyenii. J. Nat. Prod. 2003, 66, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper Server: A web server for potential drug target identification via pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, N.; Premkumar, R.E. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Ras/raf signaling and emerging pharmacotherapeutic targets. Expert Opin. Pharmacother. 2002, 3, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.F.; Guan, K. Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J. Biol. Chem. 1993, 268, 11435–11439. [Google Scholar] [PubMed]
- Sale, M.J.; Cook, S.J. Intrinsic and acquired resistance to MEK1/2 inhibitors in cancer. Biochem. Soc. Trans. 2014, 42, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; Licinio, R.; Carr, B.I.; Di, L.A.; Barone, M. Mek 1/2 inhibitors in the treatment of hepatocellular carcinoma. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Yang, X.D.; Zeng, X.H.; Xu, X.L.; Zhang, G.L.; Zhang, H.B. Synthesis and cytotoxic activities of novel hybrid compounds of imidazole scaffold-based 2-substituted benzofurans. Rsc. Adv. 2012, 2, 4612–4615. [Google Scholar] [CrossRef]
- Pauli, I.; Timmers, L.F.; Caceres, R.A.; Soares, M.B.; de Azevedo, W.F., Jr. In silico and in vitro: Identifying new drugs. Curr. Drug Targets 2008, 9, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Ohren, J.F.; Chen, H.; Pavlovsky, A.; Whitehead, C.; Zhang, E.; Kuffa, P. Structures of human map kinase kinase 1 (mek1) and mek2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192. [Google Scholar] [CrossRef] [PubMed]
- Lammers, A.; Weekes, C.D. Refametinib: Dual MEK 1/2 inhibitor oncolytic. Drug Future 2015, 38, 19–26. [Google Scholar] [CrossRef]
- Lim, H.Y.; Heo, J.; Choi, H.J.; Lin, C.Y.; Yoon, J.H.; Hsu, C. A phase II study of the efficacy and safety of the combination therapy of the mek inhibitor refametinib (bay 86–9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5976–5985. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (comsia) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Bruno, O.; Fossa, P. Docking-based CoMFA and CoMSIA analyses of tetrahydro-β-carboline derivatives as type-5 phosphodiesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D. Topomer CoMFA: A design methodology for rapid lead optimization. J. Med. Chem. 2003, 46, 374. [Google Scholar] [CrossRef] [PubMed]
- Virupaksha, B.; Alpana, G. CoMFA QSAR models of camptothecin analogues based on the distinctive SAR features of combined ABC, CD and E ring substitutions. Comput. Biol. Med. 2012, 42, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, G.; Gong, J.; Liu, X.; Li, H. Enhancing the Enrichment of Pharmacophore-Based Target Prediction for the Polypharmacological Profiles of Drugs. J. Chem. Inf. Model. 2016, 56, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Guo, S. Concise applications of molecular modeling software-MOE. Comput. Appl. Chem. 2005, 22. [Google Scholar] [CrossRef]
- Andersson, C.D.; Thysell, E.; Lindstrom, A.; Bylesjo, M.; Raubacher, F.; Linusson, A. A Multivariate Approach to Investigate Docking Parameters' Effects on Docking Performance. J. Chem. Inf. Model. 2007, 47, 1673. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E. Scalable molecular dynamics with namd. J. Comput. Chem. 2012, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Harrison, C.B.; Schulten, K.; Mccammon, J.A. Implementation of Accelerated Molecular Dynamics in NAMD. Comput. Sci. Discov. 2011, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Strahan, G.D.; Keniry, M.A.; Shafer, R.H. NMR structure refinement and dynamics of the K+-[d(G3T4G3)]2 quadruplex via particle mesh Ewald molecular dynamics simulations. Biophys. J. 1998, 75, 968–981. [Google Scholar] [CrossRef]
- Andersena, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistical Parameters | Topomer | CoMSIA |
|---|---|---|
| q2 a | 0.700 | 0.615 |
| r2 b | 0.954 | 0.897 |
| ONC c | 5 | 4 |
| SEE d | 0.058 | 0.124 |
| F e | 196.238 | - |
| rpred2 f | 0.914 | 0.852 |
| Fraction of Field contributions g | ||
| Steric | - | 0.245 |
| Electrostatic | - | 0.317 |
| Hydrophobic | - | 0.078 |
| H-bond acceptor | - | 0.124 |
| H-bond donor | - | - |
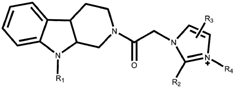
| Compound | R1 | R2 | R3 | R4 | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | H | H | Phenyl | - | >40 |
| 2 | Benzenesulfonyl | H | - | - | >40 |
| 3 * | Benzenesulfonyl | Ethyl | - | - | 17.81 |
| 4 * | Benzenesulfonyl | H | Phenyl | - | 36.37 |
| 5 | Benzenesulfonyl | H | 3,5-dimethyl-phenyl | >40 | |
| 6 | H | H | - | benzoyl | >40 |
| 7 * | H | H | - | 2-naphthoyl | >40 |
| 8 | H | H | Phenyl | benzoyl | >40 |
| 9 * | H | H | Phenyl | 4-methoxy benzoyl | >40 |
| 10 | H | H | Phenyl | 4-bromo benzoyl | 21.81 |
| 11 | H | H | Phenyl | 2-naphthoyl | 11.09 |
| 12 * | H | H | Phenyl | 4-bromo benzoyl | 10.68 |
| 13 | H | H | Phenyl | 4-nitro benzoyl | >40 |
| 14 | H | H | Phenyl | Naphthyl-2-methyl | 3.54 |
| 15 | Benzenesulfonyl | H | - | benzoyl | >40 |
| 16 | Benzenesulfonyl | Ethyl | - | 2-naphthoyl | 3.32 |
| 17 | Benzenesulfonyl | Ethyl | - | benzoyl | >40 |
| 18 | Benzenesulfonyl | Ethyl | - | 4-bromo benzoyl | 11.87 |
| 19 | Benzenesulfonyl | Ethyl | - | 2-naphthoyl | 2.47 |
| 20 | Benzenesulfonyl | Ethyl | - | 4-benzyl methyl | 2.56 |
| 21 * | Benzenesulfonyl | Ethyl | - | Naphthyl-2-methyl | 2.77 |
| 22 | Benzenesulfonyl | - | Phenyl | benzoyl | 14.39 |
| 23 | Benzenesulfonyl | - | Phenyl | 4-bromo benzoyl | 10.61 |
| 24 | Benzenesulfonyl | - | Phenyl | 4-methoxy benzoyl | 3.97 |
| 25 | Benzenesulfonyl | - | Phenyl | 2-naphthoyl | 3.24 |
| 26 * | Benzenesulfonyl | - | Phenyl | 4-benzyl methyl | 3.04 |
| 27 | Benzenesulfonyl | - | Phenyl | 4-nitrobenzol methyl | 13.58 |
| 28 | Benzenesulfonyl | - | Phenyl | Naphthyl-2-methyl | 4.34 |
| 29 | Benzenesulfonyl | - | Phenyl | benzoyl | 10.18 |
| 30 | Benzenesulfonyl | - | 3,5-dimethyl-phenyl | 4-bromo benzoyl | 3.75 |
| 31 | Benzenesulfonyl | - | 3,5-dimethyl-phenyl | 4-methoxy benzoyl | 3.39 |
| 32 | Benzenesulfonyl | - | 3,5-dimethyl-phenyl | 2-naphthoyl | 3.08 |
| 33 | Benzenesulfonyl | - | 3,5-dimethyl-phenyl | 4-benzyl methyl | 4.30 |
| 34 | Benzenesulfonyl | - | 3,5-dimethyl-phenyl | 4-nitro-4-benzyl | 12.81 |
| 35 | Benzenesulfonyl | - | 3,5-dimethyl-phenyl | Naphthyl-2-methyl | 2.61 |
Sample Availability: Samples of the compounds are not available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Wang, M.; Li, X.; He, X.; Cao, C.; Meng, F. Determination of Structural Requirements of N-Substituted Tetrahydro-β-Carboline Imidazolium Salt Derivatives Using in Silico Approaches for Designing MEK-1 Inhibitors. Molecules 2017, 22, 1020. https://doi.org/10.3390/molecules22061020
Liang J, Wang M, Li X, He X, Cao C, Meng F. Determination of Structural Requirements of N-Substituted Tetrahydro-β-Carboline Imidazolium Salt Derivatives Using in Silico Approaches for Designing MEK-1 Inhibitors. Molecules. 2017; 22(6):1020. https://doi.org/10.3390/molecules22061020
Chicago/Turabian StyleLiang, Jingwei, Mingyang Wang, Xinyang Li, Xin He, Chong Cao, and Fanhao Meng. 2017. "Determination of Structural Requirements of N-Substituted Tetrahydro-β-Carboline Imidazolium Salt Derivatives Using in Silico Approaches for Designing MEK-1 Inhibitors" Molecules 22, no. 6: 1020. https://doi.org/10.3390/molecules22061020